
Fascin1-Dependent Filopodia are Required for Directional Migration of a Subset of Neural Crest Cells
During vertebrate embryogenesis, neural crest (NC) cells migrate extensively along stereotypical migration routes and differentiate into diverse derivatives, including the craniofacial skeleton and peripheral nervous system. While defects in NC migration underlie many human birth defects and may be coopted during cancer metastasis, the genetic pathways controlling directional NC migration remain incompletely understood. Filopodia protrusions are thought to act as “cellular antennae” that explore the environment for directional cues to ensure NC cells reach their correct location. To test this idea, we generated zebrafish fascin1a (fscn1a) mutants that have severe loss of filopodia. Surprisingly, we found that most NC cells migrate to their correct locations without robust filopodial protrusions. We found that fscn1a embryos have directional migration defects in a subset of NC cells, resulting in loss of specific craniofacial elements and peripheral neurons. Interestingly, these defects were only observed in ∼20% of fscn1a embryos, but were significantly enhanced by partial loss of the chemokine receptor Cxcr4a or disruption of the localized expression of its ligand Cxcl12b. Our data show that subsets of skeletal and neurogenic NC cells require filopodia to migrate and that fscn1a-dependent filopodia cooperate with chemokine signaling to promote directional migration of a subset of NC cells.
Published in the journal:
. PLoS Genet 11(1): e32767. doi:10.1371/journal.pgen.1004946
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004946
Summary
During vertebrate embryogenesis, neural crest (NC) cells migrate extensively along stereotypical migration routes and differentiate into diverse derivatives, including the craniofacial skeleton and peripheral nervous system. While defects in NC migration underlie many human birth defects and may be coopted during cancer metastasis, the genetic pathways controlling directional NC migration remain incompletely understood. Filopodia protrusions are thought to act as “cellular antennae” that explore the environment for directional cues to ensure NC cells reach their correct location. To test this idea, we generated zebrafish fascin1a (fscn1a) mutants that have severe loss of filopodia. Surprisingly, we found that most NC cells migrate to their correct locations without robust filopodial protrusions. We found that fscn1a embryos have directional migration defects in a subset of NC cells, resulting in loss of specific craniofacial elements and peripheral neurons. Interestingly, these defects were only observed in ∼20% of fscn1a embryos, but were significantly enhanced by partial loss of the chemokine receptor Cxcr4a or disruption of the localized expression of its ligand Cxcl12b. Our data show that subsets of skeletal and neurogenic NC cells require filopodia to migrate and that fscn1a-dependent filopodia cooperate with chemokine signaling to promote directional migration of a subset of NC cells.
Introduction
Directional cell migration is used by a variety of cell types throughout embryonic development, wound healing, and cancer metastasis [1]. Distinct chemo-attractive/repulsive mechanisms, involving cell surface receptors and their secreted ligands, are common mechanisms that guide cells to their correct locations. Filopodial protrusions at the leading edges of migrating cells are thought to facilitate directional migration by acting as cellular antennae that actively monitor the distribution of guidance molecules in the environment [2,3]. Indeed, disrupting filopodia dynamics reduces the directional efficiency of migrating cells in culture [4–6] and selectively restricts cell migration in hematopoietic and neuronal lineages in vivo [7–9]. In contrast, recent studies have shown filopodia are dispensable for endothelial tip cell guidance during angiogenesis [10,11]. These studies suggest that filopodia have unique functions in a subset of cell types and/or may act redundantly with other guidance mechanisms to promote cell migration in vivo, although the selective nature of these mechanisms remains largely unknown.
Neural crest (NC) development is a dramatic example of directional cell migration in vertebrate embryos. Following delamination from the neural tube, multipotent NC cells migrate extensively throughout the embryo along stereotypical pathways to form a variety of cell types [12]. Cranial NC cells that form the craniofacial skeleton migrate as collective cell populations from the developing midbrain and hindbrain into pharyngeal arches, while other cranial NC cells migrate less collectively to generate peripheral neurons and glia of the cranial nerves and pigment cells, among other derivatives [13]. Vagal/cardiac and trunk NC cells initially migrate along a medial pathway to form ganglia of the peripheral nervous system, as well as connective tissue of the heart and outflow tract. At later stages, single trunk NC cells migrate along a lateral pathway to produce pigment cells in the skin [12]. Thus, the mode of NC migration (collective versus individual) and pathway selection (medial versus lateral) correlates with axial position, time of delamination and ultimate fate. Coordination of these migration paths is essential for patterning the vertebrate body plan and defects in NC migration mechanisms are associated with congenital defects and pediatric cancers, collectively referred to as neurocristopathies [14].
Premigratory NC cells display a polarized morphology associated with Rho GTPase activation in retracting NC cell tails and Rac1 GTPase activation in the direction of cell migration, which promotes F-actin polymerization and formation of cell membrane protrusions, including lamellipodia and filopodia [15–17]. Filopodial protrusions are dynamic cellular processes; in vivo imaging of migrating NC cells shows that filopodia are rapidly generated in the direction of chemo-attractive cues but collapse when exposed to repulsive cues [18]. In addition, NC protrusions are evident during contact inhibition of locomotion and coattraction, behaviors associated with cell-cell repulsion and adhesion that are proposed to drive the overall direction of some collective NC streams [19,20]. Most, if not all, of these mechanisms implicate dynamic filopodia extension and retraction as essential mediators of the cellular behaviors observed during directional NC migration, however this has not been directly tested.
Fascin1 (Fscn1) is required for F-actin bundling, filopodia formation and migration in a variety of metazoan cell types [21]. Fscn1 is highly upregulated in aggressive tumors, where it promotes cell migration when overexpressed and blocks migration and invasion when inhibited [22,23]. In vivo studies in Drosophila and mouse have demonstrated requirements for Fscn1 during individual-cell migration of hemocytes, neuroblasts, dendritic cells and melanoblasts [8,9,24,25]. However, the role of Fscn1 in early vertebrate embryogenesis and collective cell migration in vivo remains unknown, due in part to the fact that it is not known if the first intron retroviral insertion allele of Fscn1 affects mRNA or protein expression in the early mouse embryo [25,26]. In addition, the molecular mechanism(s) by which Fscn1 promotes cell migration in vivo is still poorly understood, likely due to redundancy with other directional cell migration mechanisms.
To determine if Fscn1-dependent filopodia are required for NC cell migration, we generated TALEN-induced null mutations in zebrafish fscn1a, the only fascin gene expressed in zebrafish NC cells. Surprisingly, homozygous fscn1a null mutants have no defects in NC filopodia formation and are viable and fertile. Analysis of protein levels in oocytes and zygotic null fscn1a mutants reveals that Fscn1a protein is maternally deposited and remarkably stable (up to 10 days post fertilization), lasting throughout embryonic development and organogenesis and masking potential zygotic functions of fscn1a in NC migration. In contrast, maternal/zygotic fscn1a (fscn1a MZ) mutant embryos show severe defects in the number, length and dynamics of NC filopodia and selective defects in migration of a subset of cranial NC streams. The fscn1a null NC phenotypes are partially penetrant and often asymmetric, leading to the loss of single cartilage elements on one side of the face. fscn1a MZ mutants also have selective loss of NC-derived peripheral sympathetic and enteric neurons, but not dorsal root ganglia. Importantly, while depletion of residual filopodia in fscn1a null mutants with the F-actin polymerizing inhibitor Latrunculin B enhanced fscn1a-dependent NC derivative defects, most NC cells still migrated normally, showing that filopodia are largely dispensable for NC migration. We also demonstrate that fscn1a controls directional migration of the first cranial NC stream through interactions with the chemokine receptor chemokine (C-X-C motif) receptor 4a (cxcr4a) and its ligand chemokine (C-X-C motif) ligand 12b (cxcl12b, or sdf1b). Together, these data show that perdurance of stable maternal proteins can mask essential zygotic gene functions in organogenesis, and demonstrate differential requirements for filopodia in directional NC migration and subsequent NC derivative formation.
Results
fscn1a expression in migrating NC cells requires tfap2a
Fascin proteins are required for filopodia formation [21]. To determine which fascins in zebrafish might regulate filopodia during NC migration, we evaluated the spatiotemporal expression of fascin1a (fscn1a) and fascin1b (fscn1b) by whole-mount RNA in situ hybridization (ISH). Only fscn1a mRNA was detected in the NC (Fig. 1), whereas fscn1b expression was restricted to the telencephalon from 36 hpf onwards (S1 Fig.). Maternal fscn1a mRNA is ubiquitously expressed (Fig. 1A), but by 6 hpf (50% epiboly) is restricted to the involuting blastoderm margin (Fig. 1B). At 11 hpf, fscn1a is expressed in rhombomere 2 (r2) of the hindbrain and at lower levels in adjacent neural tube and along the neural plate border (Fig. 1C), where its expression partially overlaps with foxd3 in NC (Fig. 1D) [27]. During cranial NC migration (12–24 hpf), fscn1a is expressed in migrating NC streams (Fig. 1C) and co-localizes with the NC marker crestin (Fig. 1E) [28]. In 18 and 24 hpf embryos, fscn1a is expressed in spinal cord neurons and crestin+ trunk NC cells, as well as in somites and vasculogenic mesoderm (S2 Fig.).

In zebrafish, loss of foxd3 and tfap2a causes a complete absence of NC cells [29,30]. To confirm fscn1a expression in NC cells, we injected antisense morpholino (MO) oligonucleotides targeting foxd3 (foxd3MO) and/or tfap2a (tfap2aMO) and performed ISH to detect fscn1a mRNA (Fig. 1F). As expected, ablation of NC by co-injecting tfap2aMO and foxd3MO caused loss of fscn1a expression only in the dorsal neural tube, but not in other cell types in the embryo such as vasculogenic mesoderm, consistent with the hypothesis that fscn1a is expressed in NC. Injection of foxd3MO or tfap2aMO alone showed that the NC-specific expression pattern of fscn1a depends on tfap2a (Fig. 1F).
Maternal Fscn1a protein persists in fscn1a mutants throughout embryonic development
Having established fscn1a expression in the NC, we next wanted to determine if fscn1a was required for NC filopodia formation and cell migration. We generated several null alleles of fscn1a using first exon-targeted TALENs (S3A-S3C Fig.). The fscn1aΔ7, fscn1aΔ10, fscn1a+17 alleles all harbor frame-shift deletions in exon 1 that generate premature stop codons immediately upstream of the highly conserved F-actin binding site required for actin bundling (S3C-S3D Fig.) [31].
We incrossed fscn1a heterozygous adults to generate homozygous fscn1a zygotic (zyg) mutant embryos, which were viable with no obvious morphological abnormalities (Fig. 2A). As fscn1a is expressed maternally (Fig. 1A, S4 Fig.), we hypothesized that maternally deposited Fscn1a protein may mask essential early functions for zygotic fscn1a during development. We analyzed Fscn1a protein levels in fscn1a zyg embryos at 24, 48 and 72 hpf, and 10 days post fertilization (dpf) by immunoblot analysis (Fig. 2B). Unexpectedly, when NC cells are actively migrating (12–72 hpf), maternally deposited Fscn1a protein is present in fscn1a zyg embryos at levels comparable to wild type siblings and persists at moderate levels through 10 dpf (Fig. 2B). Thus, maternal Fscn1a protein lasts throughout all organogenesis stages, potentially masking roles for zygotic fscn1a in NC development.
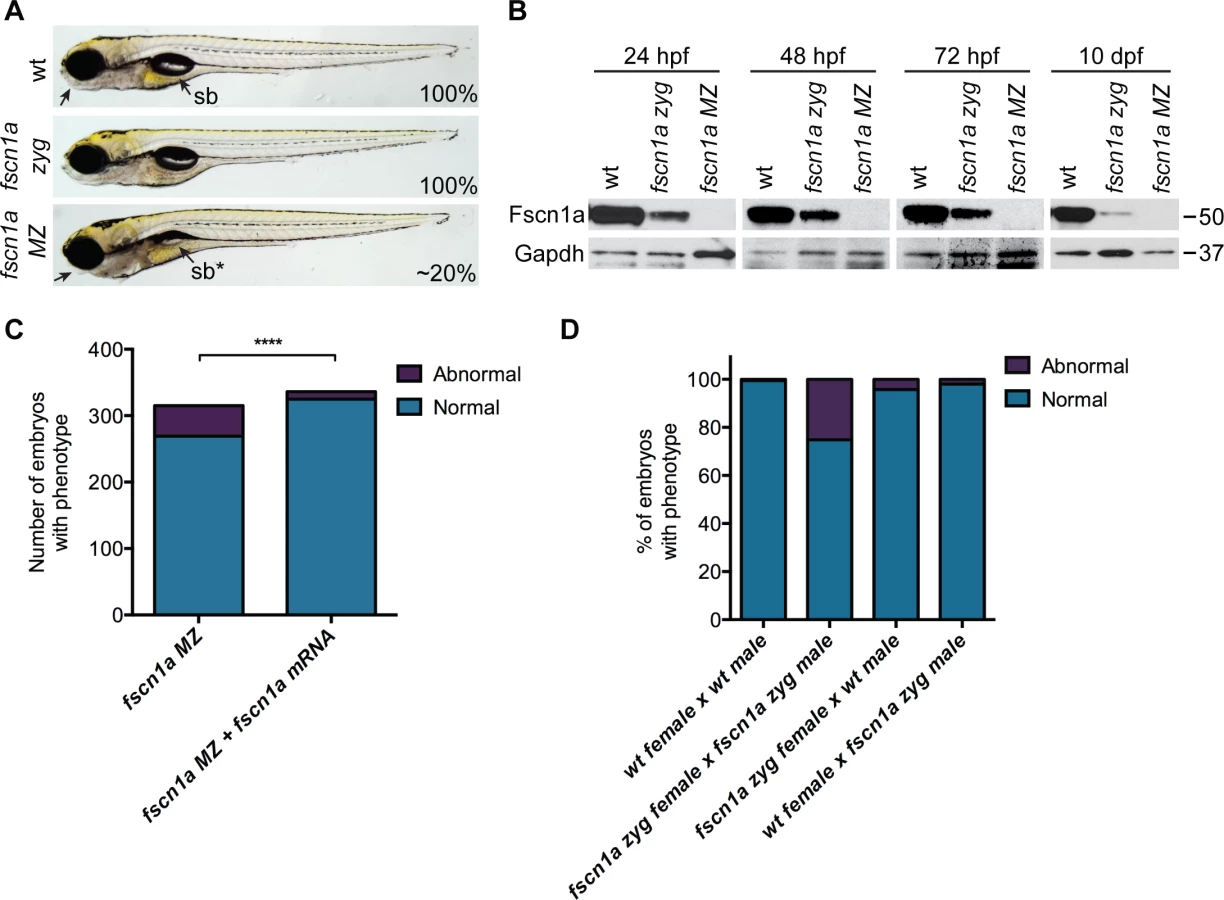
To reveal potential embryonic roles for zygotic fscn1a expression, we incrossed homozygous fscn1a zyg animals to generate maternal/zygotic fscn1a mutant embryos (fscn1a MZ, S3B Fig.). fscn1a MZ oocytes were viable and, following fertilization, embryos showed no obvious defects in cleavage or gastrulation. However, at 5 dpf fscn1a MZ embryos displayed gross morphological abnormalities in the jaw, heart, and swim bladder with partial penetrance ranging from 0–50%, with an average of ∼20% (Fig. 2A). The remaining fscn1a MZ embryos were morphologically normal and survived to adulthood, suggesting possible redundancy with other fscn zebrafish paralogs. Therefore, we analyzed the temporal expression of fscn1b, fscn2a, and fscn2b in fscn1a MZ mutants and observed no aberrant expression (e.g. upregulation) that might explain the partial penetrance (S5 Fig.). In addition, MO knockdown of all other fscn paralogs (which are not maternally expressed) did not affect the penetrance of the fscn1a MZ mutant phenotypes (S6 Fig.). Importantly, the fscn1a MZ morphological defects were fully rescued by injecting fscn1a mRNA (Fig. 2C). Combined, these data show that fscn1a function is essential for normal embryonic development, but is partially penetrant.
To determine the contribution of paternal (zygotic only) fscn1a expression to embryonic development, homozygous fscn1a zyg females were bred to wild type males. 96% of embryos derived from this cross did not display obvious morphological defects (Fig. 2D), demonstrating that paternal (zygotic) fscn1a expression is sufficient to rescue fscn1a MZ morphological defects, and that perdurance of maternal Fscn1a protein masks essential zygotic functions of fscn1a in embryogenesis.
fscn1a is required for filopodia formation and dynamics in NC cells
To determine if fscn1a is required for filopodia formation in NC cells, we analyzed Tg(sox10:rfpmb) and Tg(sox10:rfpmb); fscn1a MZ embryos in which sox10-expressing NC cells are labeled with a membrane-bound RFP, enabling visualization of membrane protrusions [32]. In 26 hpf wild type embryos, dynamic filopodial projections were visible at the leading edge of all cranial and trunk NC streams (Fig. 3A-C and S1 Movie). In contrast, filopodia at the leading edge of fscn1a MZ NC streams were significantly reduced in number, length, and change in length over time (Fig. 3A-D and S2 Movie). In addition, compared to wild type NC cells, the morphology of fscn1a MZ NC cells appeared flatter and less polarized (S1–S2 Movies). This phenotype was particularly evident in small cohorts of migrating trunk NC cells (Fig. 3C). Importantly, the penetrance of filopodia defects in fscn1a MZ embryos was 100% (n = 6 cranial, n = 6 trunk), in contrast to the partially penetrant morphological defects (Fig. 2A). These results show that, similar to other organisms, zebrafish fscn1a has an essential role in generating robust filopodia in vivo.

fscn1a is required for migration of a subset of cranial NC streams
We next asked if fscn1a-dependent filopodia were required for NC induction and migration. Examination of foxd3 mRNA expression by ISH at 11 hpf in fscn1a MZ embryos (Fig. 4A) revealed no difference between fscn1a MZ and wild type siblings, suggesting fscn1a is not required for NC induction. We next analyzed markers of NC migration. To examine early cranial NC migration, the expression pattern of sox10 mRNA was analyzed in wild type and fscn1a MZ embryos by ISH at 15 hpf. At this time point, ∼20% of fscn1a MZ embryos displayed abnormal NC migration, including a dispersion of the most anterior sox10+ NC cells over the yolk, suggesting a primary defect in the directional migration of the first cranial NC stream (Fig. 4B). To confirm this, we examined the expression pattern of dlx2a, which is expressed in migratory NC cells that contribute to the pharyngeal arches, at 18, 28 and 36 hpf. At all time points, ∼20% of fscn1a MZ embryos showed loss or reduction of dlx2a specifically in the mandibular arch, suggesting that a subset of cranial NC cells fail to migrate to the pharyngeal arches (Fig. 4C). In addition, dlx2a+ NC cells were observed outside of the normal stream boundaries in ∼20% of fscn1a MZ embryos, often in an asymmetric manner. At 36 hpf, dlx2a+ NC cells condense within the mandibular arch primordia in wild type embryos. In contrast, dlx2a+ NC cells in ∼20% of fscn1a MZ embryos remained more dispersed over the yolk and anterior to the normal location of the mandibular primordium (Fig. 4C), showing that fscn1a function is required in a subset of NC streams to direct them to the correct location.

To determine if the fscn1a-dependent defects in directional NC migration were due to abnormal cell migration behaviors, we examined cranial NC streams in Tg(sox10:rfpmb) and Tg(sox10:rfpmb); fscn1a MZ embryos by confocal time-lapse imaging. In wild type embryos we observed that NC streams destined for the pharyngeal arches are tightly associated and dynamic cellular protrusions are predominant in cells at the leading edges and sides of the streams (S3 Movie). Overall, we observed no obvious differences in individual cell behaviors at the leading edges of NC streams between wild type and fscn1a MZ cranial NC streams (n = 20 embryos, S3–S4 Movies), although occasionally we observed a cell at the leading edge move away from the stream in the fscn1a MZ mutants (S4 Movie). These data, together with data from Fig. 4, suggest that fscn1a-dependent filopodia are not required for individual cell migration behaviors, like contact-dependent adhesion, but instead are required for directional migration of a subset of collective NC streams.
A subset of NC derivatives are lost in fscn1a MZ mutants
We next sought to understand the developmental consequence of fscn1a-dependent filopodia loss and aberrant cell migration behaviors. NC cells give rise to many cell types, including the craniofacial skeleton, heart outflow tract, neurons and glia of the peripheral nervous system, and pigment-producing melanophores [12]. The most prominent phenotype in fscn1a MZ embryos is abnormal craniofacial morphology in approximately 20% of embryos (Fig. 5A). The pharyngeal skeleton is derived from cranial NC cells that migrate as three distinct streams from the dorsal region of the hindbrain to the mandibular (stream 1), hyoid (stream 2), and branchial (stream 3) arches, with stream 3 in zebrafish further subdividing into five separate streams to form the gill arches [13,33]. In fscn1a MZ embryos stained with Alcian blue, a specific loss of elements derived from the mandibular arch was evident in ∼20% of 5 dpf embryos (Fig. 5B). Dissection of Alcian blue-stained pharyngeal cartilages, as well as live imaging of sox10+ pharyngeal cartilages in 5 dpf Tg(sox10:rfpmb); fscn1a MZ embryos, revealed symmetric or asymmetric loss of first stream derivatives, including Meckel’s and palatoquadrate cartilages (Fig. 5C, S7 Fig.). Among the ∼20% of fscn1a MZ embryos displaying abnormal craniofacial morphology, the distribution of symmetric, left-sided asymmetric, and right-sided asymmetric defects was stochastic (S7 Fig.). Defects in hyoid and branchial arch elements were never observed, consistent with normal migration of these NC streams into the pharyngeal arches. These results show that fscn1a-dependent filopodia are required for the normal development of a subset of NC-derived craniofacial cartilages, consistent with the selective defects we observed in cranial NC migration (Fig. 4B,C).

fscn1a null mutants do not completely lack filopodia, suggesting that the shorter, less abundant residual filopodia in these mutants still function to promote NC migration. To test this, we treated embryos with the F-actin polymerization inhibitor Latrunculin B (Lat. B), which at low doses (80 ng/ml) has previously been shown to selectively inhibit filopodia formation but not other F-actin structures (lamellipodia, cortical actin) in zebrafish endothelial cells [10]. At 26 hpf, wild type Tg(sox10:rfpmb) embryos treated with low doses of Lat. B displayed a severe reduction in the number and length of filopodia at the leading edge of cranial NC streams (Fig. 6A,B). Compared to DMSO-treated Tg(sox10:rfpmb); fscn1a MZ embryos, low dose Lat. B-treated Tg(sox10:rfpmb); fscn1a MZ embryos displayed a further reduction in NC cell filopodia number and length at 26 hpf, presumably through effects on residual fscn1a-independent filopodia (Fig. 6A,B). Despite reducing filopodia, Lat. B did not disrupt the development of NC-derived craniofacial elements in wild type embryos (Fig. 6C,D). Treatment of fscn1a MZ embryos with low dose Lat. B had a modest but significant effect on the penetrance of fscn1a-dependent craniofacial defects, but did not result in more severe phenotypes (e.g. loss of posterior NC-derived craniofacial cartilages) (Fig. 6C,D). In all, these data suggest that the selective defects in NC migration observed in fscn1a MZ mutant embryos are unlikely to be due to residual fscn1a-independent filopodia.

The endoderm of the pharyngeal pouches plays important roles in patterning and differentiation of skeletogenic NC cells within the arches [34]. To determine if defects in pharyngeal pouch development contribute to the migration and craniofacial phenotypes observed in fscn1a MZ embryos, we examined pharyngeal endoderm at 30 hpf in wild type and fscn1a MZ embryos by whole-mount ISH but found no obvious defects in patterning of nkx2.3+ pharyngeal endoderm (S8 Fig.). These results are consistent with the craniofacial defects in fscn1a MZ embryos resulting from abnormal NC cell migration in the mandibular stream, and supported by the observation that abnormal migration of sox10-positive NC cells is the first observed defect in fscn1a MZ embryos (Fig. 4B), which occurs approximately 4 hours before endodermal pouches are formed.
To determine if NC-derived peripheral neurons derived from the vagal NC stream are present in fscn1a MZ embryos, we examined the expression of tyrosine hydroxylase (th) and dopamine β-hydroxylase (dbh), markers of noradrenergic and dopaminergic neurons. At 3 dpf, NC-derived th+ sympathetic neurons were severely reduced in ∼20% of fscn1a MZ embryos (Fig. 7A). Visualization of sympathetic ganglia in 3 dpf Tg(dbh:gfp) embryos confirmed a reduction in dbh+ sympathetic neurons and revealed a failure of existing dbh+ sympathetic neurons to condense into ganglia (Fig. 7B). We also analyzed the development of enteric neurons, which are derived from vagal NC cells. At 3 dpf, ∼20% of fscn1a MZ embryos displayed a significant reduction in phox2b+ enteric neurons relative to wild type embryos (Fig. 7C). Interestingly, formation of trunk NC-derived dorsal root ganglia (DRG) was unaffected in 5 dpf fscn1a MZ embryos (Fig. 7D). Melanophore pigment cells were also unaffected in 5 dpf fscn1a MZ embryos (Fig. 7E). In all, these results show that fscn1a is required for the development of a subset of NC lineages, with the most severe defects observed in derivatives of the cranial/vagal NC, which migrate collectively.

fscn1a cooperates with cxcr4a/cxcl12b to promote directional migration of cranial NC cells
Fscn1a is required for filopodia formation in all cranial NC streams, however defects in cell migration appear to be restricted to the first NC stream in fscn1a MZ embryos. This suggests that the few remaining filopodia in fscn1a MZ NC are sufficient to promote migration and/or additional cell migration mechanisms compensate for loss of chemoreception, adhesion, and/or motility in these cells. To identify cooperating mechanisms that regulate NC migration with fscn1a, we took advantage of the partially penetrant NC defects in fscn1a MZ embryos, as it represents a sensitized genetic background to identify interacting pathways. We took a candidate approach and focused on the chemokine receptor cxcr4a and its ligand cxcl12b (sdf1b). The cxcr4/cxcl12 signaling axis regulates the directional migration of a variety of cell types [35,36] and has been previously demonstrated to regulate NC migration and development of NC-derived craniofacial cartilages and sympathetic ganglia [37,38]. Previous work in zebrafish demonstrated that cxcr4a is expressed in migratory cranial NC streams, while the cxcl12b ligand is expressed in pharyngeal endoderm, the target tissue of directionally migrating cranial NC cells [37]. Ectopic expression of cxcr4a or cxcl12b mRNA in zebrafish embryos uncouples cxcr4a/cxcl12b-dependent directional migration by randomizing the direction of the chemokine guidance signal to surrounding cells, thus causing cranial NC cells to migrate outside of their normal boundaries, resulting in loss of craniofacial cartilages [37]. Importantly, the craniofacial defects caused by cxcr4a or cxcl12b overexpression closely resemble the defects observed in fscn1a MZ embryos, including asymmetric loss of mandibular arch elements [37]. We first confirmed that Cxcr4a-GFP localizes to fscn1a-dependent filopodia in Tg(sox10:rfpmb) cranial NC cells (Fig. 8A). To determine if fscn1a cooperates with cxcr4a/cxcl12b to regulate directional NC migration, we ectopically expressed cxcl12b ligand in all surrounding cells or depleted cxcr4a receptor in NC cells by injecting wild type and fscn1a MZ embryos with subthreshold doses of cxcl12b mRNA or cxcr4aMO, respectively. NC cell filopodia were unaffected by misexpression of cxcl12b in 26 hpf Tg(sox10:rfpmb) and Tg(sox10:rfpmb); fscn1a MZ embryos, while low doses of cxcr4aMO reduced the length, but not number of filopodia in both Tg(sox10:rfpmb) and Tg(sox10:rfpmb); fscn1a MZ embryos (Fig. 8B,C). Low doses of cxcl12b mRNA or cxcr4aMO caused no defects in craniofacial cartilage formation (cxcl12b mRNA, 1/104) or mild defects in 20% (cxcr4aMO, 10/51) of wild type embryos (Fig. 8D-E). As previously observed, ∼20% (27/165) of fscn1a MZ embryos display defects in craniofacial development. Strikingly, the penetrance of this phenotype was increased to 40% (48/123) in fscn1a MZ embryos injected with cxcl12b mRNA or 75% (95/127) in fscn1a MZ embryos injected with cxcr4aMO (Fig. 8D-E, S9A Fig.). In addition, cxcr4a morphant/fscn1a MZ embryos displayed more severe morphological defects at 5 dpf (Fig. 8D). Importantly, injection of a control mismatch morpholino (mmMO) had no effect on the penetrance of craniofacial defects in fscn1a MZ embryos (Fig. 8E). Whole-mount ISH for th in 4 dpf embryos revealed that fscn1a and cxcr4a also function synergistically to promote development of the sympathetic ganglia (S9B-S9C Fig.). These genetic results suggest fscn1a functions in NC cells to mediate cxcr4a/cxcl12b chemokine signaling and may explain the partial penetrance of the fscn1a MZ NC phenotypes (see Discussion).

Discussion
Coordinated migration of NC cells is essential for patterning the body plan of the vertebrate embryo. Multiple guidance mechanisms are employed by NC cells to ensure that they migrate to precise locations throughout the embryo, including cell-cell adhesion, chemo-attraction/repulsion and physical barriers [17,39,40]. A fundamental feature of the chemo-attraction/repulsion mechanism is the ability to sense the environment for guidance molecules, which is proposed to act through actin-rich filopodial protrusions [3,41]. In this study we generated zebrafish fscn1a null mutants to determine the relative contribution of fscn1a-dependent filopodia in patterning different NC migration pathways and formation of derivatives. Elimination of both zygotic and maternal fscn1a caused severe loss of filopodia in all migrating NC cells. Unexpectedly, we found that most NC cells migrate normally in fscn1a MZ mutants, suggesting that robust, long and numerous filopodia are not required for NC chemotaxis, at least in a subset of NC streams. Indeed, when wild type or fscn1a MZ mutants were treated with the F-actin depolymerizing drug Latrunculin B (Lat. B), which further depleted any residual filopodia, most NC cells still migrated normally. Instead, only a subset of collectively migrating NC cells show defects in directional cell migration in fscn1a MZ or fscn1a MZ/Lat. B-treated embryos: the NC cells that form the first pharyngeal arch and the peripheral sympathetic and enteric nervous systems. Disruption of the Cxcr4a/Cxcl12b signaling pathway significantly enhanced the severity of NC cell migration and derivative defects. Thus, Fscn1a functions with Cxcr4a/Cxcl12b chemokine signaling to promote directional migration of a subset of NC cells and subsequently pattern the craniofacial skeleton and sympathetic nervous system. These results also suggest that other mechanisms that regulate directional migration function redundantly with filopodia to ensure most NC cell populations reach their correct location.
Perdurance of maternal protein rescues developmental functions of zygotic fscn1a
To determine the role of fscn1a in NC migration and development, we used TALEN-based gene targeting to generate multiple null alleles. Unexpectedly, we found that zygotic fscn1a is dispensable for development because maternal protein perdures throughout organogenesis for at least 10 days. In contrast, fscn1a MZ null mutant embryos display partially penetrant, but reproducible and heritable, defects in a subset of migrating NC cells and loss of a subset of NC-derived tissues. Thus, fscn1a mutants would have been missed in conventional forward genetic screens in zebrafish as well as maternal effect screens, which focus on mutants with early gastrula and embryonic phenotypes. As most genes required for NC development have been identified through forward genetic screens (or antisense morpholinos that do not affect oocyte-deposited proteins), we predict that additional genes required for NC development will be identified through similar TALENs and Cas9/CrispRs genome targeted approaches and subsequent examination of MZ mutants.
Filopodia are required for directional migration in a subset of NC streams
As the antennae of the cell, the primary function of filopodia is presumed to be to sense the extracellular environment for directional cues and facilitate communication between migrating cells [3,41]. Live imaging studies in vivo have demonstrated that chick NC cells display dynamic short - and long-range filopodia during migration [42]. The functions of filopodia during chick NC migration is not known, but they have been proposed to initiate cell-cell adhesion and promote directional migration [42,43]. Our studies provide evidence that support a selective role for fscn1a-dependent filopodia in NC migration to promote directional migration of the whole stream into the first pharyngeal arch.
Unexpectedly, our results also demonstrate that most NC cells migrate normally in the absence of long, robustfilopodia. This may be due to redundant genetic pathways (see below), or to residual filopodia present in fscn1a mutant embryos. For example, fewer short-range filopodia may still localize enough cell-cell adhesion and receptor molecules to promote adhesion and directionality in posterior cranial NC streams and trunk NC. To test this, we used the actin depolymerizing drug Lat. B, which has been previously used in zebrafish to eliminate filopodia in endothelial cells during angiogenic sprouting [10]. Consistent with these findings, Lat. B treatment in wild type or fscn1a MZ mutants resulted in an almost complete absence of any filopodia-like protrusions, particularly in the fscn1a MZ mutants (Fig. 6), but did not cause any additional effects on NC migration or derivative formation. The few protrusions that remained were extremely short, wide, and flat and were not dynamic. Thus, while we cannot rule out the possibility that a single short membrane protrusion is sufficient for directional migration, we favor alternative models in which either localization of guidance receptors to the leading edge membrane is sufficient to promote directional migration and/or physical barriers, such as the budding endodermal pouches, overlying ectoderm and cranial placodes, provide a permissive environment for posterior NC streams to simply be pushed into the correct location without need for chemotaxis. Consistent with the latter model, disrupting endodermal pouch formation does not alter early NC migration but instead causes fusion of NC streams at later time points, which in turn selectively disrupts formation of the posterior branchial arches [34,44]. In addition, the first cranial NC stream migrates ∼4 hours before the endodermal pouches are formed and is the only stream affected by disrupting Cxcr4a/Cxcl12b signaling alone [37]. Thus, we propose that long, robust and dynamic filopodia are preferentially required in NC cells that migrate in the absence of surrounding physical barriers and/or are most dependent on long-range directional signaling through chemotaxis. This model may also explain the differential loss of sympathetic ganglia in fscn1a MZ mutants, as previous studies have shown that in vagal NC cells, Cxcr4 is required to sense Cxcl12 at the dorsal aorta to stop migration and aggregate to form ganglia [38].
Redundant mechanisms of Fscn1 function during cell migration in vivo
Fscn1 belongs to a family of highly conserved actin bundling proteins required for filopodia formation [21]. Knockdown of Fscn1 almost always decreases cell migration capacity in vitro [21,45,46]. In contrast, fscn1 genetic mutants in Drosophila and mouse have tissue-specific phenotypes and are viable, suggesting fscn1 is dispensable for embryogenesis in vivo [8,9,24–26,47]. The lack of fscn1 embryonic phenotypes could be due to genetic redundancy with: 1) other actin-bundling proteins, such as villin, cofilin or espin, which have been shown to cooperate with singed/fscn1 in cell culture and during Drosophila embryonic development [47–50], 2) parallel pathways that promote cell migration, as we demonstrate with Cxcr4a, or 3) maternal effects as discussed above. Indeed, in Drosophila, homozygous female fscn1 mutants are sterile due to requirements for fscn1 in ring canal formation during oogenesis, while the role of Fscn1 function in the mouse germline or early embryo remains unclear because it is not known if the Fscn1 retroviral insertion affects Fscn1 mRNA or protein levels in the oocyte or embryo before E14.5 [25]. Interestingly, mouse Fscn1 mRNA is prominently expressed in the neural tube by E8.0 and pharyngeal arches by E9.5 [51] and approximately half of homozygous Fscn1 mutant mouse pups die shortly after birth without milk in their stomach, suggesting a subtle or partially penetrant feeding or craniofacial defect [26].
Our studies show that fscn1a MZ loss results in ∼20% embryonic lethality, revealing an essential role for Fscn1 in embryogenesis. However, even in the absence of both maternal and zygotic fscn1a, most embryos develop to adulthood, suggesting other mechanisms compensate for fscn1a loss during embryogenesis. Indeed, by combining fscn1a MZ loss with cxcl12b-misexpression or partial cxcr4a-deficiency, we observed a dramatic increase in embryonic lethality, which at least in part, is due to defective NC development. Thus, our studies support a model in which fscn1a functions to promote directional migration of a subset of NC cells by localizing Cxcr4a to filopodia, which in turn allows efficient responses to local chemokine signals (such as Cxcl12b) in the first pharyngeal arch (mandibular cartilage) or adjacent to the dorsal aorta (sympathetic neurons); NC migration pathways previously shown to be selectively dependent on Cxcr4 signaling in fish and chick [37,38].
In recent years, Fscn1 has gained significant attention due to its upregulation in aggressive human carcinomas [22]. Assessment of FSCN1 as a potential target for anti-metastasis therapeutics is an area of active research [23,52–54]. Based on our results, we would predict that FSCN1 inhibitors would be relatively ineffective when used alone due to redundant mechanisms promoting cell migration and invasion. Instead, our studies support the idea that FSCN1 inhibitors be used in combination with other inhibitors of cell migration, particularly inhibitors that act on directional migration molecules like CXCR4. Future studies will be important to determine if Fscn1 functions redundantly with other actin cross-linking proteins, adhesion molecules and guidance cues in development and cancer invasion and metastasis, including NC-derived cancers like melanoma and neuroblastoma. Importantly, the fscn1a MZ mutants represent a sensitized genetic background to identify such cooperating mechanisms controlling cell migration in development and cancer.
Methods
Ethics statement
All experiments involving zebrafish conformed to the regulatory standards and guidelines of the University of Utah Institutional Animal Care and Use Committee (IACUC#12-11009). In accordance with IACUC standards, zebrafish embryos and adults were euthanized by immobilization by submersion in ice water (5 parts ice/1 part water) for at least 10 minutes following cessation of opercular movement.
Animal husbandry and transgenic animals
Zebrafish were maintained and bred as described [55]. Transgenic lines used were described previously: Tg(sox10:RFPmb) [32], Tg(sox10:GFP) [56], Tg(ngn1:gfp) [57], Tg(phox2b:gfp) [58] and Tg(dbh:gfp) [59].
Molecular biology and cloning
Based on published GenBank sequences, the coding sequences of zebrafish fscn1a (Gene ID: 558271), fscn1b (Gene ID: 570314), fscn2a (Gene ID: 798075), and fscn2b (Gene ID: 393743) were PCR amplified from a 24 hpf (fscn1a) or 48 hpf (fscn1b, fscn2a, fscn2b) zebrafish cDNA library and TOPO cloned into pGEM-T Easy (Promega) or directionally cloned into pCS2.
Generation and genotyping of fscn1a mutant
A pair of TALEN plasmids targeting zebrafish fscn1a was designed and constructed by the University of Utah Mutation Generation and Detection Core (http://www.cores.utah.edu/) as previously described [60,61]. TALEN target sites were designed using the TALEN Effector Nucleotide Targeter 2.0 program [62] at https://tale-nt.cac.cornell.edu. The TALEN Golden Gate kit [63] was used with modifications to construct the fscn1a TALEN plasmids. Plasmids were linearized with Not1 and in vitro transcription was carried out using SP6 mMESSAGE mMACHINE kit (Ambion). 50 pg of each mRNA was microinjected into the yolk of 100 one-cell AB embryos.
To identify fscn1a mutant adults or embryos, genomic DNA was isolated from fin clips or tail clips, respectively. A 111 bp amplicon spanning the mutation site was isolated by PCR using LightScanner Master Mix (Biofire) and analyzed by high resolution melt analysis (HRMA) on a LightScanner (Biofire) as previously described [60,64]. To distinguish homozygous fscn1a mutants from wild type, all unidentified samples that grouped with known wild type samples were spiked with 2 μl wild type reaction, heated to 95° for 5 minutes, and re-analyzed on the LightScanner. The melt curve for homozygous fscn1a mutant samples shifted to a heterozygous fscn1a mutant melt curve, while the melt curve for wild type samples did not change. Wild type, heterozygous and homozygous fscn1a melt curves were validated by sequencing.
Preparation of embryonic protein lysates
After tail clips were taken, each embryo of unknown genotype was placed in 8 μl cold modified RIPA buffer with PI cocktail (1 : 100, Roche) on ice. Samples were briefly homogenized using a sterile pipette tip and stored at −80° until genotyping was complete. Samples of the same genotype were pooled and protein concentration was determined by BCA Protein Assay (Pierce). Protein samples were diluted to 0.4 μg/μL in 1X LDS (prepared from 4X LDS, Invitrogen). To prepare lysates from non-genotyped embryos, 10 embryos were added to 80 μl modified RIPA buffer PI cocktail and homogenized using a sterile pestle.
Alcian blue staining, whole-mount in situ hybridization and immunostaining
Cartilage staining with Alcian blue and whole-mount in situ hybridization was carried out as previously described [65,66]. Antisense RNA probes against zebrafish fscn1a or fscn1b were generated by digestion of pGEM-fscn1a with Kpn1 or Nco1 and subsequent in vitro transcription with T7 or SP6 RNA polymerase, respectively. Antisense RNA probes were generated for foxd3, crestin, dlx2a, sox10, th, and nkx2.3 as described [27,67]. Whole-mount immunostaining for GFP was performed on embryos fixed in 4% PFA overnight at 4° and permeabilized in methanol for 2 hours at −20°. Antibodies used in this study include: chicken anti-GFP (1 : 1000, Aves), goat anti-chicken 488 (1 : 250, Invitrogen), rabbit anti-FSCN1 (1 : 2000, Sigma) and mouse anti-GAPDH (1 : 1000, Abcam).
Morpholino and mRNA microinjection
Previously published antisense morpholino oligonucleotides against foxd3, tfap2a, and cxcr4a were used [27,37,68]. To generate full-length capped sense mRNA, pCS2-fscn1a, pCS2-cxcr4aGFP, or pCS2-cxcl12b was linearized with Not1 and in vitro transcription was carried out using SP6 mMESSAGE mMACHINE kit (Ambion). Morpholinos or mRNA were microinjected into the yolk of one-cell embryos at the following dosages: 1.25 ng foxd3MO, 1.25 ng tfap2aMO, 1.25 ng fscn1bMO, 1.25 ng fscn2aMO, 1.25 ng fscn2bMO, 1.5 ng cxcr4aMO, 25 pg fscn1a mRNA, 25 pg cxcr4a-GFP mRNA, 7.5 pg cxcl12b mRNA.
Latrunculin B treatment
Latrunculin B (Lat. B, Sigma-Aldrich Cat. # L5288) was reconstituted to 15 mg/mL in DMSO. A working stock was prepared fresh for each experiment by diluting Lat. B to 5 μg/ml in egg water (1 : 3000). A DMSO working stock was prepared by diluting DMSO in egg water (1 : 3000). To treat embryos, 10–15 dechorionated 10 hpf embryos were placed in 1 well of a 12 well plate with 2 mL egg water and working stocks of Lat. B or DMSO were diluted to the final experimental concentration. Guided by previous studies with Lat. B-treated zebrafish embryos [10], dose response curves were performed to determine an optimal dose of Lat. B that inhibited filopodia formation but did not affect other actin structures or cause lethality. Initial dose response experiments were performed with 25 ng/ml (10 μl working stock), 50 ng/ml (20 μl), 75 ng/ml (30 μl), 100 ng/ml (40 μl) and 150 ng/ml (60 μl) Lat. B or DMSO. A second dose response was performed with 75 ng/ml (30 μl working stock), 85 ng/ml (34 μl) and 95 ng/ml (38 μl) Lat. B or DMSO. From these dose response experiments, 80 ng/ml (32 μl) Lat. B or DMSO was determined to be the optimal dose of Lat. B for inhibiting filopodia formation in NC cells. 80 ng/ml Lat. B was used in all subsequent experiments. Selective inhibition of F-actin in residual NC cell filopodia was validated by monitoring F-actin polymerization using lifeact-GFP mRNA injected into Tg(sox10:rfpmb) embryos with low dose Lat. B or DMSO and visualizing filopodia by fluorescent confocal microscopy (S10 Fig.).
Image acquisition and processing
Confocal images were acquired using an Olympus Fluoview FV1200 confocal microscope and Olympus FV10-ASW v4.1 software. Olympus UPlanSApo 60X/1.20W and Olympus UPlanSApo 10X/0.45 objectives were used in this study. For all confocal imaging, embryos were embedded on cover slips in 1% low melt agarose. For analysis of filopodia dynamics, z-stacks of the leading edge of NC streams 1–2 (cranial, n = 6) or of trunk NC cells between somites 6–8 (trunk, n = 6) in 26 hpf Tg(sox10:rfpmb) and Tg(sox10:rfpmb); fscn1a MZ embryos were acquired every 4 minutes for one hour using the 60X water objective (S1–S2 Movies). To monitor NC migration and individual cell behaviors, z-stacks were acquired every 25 minutes for 18 hours using the 10X objective (S3–S4 Movies). Widefield fluorescent images were acquired on an Olympus SZX16 microscope configured with an Olympus DP72 camera. Brightfield images were taken using a Nikon C-DSD115 microscope configured with an Olympus DP72 camera. Prism 6, ImageJ 1.46r, Adobe Photoshop CS5 and CS6, and Adobe Illustrator CS6 were used to generate figures.
Quantification and statistical analysis
Filopodia characterization in wild type and fscn1a MZ embryos. Filopodia number and length was measured using ImageJ 1.46r. Filopodia within a 20 μm region at the leading edge of NC stream 2 were included in analyses. For each experimental condition, 5 filopodial protrusions from 6 embryos were analyzed (n = 30). Filopodia from Tg(sox10:rfpmb) and Tg(sox10:rfpmb); fscn1a MZ embryos were compared using an unpaired t-test (Prism 6). Significance is denoted with asterisks. ****p<0.0001.
Filopodia number and length in embryos injected with cxcl12b mRNA, cxcr4aMO, or treated with Latrunculin B. Filopodia number and length was measured using ImageJ 1.46r. All filopodia within a 100 μm region at the leading edge of NC stream 1 were included in analyses. Filopodia number and length were compared using an unpaired t-test (Prism 6). Significance is denoted with asterisks. *p<0.05, **p<0.005, ***p<0.001, ****p<0.0001.
Sympathetic ganglia. To detect sympathetic ganglia, ISH for th was performed on 3 dpf wt (n = 25) or fscn1a MZ (n = 24) embryos. In ImageJ, all RGB images were split into separate channels and the red channel was used for analysis. Color Threshold was adjusted using the Default method to 0–150 and the Analyze Particles feature was used to measure the area (μm2) of th+ sympathetic ganglia. Area of sympathetic ganglia was compared using an unpaired t-test (Prism 6). Significance is denoted with asterisks. *p<0.05, **p<0.005, ***p<0.001, ****p<0.0001, ns = not significant.
Enhanced penetrance of fscn1a MZ craniofacial defects. Fisher’s exact test (Prism 6) was used to determine if treatment with Latrunculin B or injection of cxcl12b mRNA or cxcr4aMO enhanced the penetrance of craniofacial defects in fscn1a MZ embryos. Significance is denoted with asterisks. *p<0.05.
Enteric neurons. Tg(phox2b:gfp) (n = 18) and Tg(phox2b:gfp); fscn1aMZ (n = 15) 3 dpf embryos were imaged and used for analysis. phox2b+ enteric neurons within a 500 pixel (0.625μm) × 100 pixel (0.125μm) region were counted in ImageJ. To enable automated counting, all images were processed to Find Edges and Color Threshold was adjusted using the Yen method. The Analyze Particles feature was used to count all particles with a circularity between 0.1 and 1.0. Number of enteric neurons was compared using an unpaired t-test (Prism 6). Significance is denoted with asterisks. *p<0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Petrie RJ, Doyle AD, Yamada KM (2009) Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol 10 : 538–549. 19603038
2. Ridley AJ (2011) Life at the leading edge. Cell 145 : 1012–1022.
3. Mattila PK, Lappalainen P (2008) Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 9 : 446–454. 18464790
4. George SP, Chen H, Conrad JC, Khurana S (2013) Regulation of directional cell migration by membrane-induced actin bundling. J Cell Sci 126 : 312–326. doi: 10.1242/jcs.116244 23132923
5. Kamakura S, Nomura M, Hayase J, Iwakiri Y, Nishikimi A, et al. (2013) The cell polarity protein mInsc regulates neutrophil chemotaxis via a noncanonical G protein signaling pathway. Dev Cell 26 : 292–302. doi: 10.1016/j.devcel.2013.06.008 23891662
6. Arjonen A, Kaukonen R, Ivaska J (2011) Filopodia and adhesion in cancer cell motility. Cell Adh Migr 5 : 421–430. 21975551
7. Sinnar SA, Antoku S, Saffin JM, Cooper JA, Halpain S (2014) Capping Protein Is Essential for Cell Migration in Vivo and for Filopodial Morphology and Dynamics. Mol Biol Cell.
8. Zanet J, Stramer B, Millard T, Martin P, Payre F, et al. (2009) Fascin is required for blood cell migration during Drosophila embryogenesis. Development 136 : 2557–2565. doi: 10.1242/dev.036517 19592575
9. Sonego M, Gajendra S, Parsons M, Ma Y, Hobbs C, et al. (2013) Fascin regulates the migration of subventricular zone-derived neuroblasts in the postnatal brain. J Neurosci 33 : 12171–12185. doi: 10.1523/JNEUROSCI.0653-13.2013 23884926
10. Phng LK, Stanchi F, Gerhardt H (2013) Filopodia are dispensable for endothelial tip cell guidance. Development 140 : 4031–4040. doi: 10.1242/dev.097352 24046319
11. Ma Y, Reynolds LE, Li A, Stevenson RP, Hodivala-Dilke KM, et al. (2013) Fascin 1 is dispensable for developmental and tumour angiogenesis. Biol Open 2 : 1187–1191. doi: 10.1242/bio.20136031 24244855
12. Le Douarin N, Kalcheim C (1999) The Neural Crest. Cambridge University Press.
13. Santagati F, Rijli FM (2003) Cranial neural crest and the building of the vertebrate head. Nat Rev Neurosci 4 : 806–818. 14523380
14. Etchevers HC, Amiel J, Lyonnet S (2006) Molecular bases of human neurocristopathies. Adv Exp Med Biol 589 : 213–234. 17434298
15. Shoval I, Kalcheim C (2012) Antagonistic activities of Rho and Rac GTPases underlie the transition from neural crest delamination to migration. Dev Dyn 241 : 1155–1168. doi: 10.1002/dvdy.23799 22553120
16. Clay MR, Halloran MC (2013) Rho activation is apically restricted by Arhgap1 in neural crest cells and drives epithelial-to-mesenchymal transition. Development 140 : 3198–3209. doi: 10.1242/dev.095448 23804498
17. Theveneau E, Mayor R (2012) Neural crest migration: interplay between chemorepellents, chemoattractants, contact inhibition, epithelial-mesenchymal transition, and collective cell migration. Wiley Interdiscip Rev Dev Biol 1 : 435–445. doi: 10.1002/wdev.28 23801492
18. Clay MR, Halloran MC (2010) Control of neural crest cell behavior and migration: Insights from live imaging. Cell Adh Migr 4 : 586–594. doi: 10.4161/cam.4.4.12902 20671421
19. Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, et al. (2008) Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature 456 : 957–961. doi: 10.1038/nature07441 19078960
20. Carmona-Fontaine C, Theveneau E, Tzekou A, Tada M, Woods M, et al. (2011) Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev Cell 21 : 1026–1037. doi: 10.1016/j.devcel.2011.10.012 22118769
21. Hashimoto Y, Kim DJ, Adams JC (2011) The roles of fascins in health and disease. J Pathol 224 : 289–300. doi: 10.1002/path.2894 21618240
22. Machesky LM, Li A (2010) Fascin: Invasive filopodia promoting metastasis. Commun Integr Biol 3 : 263–270. 20714410
23. Chen L, Yang S, Jakoncic J, Zhang JJ, Huang XY (2010) Migrastatin analogues target fascin to block tumour metastasis. Nature 464 : 1062–1066. doi: 10.1038/nature08978 20393565
24. Yamakita Y, Matsumura F, Lipscomb MW, Chou PC, Werlen G, et al. (2011) Fascin1 promotes cell migration of mature dendritic cells. J Immunol 186 : 2850–2859. doi: 10.4049/jimmunol.1001667 21263068
25. Ma Y, Li A, Faller WJ, Libertini S, Fiorito F, et al. (2013) Fascin 1 is transiently expressed in mouse melanoblasts during development and promotes migration and proliferation. Development 140 : 2203–2211. doi: 10.1242/dev.089789 23633513
26. Yamakita Y, Matsumura F, Yamashiro S (2009) Fascin1 is dispensable for mouse development but is favorable for neonatal survival. Cell Motil Cytoskeleton 66 : 524–534. doi: 10.1002/cm.20356 19343791
27. Stewart RA, Arduini BL, Berghmans S, George RE, Kanki JP, et al. (2006) Zebrafish foxd3 is selectively required for neural crest specification, migration and survival. Dev Biol 292 : 174–188. 16499899
28. Luo R, An M, Arduini BL, Henion PD (2001) Specific pan-neural crest expression of zebrafish Crestin throughout embryonic development. Dev Dyn 220 : 169–174. 11169850
29. Wang WD, Melville DB, Montero-Balaguer M, Hatzopoulos AK, Knapik EW (2011) Tfap2a and Foxd3 regulate early steps in the development of the neural crest progenitor population. Dev Biol 360 : 173–185. doi: 10.1016/j.ydbio.2011.09.019 21963426
30. Arduini BL, Bosse KM, Henion PD (2009) Genetic ablation of neural crest cell diversification. Development 136 : 1987–1994. doi: 10.1242/dev.033209 19439494
31. Ono S, Yamakita Y, Yamashiro S, Matsudaira PT, Gnarra JR, et al. (1997) Identification of an actin binding region and a protein kinase C phosphorylation site on human fascin. J Biol Chem 272 : 2527–2533. 8999969
32. Kirby BB, Takada N, Latimer AJ, Shin J, Carney TJ, et al. (2006) In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat Neurosci 9 : 1506–1511. 17099706
33. Schilling T, Le Pabic P (2013) Neural crest cells in craniofacial skeletal development. Neural Crest Cell Differentiation and Disease.
34. Piotrowski T, Nusslein-Volhard C (2000) The endoderm plays an important role in patterning the segmented pharyngeal region in zebrafish (Danio rerio). Dev Biol 225 : 339–356. 10985854
35. Raz E, Mahabaleshwar H (2009) Chemokine signaling in embryonic cell migration: a fisheye view. Development 136 : 1223–1229. doi: 10.1242/dev.022418 19304885
36. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, et al. (2013) A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer 49 : 219–230. doi: 10.1016/j.ejca.2012.05.005 22683307
37. Olesnicky Killian EC, Birkholz DA, Artinger KB (2009) A role for chemokine signaling in neural crest cell migration and craniofacial development. Dev Biol 333 : 161–172. doi: 10.1016/j.ydbio.2009.06.031 19576198
38. Kasemeier-Kulesa JC, McLennan R, Romine MH, Kulesa PM, Lefcort F (2010) CXCR4 controls ventral migration of sympathetic precursor cells. J Neurosci 30 : 13078–13088. doi: 10.1523/JNEUROSCI.0892-10.2010 20881125
39. Kulesa PM, Bailey CM, Kasemeier-Kulesa JC, McLennan R (2010) Cranial neural crest migration: new rules for an old road. Dev Biol 344 : 543–554. doi: 10.1016/j.ydbio.2010.04.010 20399765
40. Cerny R, Meulemans D, Berger J, Wilsch-Brauninger M, Kurth T, et al. (2004) Combined intrinsic and extrinsic influences pattern cranial neural crest migration and pharyngeal arch morphogenesis in axolotl. Dev Biol 266 : 252–269. 14738875
41. Gupton SL, Gertler FB (2007) Filopodia: the fingers that do the walking. Sci STKE 2007: re5. 17712139
42. Teddy JM, Kulesa PM (2004) In vivo evidence for short - and long-range cell communication in cranial neural crest cells. Development 131 : 6141–6151. 15548586
43. Vasioukhin V, Fuchs E (2001) Actin dynamics and cell-cell adhesion in epithelia. Curr Opin Cell Biol 13 : 76–84. 11163137
44. Choe CP, Collazo A, Trinh le A, Pan L, Moens CB, et al. (2013) Wnt-dependent epithelial transitions drive pharyngeal pouch formation. Dev Cell 24 : 296–309. doi: 10.1016/j.devcel.2012.12.003 23375584
45. Vignjevic D, Kojima S, Aratyn Y, Danciu O, Svitkina T, et al. (2006) Role of fascin in filopodial protrusion. J Cell Biol 174 : 863–875. 16966425
46. Hashimoto Y, Parsons M, Adams JC (2007) Dual actin-bundling and protein kinase C-binding activities of fascin regulate carcinoma cell migration downstream of Rac and contribute to metastasis. Mol Biol Cell 18 : 4591–4602. 17855511
47. Okenve-Ramos P, Llimargas M (2014) Fascin links Btl/FGFR signalling to the actin cytoskeleton during Drosophila tracheal morphogenesis. Development 141 : 929–939. doi: 10.1242/dev.103218 24496629
48. Wulfkuhle JD, Petersen NS, Otto JJ (1998) Changes in the F-actin cytoskeleton during neurosensory bristle development in Drosophila: the role of singed and forked proteins. Cell Motil Cytoskeleton 40 : 119–132. 9634210
49. Cant K, Knowles BA, Mahajan-Miklos S, Heintzelman M, Cooley L (1998) Drosophila fascin mutants are rescued by overexpression of the villin-like protein, quail. J Cell Sci 111 (Pt 2): 213–221. 9405306
50. Breitsprecher D, Koestler SA, Chizhov I, Nemethova M, Mueller J, et al. (2011) Cofilin cooperates with fascin to disassemble filopodial actin filaments. J Cell Sci 124 : 3305–3318. doi: 10.1242/jcs.086934 21940796
51. De Arcangelis A, Georges-Labouesse E, Adams JC (2004) Expression of fascin-1, the gene encoding the actin-bundling protein fascin-1, during mouse embryogenesis. Gene Expr Patterns 4 : 637–643. 15465486
52. Kraft R, Kahn A, Medina-Franco JL, Orlowski ML, Baynes C, et al. (2013) A cell-based fascin bioassay identifies compounds with potential anti-metastasis or cognition-enhancing functions. Dis Model Mech 6 : 217–235. 22917928
53. Santos RN, Guido RV, Oliva G, Dias LC, Andricopulo AD (2011) Quantitative structure-activity studies on a series of migrastatin analogs as inhibitors of cancer cell metastasis. Med Chem 7 : 155–164. doi: 10.2174/157340611795564240
54. Majchrzak K, Lo Re D, Gajewska M, Bulkowska M, Homa A, et al. (2013) Migrastatin analogues inhibit canine mammary cancer cell migration and invasion. PLoS One 8: e76789. doi: 10.1371/journal.pone.0076789 24116159
55. Westerfield M (1993) The Zebrafish Book. University of Oregon Press, Eugene, OR.
56. Hoffman TL, Javier AL, Campeau SA, Knight RD, Schilling TF (2007) Tfap2 transcription factors in zebrafish neural crest development and ectodermal evolution. J Exp Zool B Mol Dev Evol 308 : 679–691. 17724731
57. Blader P, Plessy C, Strahle U (2003) Multiple regulatory elements with spatially and temporally distinct activities control neurogenin1 expression in primary neurons of the zebrafish embryo. Mech Dev 120 : 211–218. 12559493
58. Nechiporuk A, Linbo T, Poss KD, Raible DW (2007) Specification of epibranchial placodes in zebrafish. Development 134 : 611–623. 17215310
59. Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, et al. (2012) Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 21 : 362–373. doi: 10.1016/j.ccr.2012.02.010 22439933
60. Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, et al. (2012) Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet 8: e1002861. doi: 10.1371/journal.pgen.1002861 22916025
61. Hu R, Wallace J, Dahlem TJ, Grunwald DJ, O’Connell RM (2013) Targeting human microRNA genes using engineered Tal-effector nucleases (TALENs). PLoS One 8: e63074. doi: 10.1371/journal.pone.0063074 23667577
62. Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, et al. (2012) TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res 40: W117–122. doi: 10.1093/nar/gks608 22693217
63. Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, et al. (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39: e82. doi: 10.1093/nar/gkr218 21493687
64. Xing L, Quist TS, Stevenson TJ, Dahlem TJ, Bonkowsky JL (2014) Rapid and efficient zebrafish genotyping using PCR with high-resolution melt analysis. J Vis Exp: e51138.
65. Thisse C, Thisse B, Schilling TF, Postlethwait JH (1993) Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119 : 1203–1215. 8306883
66. Kimmel CB, Miller CT, Kruze G, Ullmann B, BreMiller RA, et al. (1998) The shaping of pharyngeal cartilages during early development of the zebrafish. Dev Biol 203 : 245–263. 9808777
67. Lee KH, Xu Q, Breitbart RE (1996) A new tinman-related gene, nkx2.7, anticipates the expression of nkx2.5 and nkx2.3 in zebrafish heart and pharyngeal endoderm. Dev Biol 180 : 722–731. 8954740
68. Knight RD, Nair S, Nelson SS, Afshar A, Javidan Y, et al. (2003) lockjaw encodes a zebrafish tfap2a required for early neural crest development. Development 130 : 5755–5768. 14534133
69. Aanes H, Winata CL, Lin CH, Chen JP, Srinivasan KG, et al. (2011) Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res 21 : 1328–1338. doi: 10.1101/gr.116012.110 21555364
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The Global Regulatory Architecture of Transcription during the Cell Cycle
- A Truncated NLR Protein, TIR-NBS2, Is Required for Activated Defense Responses in the Mutant
- Proteasomes, Sir2, and Hxk2 Form an Interconnected Aging Network That Impinges on the AMPK/Snf1-Regulated Transcriptional Repressor Mig1
- The SWI2/SNF2 Chromatin Remodeler BRAHMA Regulates Polycomb Function during Vegetative Development and Directly Activates the Flowering Repressor Gene
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy