
A MATE-Family Efflux Pump Rescues the 8-Oxoguanine-Repair-Deficient Mutator Phenotype and Protects Against HO Killing
Hypermutation may accelerate bacterial evolution in the short-term. In the long-term, however, hypermutators (cells with an increased rate of mutation) can be expected to be at a disadvantage due to the accumulation of deleterious mutations. Therefore, in theory, hypermutators are doomed to extinction unless they compensate the elevated mutational burden (deleterious load). Different mechanisms capable of restoring a low mutation rate to hypermutators have been proposed. By choosing an 8-oxoguanine-repair-deficient (GO-deficient) Escherichia coli strain as a hypermutator model, we investigated the existence of genes able to rescue the hypermutable phenotype by multicopy suppression. Using an in vivo-generated mini-MudII4042 genomic library and a mutator screen, we obtained chromosomal fragments that decrease the rate of mutation in a mutT-deficient strain. Analysis of a selected clone showed that the expression of NorM is responsible for the decreased mutation rate in 8-oxoguanine-repair-deficient (mutT, mutY, and mutM mutY) strains. NorM is a member of the multidrug and toxin extrusion (MATE) family of efflux pumps whose role in E. coli cell physiology remains unknown. Our results indicate that NorM may act as a GO-system backup decreasing AT to CG and GC to TA transversions. In addition, the ability of NorM to reduce the level of intracellular reactive oxygen species (ROS) in a GO-deficient strain and protect the cell from oxidative stress, including protein carbonylation, suggests that it can extrude specific molecules—byproducts of bacterial metabolism—that oxidize the guanine present in both DNA and nucleotide pools. Altogether, our results indicate that NorM protects the cell from specific ROS when the GO system cannot cope with the damage.
Published in the journal:
. PLoS Genet 6(5): e32767. doi:10.1371/journal.pgen.1000931
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000931
Summary
Hypermutation may accelerate bacterial evolution in the short-term. In the long-term, however, hypermutators (cells with an increased rate of mutation) can be expected to be at a disadvantage due to the accumulation of deleterious mutations. Therefore, in theory, hypermutators are doomed to extinction unless they compensate the elevated mutational burden (deleterious load). Different mechanisms capable of restoring a low mutation rate to hypermutators have been proposed. By choosing an 8-oxoguanine-repair-deficient (GO-deficient) Escherichia coli strain as a hypermutator model, we investigated the existence of genes able to rescue the hypermutable phenotype by multicopy suppression. Using an in vivo-generated mini-MudII4042 genomic library and a mutator screen, we obtained chromosomal fragments that decrease the rate of mutation in a mutT-deficient strain. Analysis of a selected clone showed that the expression of NorM is responsible for the decreased mutation rate in 8-oxoguanine-repair-deficient (mutT, mutY, and mutM mutY) strains. NorM is a member of the multidrug and toxin extrusion (MATE) family of efflux pumps whose role in E. coli cell physiology remains unknown. Our results indicate that NorM may act as a GO-system backup decreasing AT to CG and GC to TA transversions. In addition, the ability of NorM to reduce the level of intracellular reactive oxygen species (ROS) in a GO-deficient strain and protect the cell from oxidative stress, including protein carbonylation, suggests that it can extrude specific molecules—byproducts of bacterial metabolism—that oxidize the guanine present in both DNA and nucleotide pools. Altogether, our results indicate that NorM protects the cell from specific ROS when the GO system cannot cope with the damage.
Introduction
Maintaining the integrity of genetic information is crucial for all living organisms. Mutations originate from replication errors and DNA damage from endogenous and exogenous origin. Evolution, through natural selection, has produced a number of systems to prevent or repair these errors. The post-replication mismatch repair system (MMR) repairs mainly replication errors (for a review see [1]). Endogenous damage to DNA bases are repaired primarily by base excision repair (BER) (for a review see [2]). Of particular importance are oxidative DNA lesions which play a major role in spontaneous mutagenesis, because oxidized bases can mispair with noncognate ones [2]. Especially noteworthy amid these oxidative lesions is oxidation of guanine to 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-dG). Guanine is particularly susceptible to oxidation on account of its low redox potential. If 8-oxo-dG is not repaired, it can be bypassed by DNA polymerases and pair with either C or A, causing GC to TA transversions [2].
Because 8-oxo-dGTP is highly mutagenic, efficient sanitizing mechanisms have evolved in all living cells to mitigate its highly mutagenic potential. In E. coli there are at least three proteins, MutM, MutY and MutT, engaged in avoiding the mutagenicity of 8-oxo-dGTP. These specialized proteins are known as the GO system [3]. MutM removes 8-oxoG paired with C in DNA, while the MutY protein removes A opposite 8-oxoG resulting from A mispaired with unrepaired 8-oxoG during replication. E. coli mutants defective in MutM or MutY exhibit higher than wild-type GC to TA spontaneous transversions [3]–[5]. The MutT enzyme is a nucleoside triphosphate pyrophosphohydrolase that converts 8-oxo-dGTP to 8-oxo-dGMP and pyrophosphate, thereby inactivating this mutagenic activity. In the absence of MutT there is an increase in AT to CG mutations [3].
Despite the high mutational burden produced by the absence of MMR or GO systems, naturally-occurring hypermutable E. coli and Pseudomonas aeruginosa isolates deficient in these systems have been found [6]–[10]. Hypermutable E. coli strains (mutators) can increase in frequency in laboratory bacterial populations under specific conditions that select rapid adaptation to environmental changes [11]–[13]. However, cells with the mutator phenotype pay a high biological price of deleterious mutations in the long run, which may result in extinction [14], [15]. Theoretical and experimental evidence of this cost has been obtained in bacterial populations propagated in the laboratory when submitted to severe bottlenecks [16]–[18].
Several possible ways to reduce the mutation rates of mutator strains can be envisaged, among these, mutations in additional loci that are able to reduce the mutation rate have been obtained in laboratory populations of mutT mutators submitted to long-term evolution [19]. However, after more than 20 years, the molecular mechanisms responsible for these compensatory phenotypes have not yet been described. Thus, our initial hypothesis raised the question of whether some back-up mechanisms may have evolved to alleviate the high cost of hypermutability in the absence of the original antimutator function.
In this work, we looked for genes that, when over-expressed, reversed or reduced the mutT mutator phenotype. To this end, we constructed and explored an in vivo library of E. coli genomic DNA fragments using a mini-Mu transposable element and the Mu phage [20]. We found that multicopy expression of norM is able to decrease the mutation rate in GO-repair deficient strains. NorM is the prototype of the multidrug and toxin-extrusion (MATE) family of cation-coupled efflux pumps, which includes many bacterial and eukaryotic members [21].
Results
Search for multicopy suppressors of the mutator phenotype of a mutT strain
As stated in the Materials and Methods section, the mini-Mu system produces a plasmid overexpression library of chromosomal fragments by homologous recombination between two adjacent mini-Mu transposons. We screened for GO− strains with a reduced mutation rate using the colony papillation screen described in figure 1. The screen of the mini-Mu library (about 1,500 clones) yielded several colonies with a clearly reduced number of white Ara+ papillae on agar plates containing arabinose and tetrazolium chloride. One of them, showing a papillation pattern similar to the wild-type strain was chosen for further study (Figure 1). Plasmid DNA was purified, retransformed into the original ΔmutT strain and retested with the Ara−→Ara+ reversion assay. The ends of the fragment contained in the mini-Mu plasmid were sequenced and the chromosomal region between them inferred. This region included 16 genes, 8 with assigned functions (gloA, rnt, lhr, sodB, purR, cfa, ribC and norM) (Figure 2). In principle, we considered sodB, which encodes the Fe-dependent superoxide dismutase, and norM, which encodes an orthologue of the MATE family [21]–[23], as major candidates responsible for the decreased mutation rate in the mutT-deficient background. Overexpression of sodB may reduce the level of reactive oxygen species (ROS) in the cell, leading to a reduced level of 8-oxo-dG, which consequently compensates for the absence of MutT activity. On the other hand, NorM is the prototype of the MATE family of cation-coupled transporters, which characteristically possess 12 putative transmembrane domains and have been reported in all three kingdoms of life [21]. Expression of NorM conferred resistance to several agents, such as norfloxacin, aminoglycosides and ethidium bromide, via a mechanism requiring the proton motive force [24]. Interestingly, MATE proteins have been described as exporters of toxic organic cations and guanidine [25], rendering NorM an excellent candidate for the export of oxidative precursor molecules.


norM is the antimutator gene
Plasmids containing the genes sodB [pCsodB] and norM [pCnorM] were obtained from the Complete Set of E. coli K-12 Open Reading Frame Archive (ASKA) library [26]. These plasmids, harboring the two cited genes cloned into the pCA24N vector, were transformed into the host strain GLF1 ΔmutT::kan [F′CC101] (Table 1), a strain that assays AT to CG Lac+ mutations. Several transformants carrying either pCsodB or pCnorM, and the parental vector pCA24N, were analyzed by a Lac+ reversion papillation assay. A clear decrease in the number of Lac+ papillae was observed in the strain harboring pCnorM, but not in those harboring pCsodB or the vector pCA24N (data not shown).

Table 2 shows the mutation rates of strains NR10831, GLF0 (ΔnorM::kan) and GLF1 (ΔmutT::kan) harboring plasmids pCA24N or pCnorM. No differences were observed between the wild-type and the mutant ΔnorM::kan, and the presence of plasmid pCnorM did not modify mutation rates. However, the plasmid pCnorM produced a dramatic decrease in the mutation rate when introduced into the ΔmutT strain (Table 2). These results suggest that, in the absence of additional damage, other protective mechanisms may suffice to cope with this damage, and that the anti-mutator effect of NorM can be observed only when mutagenesis is increased by the absence of a key antimutator mechanism such as MutT.

Effect of norM on the Val-R mutation rate
The norM antimutator effect was observed for a lac marker on the F′pro-lac episome. This may indicate a phenomenon similar to the process of “stress-induced mutation”, which may occur preferentially in the F′ episome [27]. To test this possibility, we used acquisition of valine-resistance (Val-S to Val-R) to verify the antimutator properties of NorM for a chromosomal marker. The valine-resistance (Val-R) mutation assay has been used previously by others [28], [29]. Table 3 shows that plasmid pCnorM is able to reduce the frequency of Val-R mutants of the strain GLF9 ΔmutT by two orders of magnitude, as in the case of the lac reversion assay.

In conclusion, the experiments described above show that the expression of NorM in a multicopy plasmid reverses or reduces the mutator phenotype caused by the lack of MutT activity at both episomal and chromosomal markers.
Effect of norM expression on the mutator phenotype of other GO-repair-deficient strains
The norM effect might be due to the active extrusion of toxic metabolites involved in the oxidative damage of guanine. In this case, the same effect, i.e., a decrease in mutation rate, should also be observed in the mutM mutY background, because both MutM and MutY remove errors caused by the presence of 8-oxodG in the DNA. This double mutant has an elevated rate of GC to TA transversions [3]. The frequency of Lac+ mutants was measured for strain NR10834 and its mutant derivatives GLF6 (mutM), GLF7 (mutY) and GLF8 (mutM mutY) containing either pCA24N or pCnorM (Table 4). All these strains carry the lacZ missense allele present in the episome F′CC104 [30], which reverts to Lac+ uniquely by a GC to TA transversion mutation. The mutM mutant shows a slight increase in mutant frequency, and both the mutY and the double mutM mutY mutants show a high mutant frequency (one and two orders of magnitude, respectively, in relation to the wild-type strain NR10834) (Table 4). The expression of norM from the multicopy plasmid decreased the mutant frequency of these three mutants significantly (P values<0.05 in all cases; see Table 4 for values). These mutant frequencies were even below that of the wild-type. At this stage, unfortunately, we have no satisfactory explanation for this phenomenon, although the transcriptional or postranscriptional regulation of the activity of other systems cannot be ruled out.
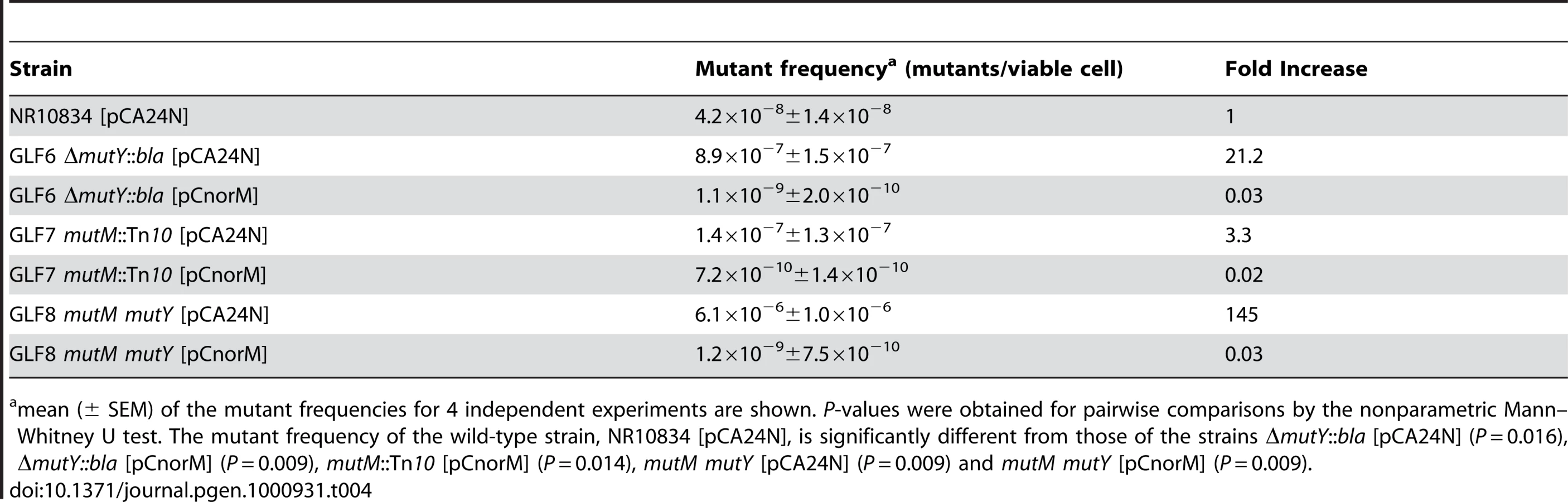
These results strongly suggest that the norM effect is due to an active extrusion of toxic metabolites involved in the oxidative damage of guanine, even when 8-oxodG is incorporated into the DNA.
Multicopy expression of norM protects against killing by hydrogen peroxide in a mutT-deficient background
GO-deficient cells are more sensitive to H2O2-induced killing than those of the wild-type [31], [32] via a mechanism that is still unknown. This has led us to investigate the effect of norM expression on cell protection against H2O2-induced killing. Figure 3 shows that norM expression in the multicopy plasmid promotes evident protection for mutT cells from H2O2-induced killing (P = 0.009). However, this protection is not statistically significant in the wild-type background (P = 0.6) (Figure 3). As in the case of mutagenesis, these results suggest that NorM protects cells from oxidative damage mainly in the absence of a key protective mechanism and when this damage is increased, e.g. in the presence of ROS-generating substances. Therefore, it is conceivable that NorM could protect cells from oxidative damage in the absence of a key cellular ROS-protective mechanism, such as superoxide dismutases [33]. When we tested the effect of norM expression in a sodB mutant background (lacking the Fe-dependent form of superoxide dismutase), we found that norM expression promotes only a minor protection, statistically not significant (P = 0.12), against H2O2 in this sensitive background (Figure 3).

Altogether these results show that NorM offers evident protection against H2O2-killing when the GO system cannot cope with the damage, although it provides no protection in the absence of other ROS protective mechanisms, such as superoxide dismutase SodB, or in the wild-type background. This suggests that NorM acts almost exclusively as a backup of the GO system, and is capable of alleviating both the increased oxidative DNA damage as well as the mutagenesis produced by the lack of this key system.
Effect of norM expression on intracellular ROS levels
All the previous results suggest that norM expression can reduce the intracellular ROS levels under specific circumstances. We have examined ROS levels qualitatively via the use of dihydrorhodamine 123 (DHR) and flow cytometry. DHR is a probe for the detection of intracellular reactive oxygen species. It is oxidized into rhodamine 123, which produces a maximal emission at 529 nm when excited at 507 nm (Enzo Life Sciences). Figure 4 shows that norM expression in the multicopy plasmid pCNorM can reduce the amount of reactive oxygen species slightly, but consistently, in the wild-type strain (Figure 4A). However, the same plasmid produced a greater reduction in the ROS intracellular level of the ΔmutT strain compared to the wild-type (Figure 4C) (note that the ROS intracellular levels, measured as oxidized rhodamine 123, are represented in a logarithmic scale). Expression of norM reduces the ROS level only slightly in the sodB-defective background (Figure 4D). In parallel experiments, ROS intracellular levels were measured after H2O2 treatment for 30 min. As expected, treated cells produced higher levels of ROS in all cases (the histograms are shifted slightly to the right), with minor reductions produced by the expression of norM, compared to values from cells harboring the empty vector (Figure 4E–G). The higher effect was observed in the mutT-deficient background (Figure. 4G). Once again, and in agreement with the data from the H2O2-protection experiments, our results suggest that the expression of norM acts almost exclusively as a back-up mechanism of the GO-system.
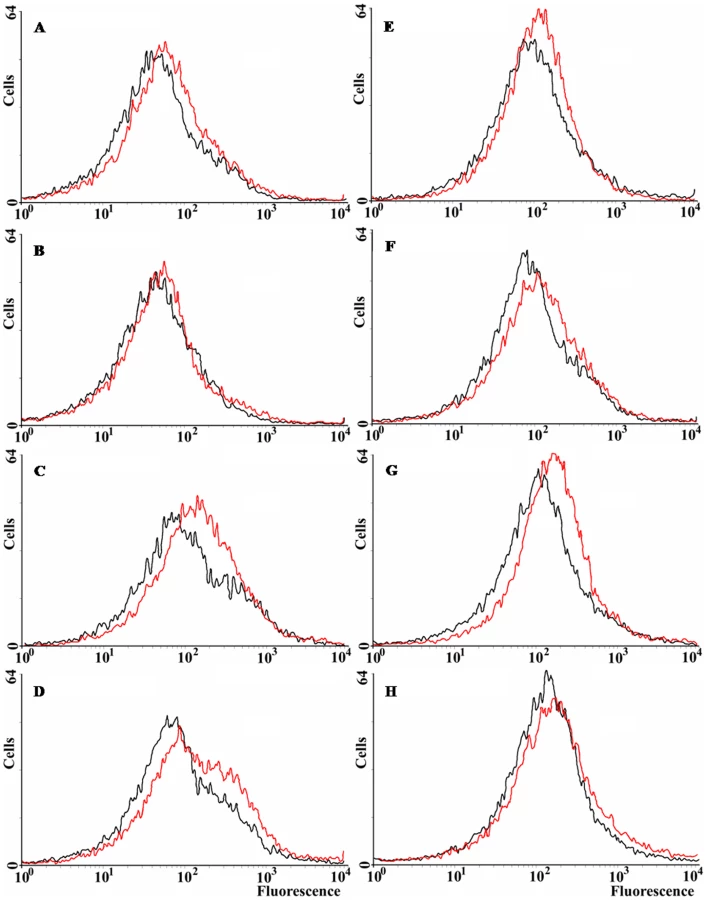
Effect of norM expression on protein carbonylation
One of the most important effects of an increased intracellular ROS level is protein carbonylation. Thus, we studied the effect of norM expression on protein carbonylation in H2O2-treated and non-treated wild-type and ΔmutT cells. The level of spontaneous protein carbonylation in both the wild-type and mutT non-treated cells growing in the exponential phase was undetectable with the OxyBlot kit. Nevertheless, when submitted to H2O2 pre-treatment, the expression of norM in the multicopy plasmid pCNorM produced a consistent decrease in the amount of carbonylated proteins in both the wild-type NR10831 and the ΔmutT strains (Figure 5A and 5B).

Discussion
The integrity of genetic information is a critical process for life. Consequently, a number of systems have evolved to prevent or repair replicative and postreplicative errors. The GO system, which is able to prevent and repair oxidative damage produced by the oxidation of deoxy-guanine to 8-oxo-dG, is a key component of the antimutation cell machinery. However, despite the great mutational burden produced by the absence of the GO system, naturally-occurring hypermutable E. coli and P. aeruginosa isolates deficient in mutT have been found [6], [10].
Hypermutable E. coli strains (mutators) show a selective advantage over the wild-type as they can produce more favorable mutations. In fact, it has been demonstrated that mutators can increase in frequency under specific conditions of environmental change in laboratory bacterial populations [11]–[13].
However, hypermutation may represent a colossal evolutionary cost for bacteria because most of the mutations are neutral or deleterious [14], [15]. Theoretical and experimental evidence of this cost has been obtained in laboratory propagated bacterial populations when they were submitted to severe bottlenecks [16]–[18]. The accumulation of deleterious mutations in genes experiencing relaxed selection or no selection at all will cause a more rapid fitness loss if mutators later encounter environments in which those genes are important [34], [35]. Thus, from a theoretical stand-point, hypermutator populations must reduce the mutation rate or face the possibility of extinction, at least under specific conditions. There are three possible ways to reduce the mutation rate for a mutator: (i) reverse the mutation that produced the mutator allele; (ii) replace the mutator allele with a wild-type gene from a non-mutator cell via horizontal gene transfer; and (iii) compensatory mutations at additional loci. Two studies strongly suggest that the replacement of the mutator allele with a non-mutator by horizontal transfer may have occurred in nature [36], [37]. Evidence of the third way, i.e., mutations at additional loci that reduce the mutation rate, has been obtained in laboratory populations of mutators submitted to long-term evolution, although the genes responsible for this have not been characterized [19]. Moreover, some antimutator mutations in the α catalytic subunit of the DNA-polymerase III compensate the high mutation rate of mutT-deficient strains and dnaQ (proofreading-deficient) mutators, such as mutD5, by increasing replication fidelity [38]–[40]. Finally, a different mode is to reduce the cost of accumulating deleterious mutations (deleterious load). Increased levels of the heat-shock chaperones, DnaK and GroEL, in lineages that accumulate many mutations, reduce the fitness cost produced by deleterious load [41].
The hypothesis we tested was that some back-up mechanisms could alleviate the high cost of hypermutability in the absence of the original antimutator function. We chose deficiency in the GO-repair system as a model. Our strategy was to look for genes which, when over-expressed, rescue or reduce the mutT mutator phenotype. Our results indicate that the expression of a pump from the multidrug and toxin extrusion (MATE) family is able to counteract the mutagenic effect of the lack of mutT, completely restoring the basal levels of AT to CG transversions. Interestingly, Yang et al [42] found that the overproduction of emrR, encoding a negative regulator of the multidrug resistance pump EmrAB, led to a mutator phenotype, suggesting a link between multidrug efflux and mutagenesis.
NorM is the prototype of the MATE family of cation-coupled transporters, which include many bacterial members [21]. Curiously, one of these members is E. coli DinF, which also seems to be involved in the efflux of DNA damage-inducing compounds (Guelfo, J.R. et al, unpublished data). Based on sequence similarity, two human MATE transporter genes, hMATE1 and hMATE2, have been described [43]. hMATE1 is primarily expressed in the liver, skeletal muscle and the kidney, whereas hMATE2 is expressed in testis. It is thought that mammalian MATE transporters mediate the final step in the excretion of toxic organic cations [43]. The existence of these NorM orthologues in mammalian cells suggests that they might play a similar antimutator role, protecting the cells from endogenous and exogenous mutagenic metabolites.
Several lines of evidence strongly suggest that NorM extrudes molecules that oxidize dG to 8-oxo-dG, when present in both the DNA and the dNTP pool: (i) overexpression of norM is also able to reduce the elevated rate of GC to TA transversions induced by the lack of both MutM and MutY activities, which act on errors produced when 8-oxo-dG is present in the DNA; (ii) there is a decrease of intracellular ROS and carbonylated protein levels in the mutT background; and (iii) there is significant protection of mutT-deficient cells from H2O2-induced killing.
The fact that overexpression of NorM reduces mutagenesis in MutM MutY-deficient cells (which do not have increased ROS levels but rather decreased removal of 8-oxo-dG from DNA), but not in the wild-type cells, is intriguing. An alternative hypothesis is that in wild-type cells most spontaneous mutagenesis is not caused by 8-oxo-dG, but is rather due to other problems and pathways. Therefore, lowering ROS levels in the wild-type cells does not affect those pathways or spontaneous mutant frequencies. However, in MutM MutY-deficient cells, the mutagenesis caused by 8-oxo-dG exceeds the levels from the normally main spontaneous mutation pathways. Once this happens, either because cells are mutM mutY or because they were treated with H2O2, GO-mediated mutations dominate and these are reduced by NorM.
Altogether our results suggest that NorM may act as a specific backup mechanism able to alleviate both oxidative DNA damage and mutagenesis when the GO system is impaired. The nature of the oxidizing molecules putatively extruded by NorM remains unknown. However, the apparent substrate specificity of NorM provides a good starting point to understanding why GO-deficient cells are more sensitive to H2O2-induced killing than those of the wild-type.
Concerning the evolutionary aspect, the major goal of this work is to demonstrate that a mutator phenotype can be reverted by extragenic-based mutation. To our knowledge, this kind of mutator-phenotype rescue has not been described before and may provide an explanation as to how some naturally-occurring hypermutator populations can avoid losing fitness by deleterious load. This striking discovery suggests that the surprisingly high proportion of E. coli mutators deficient in a repair pathway (up to 1%) in nature [6]–[8] could have been even higher, because some of the mutator phenotypes may have been hidden by extragenic “compensation”. In any case, irrespective of possible overproduction of NorM in nature, this overproduction has been necessary to discover the antimutator and ROS-protective effect of NorM.
The over-expression of norM in bacteria has been associated with multiple drug resistance [25], [44]. Here we reveal that this over-expression may confer several advantageous phenotypes simultaneously, such as antibiotic resistance, protection against ROS and antimutability.
Materials and Methods
Strains and constructions
The strains, phages and plasmids used in this study are listed in Table 1. The strains with the F′pro-lac episomes NR10831 [F′CC101] and NR10834 [F′CC104] carry the lacZ marker on these F′ episomes containing specific mutations that can revert to Lac+ by only one defined mutational event [30], [45]. These strains were kindly provided by Dr. I. Fijalkowska (Institute of Biochemistry and Biophysics, Warsaw, Poland). Strains NR10831ΔnorM::Kan (GLF0), NR10831ΔmutT::Kan (GLF1) and NR10831ΔsodB::Kan (GLF10) were constructed by P1 transduction of the alleles from strains BW25113ΔnorM::Kan (JW1655), BW25113ΔmutT::Kan (JW0097) and BW25113ΔsodB::Kan (JW1648), respectively, obtained from the Keio collection, NARA Institute (http://ecoli.aist-nara.ac.jp) [46]. Strains NR10834 ΔmutM::bla (AmpR) and NR10834 mutY::miniTn10 (TetR) were constructed by P1 transduction from strains BW853Δfpg::bla and CSH11 mutY::miniTn10 [47], respectively.
The vector used in this study was pCA24N, harboring a ColE1 replicon and a predicted copy number per cell of around 20 [26] (Table1). The plasmids pCnorM and pCsodB contain the wild-type norM and sodB genes, respectively, cloned in the vector pCA24N. These genes are transcribed from the pT5/lac promoter, which is repressed by the product of the lacI gene. Despite the predicted strict repression, our data with GFP fusions indicate that most genes cloned in this plasmid are transcribed, even in the absence of IPTG (data not shown). Plasmids were obtained from the Complete Set of E. coli K-12 Open Reading Frame Archive (ASKA) library [26].
Media
All strains were grown routinely in Luria-Bertani (LB) [48]. The arabinose papillation assay was carried out in tetrazolium arabinose, which contains (per litre) tryptone (10 g), yeast extract (1 g), NaCl (5 g), agar (16 g), arabinose (10 g) and tetrazolium chloride (0.05 g). Lactose reversion and valine-resistance assays were conducted in M9 minimal medium, as described below. For M9 minimal medium (MM) preparation, M9 salts (Sigma) were supplemented with thiamine (2.5 mg/l), MgSO4 (1 mM) and amino acids (0.04 mg/ml), when required. Glucose or lactose were used as carbon source at 2 mg/ml final concentration. Solid and soft medium contained 15 g/l and 7 g/l of agar, respectively.
Valine resistance assays were performed in glucose MM and the plates were supplemented with valine (0.04 mg/ml). Lac reversion assays were carried out in MM. Inocula were grown with glucose as carbon source and the plates were supplemented with lactose as unique carbon source according to Miller [48]. The scavenger strain MEC222 [49], harboring a truncated lacZ allele (lacZΔT::cat) with the C-terminal region replaced by the cat cassette (Table 1), was added to the lactose agar MM before being spread (40 µl of a stationary-phase culture per litre of media, approximately 107 cells/plate). Plates were stored overnight at room temperature. The desired cultures for Lac reversion assays were spread in a M9 top-agar layer, without carbon source and supplemented with 5-bromo-4-chloro-3-indolyl-3-D-galactoside (X-Gal), as described by Miller [48].
When required, antibiotics were added to the media: kanamycin (Km) 50 µg/ml, chloramphenicol (Cm) 40 µg/ml, ampicillin (Amp) 100 µg/ml and tetracycline (Tet) 20 µg/ml.
DNA manipulations and phages handling
DNA manipulations have been described previously [50]. Plasmid DNA was routinely extracted by alkaline lysis and transformed into E. coli strains by the CaCl2 method [50]. Procedures for handling bacteriophages Mu and P1 have been described [48].
Antimutator screen
MudII4042 was used to construct an in vivo random library of E. coli chromosomal fragments into a multicopy plasmid [51]. MudII4042 is a derivative of the Mu bacteriophage that contains the P15A replication origin and the chloramphenicol-resistance gene. This mini-Mu element can transpose at high frequency when de-repressed and it can be replicated in a lytic growth when present with a helper Mu cts phage. The heat-induced lysate of a MudII4042 Mu cts strain, Pop3001.6 in this case, produces a variety of packaged DNA. Sequences flanked by two copies of this mini-Mu can be packaged along with them. After infection, homologous recombination can occur between the mini-Mu sequences, resulting in the formation of plasmids carrying the transduced sequences. This library was transduced into the GLF1 (ΔmutT) strain (Table 1) and plated onto arabinose-tetrazolium agar plates with chloramphenicol. The hypermutable ΔmutT strain produces red colonies with a high number of white papillae as a result of the spontaneous reversion of Ara− to Ara+. The plates were incubated for a total of 7 days and examined at daily intervals for colonies with decreased reversion to Ara+, as visualized by the number of white papillae appearing per colony. About 1,500 clones were analyzed for the Ara−→Ara+ reversion rate. The clones with an evident decrease in this rate (low number of Ara+ papillae) were selected, their plasmids purified, retransformed into the original ΔmutT strain, retested and preserved for further analysis. The plasmid from one of them, presenting a papillation pattern similar to the wild-type (mutT+) strain, was sequenced and analyzed in detail. The sequences at the ends of the cloned fragments were analyzed using BLAST searches, thereby identifying the region cloned in the mini-Mu-derived plasmid. This plasmid contained several genes, some of which were considered by us as better candidates to reduce mutation rate. To find the gene responsible for this decreased papillation we used the appropriate plasmids, pCsodB and pCnorM, from the Complete Set of E. coli K-12 Open Reading Frame Archive (ASKA) library. This library contains each E. coli open reading frame cloned into the pCA24N vector [26]. Plasmids harboring the candidate genes were taken from this collection and transformed into the host strain GLF1 (Table 1). All transformants were analyzed by Lac+ reversion frequency.
Mutation rate and mutant frequency measurements
To calculate mutation rates pre-inocula were initiated in tubes with 3 ml of M9 glucose directly from frozen samples. The pre-inocula were grown at 37°C overnight to stationary phase. From each culture less than 105 cells were inoculated in 100 ml of M9 glucose and divided into 10 independent cultures, 10 ml each and less than 104 cells/culture. These inocula were grown for 24 hours. Appropriate dilutions of the saturated cultures were plated on selective media valine-MM or Lac-X-gal-MM to determine the number of valine resistant mutants or Lac+ mutants, respectively. LB plates were used to determine the total colony-forming units (cfu). Mutation rates (number of mutants per cell per division) were estimated by the method described [52]. To calculate mutant frequencies (number of mutants per total cell count), the mean number of mutants per millilitre was determined and divided by the average number of cfu per ml. Experiments were repeated at least three times.
Flow cytometry
Flow cytometry analysis was performed using the H2O2-activated fluorescent dye Dihydrorhodamine 123 (DHR) (Enzo Life Sciences). Wild-type and mutant derivatives were grown in M9 at 37°C, as described above, and then each one was split into two cultures (one control and one treated with 50 mM H2O2) and incubated for 30 min. Cells (0.5 ml/culture) were pelleted by centrifugation, resuspended in saline containing 15 µM DHR, and then incubated for 15 min and diluted 1 : 50 in phosphate-buffered saline. The fluorescence levels (excitation 488 nm and emission 530 nm) of 15,000 cells were then counted for each strain under each condition using a FACSCalibur cytometer (BD Biosciences). WinMDI (The Scripps Institute, Purdue University, USA) was used for data analysis and generation of histograms.
Determination of the cellular level of protein carbonylation
Wild-type and mutant derivatives were grown, as described above, and then each was split into two cultures (one control and one treated with 50 mM H2O2) and incubated for 30 min. After the time indicated, peroxide was removed by centrifugation. Subsequently, the cells were washed and resuspended in M9 medium preheated to 37°C and further incubated. Cells were lysed as follows: 1 ml of the culture was washed with 50 mM Tris buffer (pH 7.5) and centrifuged for 10 min at 14,000 rpm. The pellet was re-suspended in 150 µl lysis buffer containing 0.5 mg/ml lysosyme, 20 µg/ml DNAse, 50 µg/ml RNAse, 1 mM EDTA, and 10 mM Tris (pH 8). 15µl of 10% SDS solution was added and the cells were incubated at 100°C for 5 min. In order to examine the level of protein carbonylation in these lysates, we used the Chemicon OxyBlot kit (Chemicon) to derivatize the carbonyl groups in the protein side chains to 2,4-dinitrophenylhydrazone (DNP-hydrazone) by reaction with 2,4-dinitrophenylhydrazine. These DNP derivative crude protein extracts were dot blotted onto a nitrocellulose membrane, which was incubated with primary antibody specific to the DNP moiety of the proteins, and subsequently incubated with secondary (goat anti-rabbit) horseradish peroxidase-antibody conjugate directed against the primary antibody. Carbonylation was observed by the ECL, enhanced chemiluminiscence, reagent (Amersham Pharmacia Biotec). The intensity of each dot was quantified by densitometry analysis using the Image Master VPS-CL software. The intensity of each dot was normalized to equal levels of protein, which were determined using Bradford reagent (Bio-Rad) and expressed in femtomoles of DNP, according to the control of the OxyBlot kit.
Estimation of H2O2-induced cell death
The strains were grown at 37°C in M9 supplemented with appropriate antibiotics to mid-exponential phase and washed with 0.9% NaCl. The cells were treated with 50 mM H2O2 for 30 min at 37°C and washed with 1 ml of 0.9% NaCl. A non-treated control was also included. Appropriate dilutions were immediately plated onto LB plates and incubated overnight at 37°C to determine viability. Experiments consisted of five independent cultures for each strain. Cell survival was calculated by comparing the number of cfus of treated cells to those of the cells not treated.
Statistical analysis
The statistical signification for pairwise comparisons was estimated by the Mann-Whitney U test. P values≤0.05 were considered to be statistically significant.
Zdroje
1. ModrichP
LahueR
1996 Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 65 101 133
2. FriedbergE
WalkerG
SeideW
WoodR
SchultzR
2006 DNA Repair and Mutagenesis Washington DC, USA American Society of Microbiology
3. MichaelsML
MillerJH
1992 The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J Bacteriol 174 6321 6325
4. WoodML
EsteveA
MorningstarML
KuziemkoGM
EssigmannJM
1992 Genetic effects of oxidative DNA damage: comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine in Escherichia coli. Nucleic Acids Res 20 6023 6032
5. ChengKC
CahillDS
KasaiH
NishimuraS
LoebLA
1992 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G-T and A-C substitutions. J Biol Chem 267 166 172
6. GrossMD
SiegelEC
1981 Incidence of mutator strains in Escherichia coli and coliforms in nature. Mutat Res 91 107 110
7. LeClercJE
LiB
PayneWL
CebulaTA
1996 High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274 1208 1211
8. MaticI
RadmanM
TaddeiF
PicardB
DoitC
1997 Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277 1833 1834
9. OliverA
CantonR
CampoP
BaqueroF
BlazquezJ
2000 High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288 1251 1254
10. MandsbergLF
CiofuO
KirkbyN
ChristiansenLE
PoulsenHE
2009 Antibiotic resistance in Pseudomonas aeruginosa strains with increased mutation frequency due to inactivation of the DNA oxidative repair system. Antimicrob Agents Chemother 53 2483 2491
11. GibsonTC
ScheppeML
CoxEC
1970 Fitness of an Escherichia coli mutator gene. Science 169 686 688
12. CoxEC
GibsonTC
1974 Selection for high mutation rates in chemostats. Genetics 77 169 184
13. ChaoL
CoxEC
1983 Competition between high and low mutating strains of Escherichia coli. Evolution 37 125 134
14. KimuraM
1967 The evolution of mutation rates. Genetics 73 1 18
15. LeighEGJr
1973 The evolution of mutation rates. Genetics 73: Suppl 73 71 18
16. AnderssonDI
HughesD
1996 Muller's ratchet decreases fitness of a DNA-based microbe. Proc Natl Acad Sci U S A 93 906 907
17. KibotaTT
LynchM
1996 Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature 381 694 696
18. FunchainP
YeungA
StewartJL
LinR
SlupskaMM
2000 The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics 154 959 970
19. TrobnerW
PiechockiR
1984 Selection against hypermutability in Escherichia coli during long term evolution. Mol Gen Genet 198 177 178
20. GroismanEA
CasadabanMJ
1987 Cloning of genes from members of the family Enterobacteriaceae with mini-Mu bacteriophage containing plasmid replicons. J Bacteriol 169 687 693
21. BrownMH
PaulsenIT
SkurrayRA
1999 The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol 31 394 395
22. PutmanM
van VeenHW
KoningsWN
2000 Molecular properties of bacterial multidrug transporters. Microbiol Mol Biol Rev 64 672 693
23. HvorupRN
WinnenB
ChangAB
JiangY
ZhouXF
2003 The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur J Biochem 270 799 813
24. LongF
Rouquette-LoughlinC
ShaferWM
YuEW
2008 Functional cloning and characterization of the multidrug efflux pumps NorM from Neisseria gonorrhoeae and YdhE from Escherichia coli. Antimicrob Agents Chemother 52 3052 3060
25. MoritaY
KodamaK
ShiotaS
MineT
KataokaA
1998 NorM, a putative multidrug efflux protein, of Vibrio parahaemolyticus and its homolog in Escherichia coli. Antimicrob Agents Chemother 42 1778 1782
26. KitagawaM
AraT
ArifuzzamanM
Ioka-NakamichiT
InamotoE
2005 Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12 291 299
27. FosterPL
2007 Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol 42 373 397
28. Al MamunAA
HumayunMZ
2009 Spontaneous mutagenesis is elevated in protease-defective cells. Mol Microbiol 71 629 639
29. LawtherRP
CalhounDH
AdamsCW
HauserCA
GrayJ
1981 Molecular basis of valine resistance in Escherichia coli K-12. Proc Natl Acad Sci U S A 78 922 925
30. CupplesCG
CabreraM
CruzC
MillerJH
1990 A set of lacZ mutations in Escherichia coli that allow rapid detection of specific frameshift mutations. Genetics 125 275 280
31. AlhamaJ
Ruiz-LagunaJ
Rodriguez-ArizaA
ToribioF
Lopez-BareaJ
1998 Formation of 8-oxoguanine in cellular DNA of Escherichia coli strains defective in different antioxidant defences. Mutagenesis 13 589 594
32. SandersLH
SudhakaranJ
SuttonMD
2009 The GO system prevents ROS-induced mutagenesis and killing in Pseudomonas aeruginosa. FEMS Microbiol Lett 294 89 96
33. ImlayJA
LinnS
1987 Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J Bacteriol 169 2967 2976
34. CooperVS
LenskiRE
2000 The population genetics of ecological specialization in evolving Escherichia coli populations. Nature 407 736 739
35. GiraudA
RadmanM
MaticI
TaddeiF
2001 The rise and fall of mutator bacteria. Curr Opin Microbiol 4 582 585
36. DenamurE
LecointreG
DarluP
TenaillonO
AcquavivaC
2000 Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell 103 711 721
37. BrownEW
LeClercJE
LiB
PayneWL
CebulaTA
2001 Phylogenetic evidence for horizontal transfer of mutS alleles among naturally occurring Escherichia coli strains. J Bacteriol 183 1631 1644
38. FijalkowskaIJ
DunnRL
SchaaperRM
1993 Mutants of Escherichia coli with increased fidelity of DNA replication. Genetics 134 1023 1030
39. SchaaperRM
1993 The mutational specificity of two Escherichia coli dnaE antimutator alleles as determined from lacI mutation spectra. Genetics 134 1031 1038
40. SchaaperRM
1996 Suppressors of Escherichia coli mutT: antimutators for DNA replication errors. Mutat Res 350 17 23
41. Maisnier-PatinS
RothJR
FredrikssonA
NystromT
BergOG
2005 Genomic buffering mitigates the effects of deleterious mutations in bacteria. Nat Genet 37 1376 1379
42. YangH
WolffE
KimM
DiepA
MillerJH
2004 Identification of mutator genes and mutational pathways in Escherichia coli using a multicopy cloning approach. Mol Microbiol 53 283 295
43. OtsukaM
MatsumotoT
MorimotoR
AriokaS
OmoteH
2005 A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci U S A 102 17923 17928
44. MiyamaeS
UedaO
YoshimuraF
HwangJ
TanakaY
2001 A MATE family multidrug efflux transporter pumps out fluoroquinolones in Bacteroides thetaiotaomicron. Antimicrob Agents Chemother 45 3341 3346
45. SchaaperRM
DanforthBN
GlickmanBW
1985 Rapid repeated cloning of mutant lac repressor genes. Gene 39 181 189
46. BabaT
AraT
HasegawaM
TakaiY
OkumuraY
2006 Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2 2006 0008
47. BlaisdellJO
HatahetZ
WallaceSS
1999 A novel role for Escherichia coli endonuclease VIII in prevention of spontaneous G>T transversions. J Bacteriol 181 6396 6402
48. MillerJH
1992 A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli related bacteria New York Cold Spring Harbor Laboratory Press
49. ElezM
RadmanM
MaticI
2007 The frequency and structure of recombinant products is determined by the cellular level of MutL. Proc Natl Acad Sci U S A 104 8935 8940
50. SambrookJ
RussellDW
2001 Molecular cloning : a laboratory manual Cold Spring Harbor [New York] Laboratory Press
51. GroismanEA
CastilhoBA
CasadabanMJ
1984 In vivo DNA cloning and adjacent gene fusing with a mini-Mu-lac bacteriophage containing a plasmid replicon. Proc Natl Acad Sci U S A 81 1480 1483
52. CraneGJ
ThomasSM
JonesME
1996 A modified Luria-Delbruck fluctuation assay for estimating and comparing mutation rates. Mutat Res 354 171 182
53. CasadabanMJ
1976 Regulation of the regulatory gene for the arabinose pathway, araC. J Mol Biol 104 557 566
54. GroismanEA
CasadabanMJ
1987 In vivo DNA cloning with a mini-Mu replicon cosmid and a helper lambda phage. Gene 51 77 84
55. DatsenkoKA
WannerBL
2000 One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 6640 6645
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Common Genetic Variants near the Brittle Cornea Syndrome Locus Influence the Blinding Disease Risk Factor Central Corneal Thickness
- All About Mitochondrial Eve: An Interview with Rebecca Cann
- Aging and Chronic Sun Exposure Cause Distinct Epigenetic Changes in Human Skin
- The Relationship among Gene Expression, the Evolution of Gene Dosage, and the Rate of Protein Evolution
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy