
Aging and Chronic Sun Exposure Cause Distinct Epigenetic Changes in Human Skin
Epigenetic changes are widely considered to play an important role in aging, but experimental evidence to support this hypothesis has been scarce. We have used array-based analysis to determine genome-scale DNA methylation patterns from human skin samples and to investigate the effects of aging, chronic sun exposure, and tissue variation. Our results reveal a high degree of tissue specificity in the methylation patterns and also showed very little interindividual variation within tissues. Data stratification by age revealed that DNA from older individuals was characterized by a specific hypermethylation pattern affecting less than 1% of the markers analyzed. Interestingly, stratification by sun exposure produced a fundamentally different pattern with a significant trend towards hypomethylation. Our results thus identify defined age-related DNA methylation changes and suggest that these alterations might contribute to the phenotypic changes associated with skin aging.
Published in the journal:
. PLoS Genet 6(5): e32767. doi:10.1371/journal.pgen.1000971
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000971
Summary
Epigenetic changes are widely considered to play an important role in aging, but experimental evidence to support this hypothesis has been scarce. We have used array-based analysis to determine genome-scale DNA methylation patterns from human skin samples and to investigate the effects of aging, chronic sun exposure, and tissue variation. Our results reveal a high degree of tissue specificity in the methylation patterns and also showed very little interindividual variation within tissues. Data stratification by age revealed that DNA from older individuals was characterized by a specific hypermethylation pattern affecting less than 1% of the markers analyzed. Interestingly, stratification by sun exposure produced a fundamentally different pattern with a significant trend towards hypomethylation. Our results thus identify defined age-related DNA methylation changes and suggest that these alterations might contribute to the phenotypic changes associated with skin aging.
Introduction
Aging is defined by characteristic phenotypic changes, but there appear to be few corresponding changes in the genotype. As such, aging represents a fundamental epigenetic phenomenon [1]. Epigenetic mechanisms regulate the interpretation of genetic information and thus have the ability to produce different phenotypes from a single genotype [2]. The corresponding regulatory mechanisms are based on two independent modification systems, the covalent modification of histones and the methylation of cytosine residues in DNA [3]. Over the past few years, numerous histone modifications have been described and it has been suggested that complex histone modification patterns encode detailed information about the regulation of associated genes and promoters [4]. Similarly, the methylation status of a gene promoter can have an important effect on the activity status of the corresponding gene and hypermethylation-associated silencing of tumor suppressor genes has been shown to play a prominent role in human cancers [5].
Epigenetic mechanisms are generally considered to represent a regulatory interface between environmental cues and the genome [6]. More specifically, it has also been suggested that age-associated epigenetic changes might restrict the phenotypic plasticity for a successful adaptation to environmental changes [7]. Due to methodological limitations, however, experimental evidence supporting these notions has remained scarce. Based on the analysis of DNA methylation patterns in peripheral blood samples from monozygotic twins it has been suggested that widespread epigenetic variations arise during the lifetime of human individuals [8]. A later study also described profound methylation changes when unfractionated peripheral blood samples were compared that had been sampled from the same individual 11–16 years apart [9]. However, it was also noted that variations in the cellular composition of tissues are prevalent in older individuals and that the observed epigenetic variations might be a consequence of the tissue heterogeneity [10]. In agreement with this notion, an independent study concluded that tissue-specific interindividual DNA methylation differences are very small and that major variations could only be observed when different tissues were compared [11]. As such, the magnitude of age-related epigenetic changes has remained unresolved.
Human skin represents an organ with several advantages for studying age-associated and environmentally-induced epigenetic changes. The skin is directly exposed to many environmental factors and shows an invariant age-dependent phenotype characterized by changes in the vascular network, reduction in the number of melanocytes and Langerhans cells, decreased thickness of the epidermis, lower levels of specific collagen types and other factors [12], [13]. Skin samples can be obtained from healthy individuals, either as suction blisters or via punch biopsies. Importantly, these samples contain defined tissue layers with a very high degree of cellular homogeneity, with epidermal tissues consisting mainly of keratinocytes and dermal tissues consisting mainly of fibroblasts. We have now used a recently developed array technology [14] for the analysis of complex DNA methylation patterns in skin samples obtained from healthy human volunteers. Our results identify a comparably small, but consistent and statistically significant trend towards DNA hypermethylation in aged samples and consistent hypomethylation of a small set of markers in sun-exposed samples.
Results
The recent development of robust arrays for comprehensive DNA methylation analysis provided a sensitive tool for the identification of limited DNA methylation changes in the human genome. We investigated DNA methylation patterns in human skin samples, because the tissue is known to undergo well-defined age-related and sun exposure-related phenotypic changes. Skin samples were obtained as suction blisters (epidermis) or as punch biopsies (epidermis and dermis, see Figure 1A). Punch biopsies were taken both from the outer forearm (sun-exposed area) and the inner arm (sun-protected area) and were separated into epidermal and dermal tissues by dispase II treatment. Samples were obtained from healthy male and female donors and from two distinct age groups to analyze the influence of various intrinsic and extrinsic factors on the genomic DNA methylation patterns (Figure 1B, see Table S1 for details). We used Illumina Infinium arrays to determine the methylation status of 27,578 CpG dinucleotides in the human genome. The analysis of 50 samples generated 50 million data points for further analysis (see Materials and Methods for details).

The robustness of array-predicted methylation patterns was characterized by an initial bisulfite sequencing experiment. Three genes with methylation scores (beta values) of 0 (unmethylated), 0.5 (partially methylated) and 1 (completely methylated) were arbitrarily chosen from a pilot array. The analysis of bisulfite sequencing results revealed a very good correlation between the array data and the bisulfite sequencing results for 2 genes (Figure S1). For the partially methylated gene (DIRAS3, Figure S1), bisulfite sequencing indicated a higher methylation level than the array, which can probably be explained by strand-specific PCR bias in the bisulfite sequencing reaction [15]. Indeed, bisulfite sequencing of a second partially methylated gene (ZIM2, Figure S1) showed a very good correlation between the array data and the bisulfite sequencing results. It should be noted that our complete bisulfite sequencing validation set consisted of 10 PCR amplicons (see below in Results) and that bisulfite sequencing confirmed the array-predicted results in 9 out of 10 cases. This strongly suggests that the array produces reliable methylation results.
In an initial step of our analysis, we analyzed methylation profiles from suction blister samples obtained from 5 healthy male donors aged 26–35 years. The methylation profiles (see Figure 1C for an example) showed that the majority (86%) of CpG island associated markers had a methylation score of 0.2 or less and were therefore considered unmethylated. Consistent with other published reports [16], [17], 5% of the CpG island associated markers showed a methylation score of 0.8 or more and were therefore considered methylated. The fraction of methylated markers was substantially higher (29%) in regions not associated with CpG islands, which is again consistent with the general patterns of human DNA methylation reported in other studies [16], [18].
Comparisons between individual methylation profiles also showed a very high degree of similarity between samples (Figure 1D), with correlation coefficients ranging from 0.97–0.98. This substantial similarity of methylation patterns was important for our overall study design, because it permitted the identification of statistically significant changes in a comparably low number of samples. The methylation pattern observed in the pilot sample set obtained from 5 male volunteers aged 26–35 years was largely confirmed in an independent set of epidermis samples obtained from punch biopsies of 5 healthy female individuals aged 19–24 years. However, we also noted a prominent group of markers that had a substantially higher methylation score in the second sample set (Figure 1E). Notably, the vast majority (547 out of 595, Figure 1E) of these markers were derived from the X-chromosome, which is known to be dosage compensated by DNA methylation in females [16], [17], [19]. The observed methylation differences therefore reflect gender differences in the donor groups (Table S1) and further illustrate the sensitivity of the methylation array.
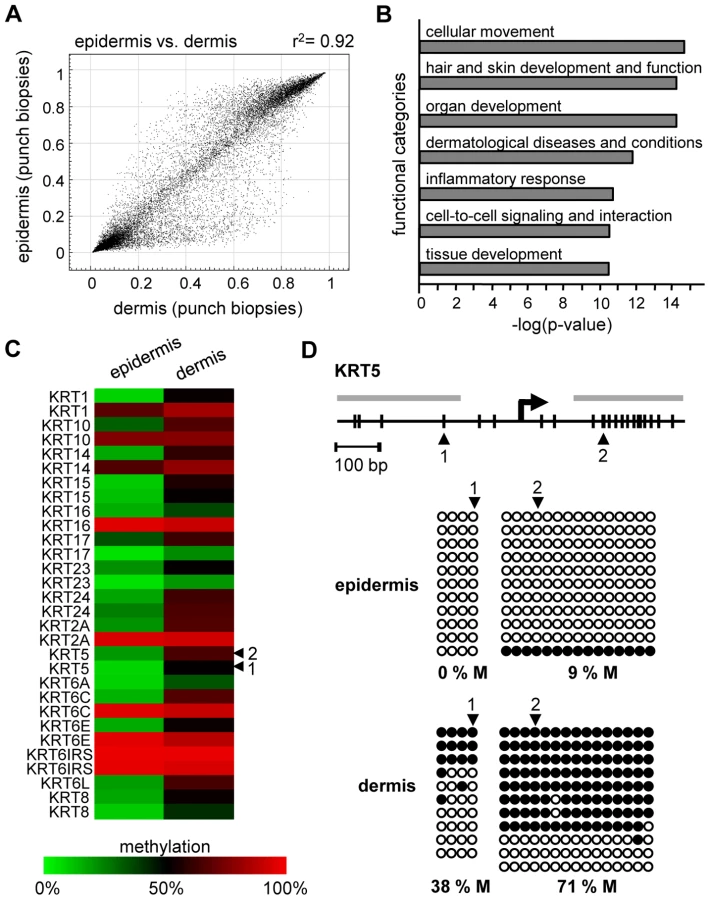
We also analyzed the methylation profiles from all dermis samples (n = 20; outer forearm and inner arm) of 10 healthy female individuals aged 18–72 years (Table S1). The results again revealed substantial interindividual similarities, with correlation coefficients between 0.95 and 0.98. However, when the dermal methylation profiles were compared to the matched epidermal methylation profiles of the punch biopsy samples (n = 20), both tissues showed remarkable differences (Figure 2A), with 742 markers being considerably more (Δ(beta)≥0.2) methylated in the epidermis and 1034 markers being more methylated in the dermis. Genes associated with differentially methylated markers were strongly enriched in functional categories associated with the molecular and cellular characteristics of (skin) tissue development (Figure 2B). These findings are consistent with a role of DNA methylation in the regulation of cell type-specific gene expression patterns and illustrate the differences in the cellular composition of epidermis (keratinocytes) and dermis (fibroblasts). In this context, several keratin genes provided a notable example for differential methylation (Figure 2C), which was validated by bisulfite sequencing of the KRT5 gene (Figure 2D). The results showed low levels of KRT5 methylation in the epidermis (0% in the promoter region, 9% in the downstream CpG island) and substantially higher methylation levels in the dermis (38% in the promoter region, 71% in the downstream CpG island). In agreement with a role of DNA methylation in the silencing of cell type-specific genes, KRT5 has been shown to be highly expressed in the epidermis, but not in the dermis [20]–[24].
We then extended our analysis to the comparison of methylation patterns of the two distinct age groups. Again, the comparison of suction blister methylation patterns from 5 healthy male individuals aged 65–71 years revealed a very high degree of similarity (Figure 3A), with correlation coefficients between 0.91 and 0.98. The establishment of average methylation values for young and old epidermis samples subsequently allowed the comparison of these data sets. To identify markers with relevant methylation changes, we applied a stringent cutoff at a beta value change of 0.2 or more, based on previously published data [14]. The results indicated that 104 markers (0.37%) showed a beta value increase by 0.2 or more in old epidermis, while only 8 markers (0.03%) showed a beta value decrease by 0.2 or more (Figure 3B). Out of the 104 hypermethylated markers, 90 were associated with CpG islands, which may be related both to the overall low methylation of CpG islands (see above) and to the overrepresentation of CpG island-associated probes on the array [14]. Together, these data suggested that skin aging is associated with predominant hypermethylation in a comparably small set of markers. Age-related hypermethylation was also confirmed by an independent statistical analysis (see Materials and Methods for details) and by inclusion of the methylation profiles from the epidermal and dermal punch biopsy samples (outer forearm and inner arm). The results showed a strong and unambiguous trend towards hypermethylation in epidermis and dermis samples from the old donor group (Figure 3C and 3D). Age-related hypermethylation was stronger in the epidermis than in the dermis (Figure 3C and 3D), which is conceivably due to the more immediate environmental exposure of the epidermis. Out of the 61 markers found to be substantially (Δ(beta)≥0.2, with a Benjamini-Hochberg adjusted P-value P(BH)<0.01) hypermethylated in epidermal punch biopsies from old female individuals, 43 were also found to be hypermethylated in suction blister samples from old male donors, that were obtained with a completely independent sampling protocol (Figure 3E). This result provided further confirmation for a distinct age-associated epigenetic shift in human skin.

To further demonstrate the specificity of the observed methylation shift, we also stratified the punch biopsy sample sets according to chronic sun exposure (outer forearm vs. inner arm). Remarkably, this comparison revealed a fundamentally different methylation change: out of the 27,578 markers analyzed, none showed a beta value increase by 0.2 or more in sun-exposed epidermis, while 14 (0.05%) showed a beta value decrease by 0.2 or more (Figure 4A). Statistical analysis confirmed the trend towards hypomethylation and also indicated that sun exposure-related hypomethylation was less pronounced than age-related hypermethylation (Figure 4B and 4C). Again, the effects appeared stronger in the epidermis than in the dermis (Figure 4B and 4C), which may be related to the more direct sun exposure of the epidermis. Importantly, global age-related and sun exposure-related DNA methylation shifts were highly significant when compared to Gaussian distributions of randomly generated methylation differences and after a global Benjamini-Hochberg adjustment of the P-values (Figure 4D). This finding further confirms our notion that aging and sun exposure cause distinct global DNA methylation changes in human skin.

Finally, to confirm the observed methylation changes on the level of individual genes we analyzed the gene-specific methylation patterns by bisulfite sequencing. Because previous reports have suggested that UV-induced mutations preferentially occur at methylated CpG dinucleotides [25], a sequencing approach also allowed us to control for the possibility that the methylation changes identified by the array might actually represent genetic polymorphisms or mutations. In a first experiment, we therefore focused on the KRT75 promoter region, which was found among the 16 markers that were substantially (Δ(beta)≤−0.2, P(BH)<0.01) hypomethylated in the sun-exposed epidermis samples. KRT75 encodes a Keratin protein that is normally not expressed in the epidermis, but can affect the intermediate filament architecture of epithelial cells [26], [27]. Results from deep sequencing of pooled samples (epidermis DNA from a sun-protected area and a sun-exposed area from 10 healthy female donors) did not reveal any genetic polymorphisms or mutations in the KRT75 5′ upstream region that could have contributed to the array result (Figure S2). Nevertheless, methylation analysis by deep bisulfite sequencing of the same region confirmed the demethylation of the CpG dinucleotide represented on the array (61% methylation in sun-protected samples, 33% in sun-exposed samples, Figure 5A). Similarly, the data also revealed demethylation for the CpG dinucleotide located immediately distal (81% methylation in sun-protected samples, 48% methylation in sun-exposed samples, Figure 5A). Together, these results validate the demethylation of sun-exposed epidermis samples at the KRT75 promoter region.

Lastly, we also sought to validate the age-associated hypermethylation shift by deep bisulfite sequencing. A probe from the SEC31L2 gene was identified among the 43 markers substantially (Δ(beta)≥0.2, P(BH)<0.01) hypermethylated both in epidermal punch biopsies and in suction blister samples from old individuals. SEC31 proteins have been shown to be important for collagen secretion in primary human fibroblasts [28] and hypermethylation-associated silencing of SEC31L2 could thus play an important role in the age-associated skin phenotype. To experimentally validate age-associated hypermethylation, we pooled equal amounts of epidermis DNA samples obtained from 5 young male (26–35 years) and 5 young female donors (19–24 years) and from 5 older male (65–71 years) and 5 older female (67–72 years) individuals, respectively, and used deep bisulfite sequencing to determine the methylation pattern of 19 CpG dinucleotides in the SEC31L2 5′ region (Figure 5B). Sequence analysis showed that SEC31L2 was mostly unmethylated in the young sample pool, but became distinctly methylated in the old sample pool (Figure 5B). Two additional, arbitrarily chosen loci that showed age-related hypermethylation on the array, DDAH2, an epigenetic marker associated with cellular differentiation [29], [30], and TET2, a putative tumor suppressor gene [31], also were substantially hypermethylated in old epidermis samples (Figure 5C and 5D). These results provide direct confirmation for the array-based findings and again illustrate that aging and sun exposure of human skin are characterized by distinct epigenetic variations.
Discussion
The molecular pathways contributing to human aging are currently being investigated in many experimental contexts. It is widely assumed that epigenetic changes play a fundamental role in establishing gene expression patterns specific for aged cells and tissues [1]. However, the experimental evidence to support this notion has been limited. For example, it was shown that global DNA methylation levels decrease during in vitro fibroblast cultivation [32], suggesting that DNA hypomethylation might be a molecular marker of aging. More recently, evidence has been provided for age-associated hypermethylation of specific loci in various model systems [1]. However, there are only few published reports that have directly analyzed this question on a genome-scale level.
A distinct age-related phenotype and a high level of cellular homogeneity establish human skin as an excellent model system to study age-related epigenetic alterations. Our results show that defined skin tissue layers (epidermis and dermis) are characterized by specific DNA methylation patterns that are highly similar between individual samples, with correlation coefficients that are commonly achieved for biological and technical replicates. Our results further show that skin aging is associated with DNA hypermethylation in less than 1% of the markers analyzed.
The availability of array-based technologies for methylation patterns analysis also allows the identification of defined epigenetic candidate biomarkers for human aging. For example, a recent study has used Illumina GoldenGate methylation arrays to interrogate the methylation status of 1505 CpG dinucleotides representing 803 cancer-associated genes [33]. While this study investigated age-related methylation changes in various primary human tissues, the analysis was restricted to cancer-associated genes and did not account for the cellular heterogeneity of the tissues analyzed. In addition, the methylation changes appeared rather minor and were only analyzed at single cytosine residues. Our data provides evidence for directed methylation shifts that were validated by bisulfite sequencing of PCR fragments containing several CpG dinucleotides beyond the cytosine residue queried by the array. Notably, the methylation changes observed by bisulfite sequencing of SEC31L2, DDAH2 and TET2 show a distinct similarity to epigenetic mutations described in human cancers [7] (see below).
The molecular steps leading to the establishment of altered methylation patterns are presently unclear. Because DNA methyltransferase mRNA levels did not show any significant differences between old and young and between sun-exposed and non-exposed samples (Figure S3), the observed DNA methylation changes do not seem to involve altered expression of DNA methyltransferase genes. An association between DNA methylation and sunlight-mediated mutagenesis has been suggested previously, based on the observation that UV-induced mutations preferentially occurred at methylated CpG dinucleotides [25]. However, when we analyzed the mutational status of KRT75 by deep sequencing, we did not detect any evidence for local genetic mutations. It is possible that the observed epigenetic changes may be influenced by more distant genetic mutations or by mutations in trans-acting epigenetic modifiers. These details will have to be investigated in future studies.
Recently, specific age-related hypermethylation has also been described at a subset of developmentally regulated genes in various human tissues [34], [35]. While the methylation differences observed in these studies were highly significant, they were also comparably small and might thus have escaped detection in our analysis. In addition, age-related hypermethylation has also been observed in the human intestine [36], [37], and has been interpreted as a preneoplastic epigenetic lesion. Age is the most significant risk factor for human cancer [38] and gene-specific hypermethylation represents one of the most consistent markers of tumorigenesis [5]. Our bisulfite sequencing results showed pronounced age-related hypermethylation of DDAH2, which encodes a key enzyme in the nitric oxide pathway. Nitric oxide plays an important role in the regulation of keratinocyte proliferation and in the development of skin cancer [39]. Similarly, our results also demonstrated age-related hypermethylation of TET2, a putative tumor suppressor gene that has recently been shown to be genetically mutated in myeloproliferative disorders [31]. These findings lend further support to the notion that age-related epigenetic changes provide a molecular link between aging and tumorigenesis.
Materials and Methods
Ethics statement
The recommendations of the current version of the Declaration of Helsinki and the guideline of the International Conference on Harmonization Good Clinical Practice (ICH GCP) were observed as applicable to a non-drug study. All donors provided written, informed consent. Punch biopsies were obtained through a clinical study approved by the Ethics Committee of the Medical Association of Hamburg (PV 3107). All volunteers provided written, informed consent.
Tissue samples
Suction blisters were obtained from the volar forearms of 10 healthy male volunteers, as described previously [40]. Suction blister roofs were taken and immediately stored at −20°C. Full-thickness skin samples (diameters: 6 mm or 4 mm) were obtained from 20 female volunteers by Sciderm GmbH (Hamburg, Germany). Biopsies were isolated from the outer forearm (sun-exposed area) and inner arm (sun-protected area), respectively. Immediately after removal, punch biopsies were transferred into DMEM medium (Gibco BRL) and stored on ice for up to 5 h. After dispase II treatment (2 U/ml, Roche) for 2 h at 37°C, epidermis and dermis were separated and stored at −80°C.
DNA and RNA isolation
Suction blister and punch biopsy samples (epidermis or dermis) were washed in DPBS (Cambrex) and homogenized using a TissueLyser (Retsch). DNA from suction blister samples was isolated using the QIAamp DNA Investigator Kit (Qiagen) according to the manufacturer's instructions. RNA and DNA from punch biopsy samples were processed with the Qiagen AllPrep DNA/RNA/Protein Mini Kit (Qiagen) as recommended by the supplier. The concentrations and purities of isolated DNA and RNA were assessed spectrophotometrically using a NanoDrop ND-1000 (Peqlab).
Array-based methylation analysis
The HumanMethylation27 BeadChip has been described previously [14]. A single BeadChip utilizes more than 1,000,000 beads per sample and generates 27,578 DNA methylation measurements. The CpGs under investigation are located in more than 13,500 promoters of well-annotated genes. Genomic DNA (500 ng) from each sample was bisulfite converted using the EZ-96 DNA Methylation Kit (Zymo Research Corporation) according to the manufacturer's recommendations. After bisulfite conversion, each sample was whole-genome amplified, enzymatically fragmented, and about 200 ng of DNA was applied to the arrays. During hybridization, the DNA molecules anneal to locus-specific DNA oligomers linked to individual bead types. After extension, the array was fluorescently stained, scanned, and the intensities of the non-methylated and methylated bead types were measured. DNA methylation values, described as beta values, were recorded for each locus in each sample and analyzed by BeadStudio (Illumina).
Data deposition
Microarray data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-202.
Statistical analysis of array data
Because the raw data from the green and the red channel of the array did not show the same shape of distribution, both channels were quantile normalized [41] separately. Subsequently, all beads of one probeID from all samples of a group were aggregated to calculate the mean values, standard deviations, detection P-values and beta values of the given probeID in the group. The detection P-value is defined as the minimum of the 2 separate P-values of the 2 variables (grn.A/B or red.A/B), where each P-value is the result of testing (Mann-Whitney-U-test) against the negative beads of each channel. Because 2 variables were measured to calculate the beta value we applied the Benjamini-Hochberg correction [42] before choosing the minimum P-value. Less than 0.1% of the probeIDs showed detection P-values >0.05 and the corresponding data were excluded from further analysis.
For differential methylation analysis, the difference of beta values was calculated by using the mean beta values of the two groups in question and the two-sample Wilcoxon test (Mann-Whitney-U-test) was used to calculate P-values. For global statements about overall methylation effects all P-values were adjusted using the Benjamini-Hochberg [42] correction.
To visualize the differential methylation trend between two groups the data points were plotted as an RS-plot (ratio over sum plot), which is a variation of the MA-plot [43]. Each point was calculated as follows: Calculate the difference (Δ(beta)) between the beta values of two groups and sort the data according to the absolute difference in decreasing order. The N-th data point for the graph is then calculated as: and on the y-Axis
and on the x-Axis.
Assuming a normal distribution of Δ(beta) (P(BH)<0.01) values, the t-test was used to test the distributions of the experimental data (sd<0.1) against a random normal distribution (mean = 0.0, sd = 0.1). This provided P-values for the significance of the observed global methylation shifts.
Bisulfite sequencing
Genes were randomly chosen from array-predicted groups. For bisulfite treatment of DNA, the EpiTect Bisulfite Kit (Qiagen) was used according to the manufacturer's instructions. Up to 300 ng DNA was used. Deaminated DNA was amplified by PCR using the following primers: Krt5_1_for GTTGTTTGGAAAAGTGTAAGAGTAGATTAT, Krt5_1_rev TACCTTACAACACTAATCTCTTAACAACAA, Krt5_2_for AGTGTTTGGTTTTTTTGTTTTATTAGG, Krt5_2_rev CCCCCAAATTATAAAAACTCC. PCR conditions were as follows: 95°C for 3 min followed by 40 cycles at 95°C for 30 sec, annealing temperature for 40 sec and 72°C for 45 sec. At last, the reaction was incubated at 72°C for 3 min. PCR products were gel-extracted using the QIAquick gel extraction Kit (Qiagen) and cloned using the TOPO TA cloning Kit for sequencing (Invitrogen), according to the manufacturer's instructions.
Deep bisulfite sequencing
Equal amounts of DNA from each skin sample were pooled according to age and sun exposure, respectively. The DNA pools were bisulfite treated by using the EpiTect Bisulfite Kit (Qiagen) according to the manufacturer's instructions. KRT75, DDAH2, TET2 and SEC31L2 fragments were amplified by PCR, using the primers KRT75_for GGTTTGTATTAATATAAGATGTTTGGATAG, KRT75_rev AACCACTAACTAATTCCCTAACACC, DDHA2_ for TAGGGTAGAAGTTAGGAATTAAGAAGG, DDAH2_rev CCAAACCCACCCAAATCTAA, TET2_for GAGAAATTTATTTTAATTTGTGAGA, TET2_rev TAAAAACCTATATTTTTAAAAACCC, SEC31L2_for GTTTGGGGTTTTTGGTAGTAGAGA, SEC31L2_rev CAACAATAAACAAAAAAACCCTCAT. PCR conditions were as follows: 95°C for 3 min followed by 40 cycles at 95°C for 30 sec, annealing temperature for 40 sec and 72°C for 45 sec, followed by a 3 min incubation at 72°C. PCR products were subsequently purified using the QIAquick gel extraction Kit (Qiagen). For sequencing, equimolar amounts of all amplicons were combined in a single tube. Ligation of adaptor sequences and pool-specific tags and Roche 454 sequencing was provided by GATC (Konstanz, Germany) and LGC Genomics (Berlin, Germany). Sequencing data were processed using DNAstar Lasergene 8.0 software.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FragaMF
EstellerM
2007 Epigenetics and aging: the targets and the marks. Trends Genet 23 413 418
2. BirdA
2007 Perceptions of epigenetics. Nature 447 396 398
3. SuzukiMM
BirdA
2008 DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9 465 476
4. KouzaridesT
2007 Chromatin modifications and their function. Cell 128 693 705
5. EstellerM
2002 CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 21 5427 5440
6. JaenischR
BirdA
2003 Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33 Suppl 245 254
7. FeinbergAP
2007 Phenotypic plasticity and the epigenetics of human disease. Nature 447 433 440
8. FragaMF
BallestarE
PazMF
RoperoS
SetienF
2005 Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102 10604 10609
9. BjornssonHT
SigurdssonMI
FallinMD
IrizarryRA
AspelundT
2008 Intra-individual change over time in DNA methylation with familial clustering. Jama 299 2877 2883
10. MartinGM
2005 Epigenetic drift in aging identical twins. Proc Natl Acad Sci U S A 102 10413 10414
11. EckhardtF
LewinJ
CorteseR
RakyanVK
AttwoodJ
2006 DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38 1378 1385
12. GilharA
UllmannY
KarryR
ShalaginovR
AssyB
2004 Ageing of human epidermis: the role of apoptosis, Fas and telomerase. Br J Dermatol 150 56 63
13. BaumannL
2007 Skin ageing and its treatment. J Pathol 211 241 251
14. BibikovaM
LeJ
BarnesB
Saedinia-MelnykS
ZhouL
2009 Genome-wide DNA methylation profiling using Infinium assay. Epigenomics 1 177 200
15. WarneckePM
StirzakerC
MelkiJR
MillarDS
PaulCL
1997 Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res 25 4422 4426
16. WeberM
HellmannI
StadlerMB
RamosL
PaaboS
2007 Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39 457 466
17. IllingworthR
KerrA
DesousaD
JorgensenH
EllisP
2008 A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol 6 e22 doi:10.1371/journal.pbio.0060022
18. BallMP
LiJB
GaoY
LeeJH
LeProustEM
2009 Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 27 361 368
19. HeardE
ClercP
AvnerP
1997 X-chromosome inactivation in mammals. Annu Rev Genet 31 571 610
20. TobinDJ
2006 Biochemistry of human skin–our brain on the outside. Chem Soc Rev 35 52 67
21. MollR
DivoM
LangbeinL
2008 The human keratins: biology and pathology. Histochem Cell Biol 129 705 733
22. ProkschE
BrandnerJM
JensenJM
2008 The skin: an indispensable barrier. Exp Dermatol 17 1063 1072
23. ByrneC
TainskyM
FuchsE
1994 Programming gene expression in developing epidermis. Development 120 2369 2383
24. TroyTC
TurksenK
2005 Commitment of embryonic stem cells to an epidermal cell fate and differentiation in vitro. Dev Dyn 232 293 300
25. IkehataH
KawaiK
KomuraJ
SakatsumeK
WangL
2008 UVA1 genotoxicity is mediated not by oxidative damage but by cyclobutane pyrimidine dimers in normal mouse skin. J Invest Dermatol 128 2289 2296
26. WinterH
LangbeinL
PraetzelS
JacobsM
RogersMA
1998 A novel human type II cytokeratin, K6hf, specifically expressed in the companion layer of the hair follicle. J Invest Dermatol 111 955 962
27. ChenJ
JaegerK
DenZ
KochPJ
SundbergJP
2008 Mice expressing a mutant Krt75 (K6hf) allele develop hair and nail defects resembling pachyonychia congenita. J Invest Dermatol 128 270 279
28. TownleyAK
FengY
SchmidtK
CarterDA
PorterR
2008 Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J Cell Sci 121 3025 3034
29. TomikawaJ
FukatsuK
TanakaS
ShiotaK
2006 DNA methylation-dependent epigenetic regulation of dimethylarginine dimethylaminohydrolase 2 gene in trophoblast cell lineage. J Biol Chem 281 12163 12169
30. BackdahlL
HerberthM
WilsonG
TateP
CamposLS
2009 Gene body methylation of the dimethylarginine dimethylamino-hydrolase 2 (Ddah2) gene is an epigenetic biomarker for neural stem cell differentiation. Epigenetics 4 248 254
31. LangemeijerSM
KuiperRP
BerendsM
KnopsR
AslanyanMG
2009 Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 41 838 842
32. WilsonVL
JonesPA
1983 DNA methylation decreases in aging but not in immortal cells. Science 220 1055 1057
33. ChristensenBC
HousemanEA
MarsitCJ
ZhengS
WrenschMR
2009 Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 5 e1000602 doi:10.1371/journal.pgen.1000602
34. RakyanVK
DownTA
MaslauS
AndrewT
YangTP
2010 Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 20 434 439
35. TeschendorffAE
MenonU
Gentry-MaharajA
RamusSJ
WeisenbergerDJ
2010 Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 20 440 446
36. AhujaN
LiQ
MohanAL
BaylinSB
IssaJP
1998 Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 58 5489 5494
37. MaegawaS
HinkalG
KimHS
ShenL
ZhangL
2010 Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20 332 340
38. ErshlerWB
LongoDL
1997 Aging and cancer: issues of basic and clinical science. J Natl Cancer Inst 89 1489 1497
39. Bruch-GerharzD
RuzickaT
Kolb-BachofenV
1998 Nitric oxide in human skin: current status and future prospects. J Invest Dermatol 110 1 7
40. SudelKM
VenzkeK
Knussmann-HartigE
MollI
StabF
2003 Tight control of matrix metalloproteinase-1 activity in human skin. Photochem Photobiol 78 355 360
41. BolstadB
2001 Probe Level Quantile Normalization of High Density Oligonucleotide Array Data. Berkeley University of California
42. BenjaminiY
HochbergY
1995 Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B 57 289 300
43. BolstadBM
IrizarryRA
AstrandM
SpeedTP
2003 A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19 185 193
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Common Genetic Variants near the Brittle Cornea Syndrome Locus Influence the Blinding Disease Risk Factor Central Corneal Thickness
- All About Mitochondrial Eve: An Interview with Rebecca Cann
- Aging and Chronic Sun Exposure Cause Distinct Epigenetic Changes in Human Skin
- The Relationship among Gene Expression, the Evolution of Gene Dosage, and the Rate of Protein Evolution
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy