
Cirkulující nádorová DNA v krvi a její využití jako potenciálního biomarkeru nádorových onemocnění
Circulating Tumor DNA in Blood and Its Utilization as a Potential Biomarker for Cancer
Pursuing sensitive methods for detection and monitoring of oncologic diseases, that would limit the stress for patients, represents a long‑standing challenge in cancer diagnostics. As an ideal non‑invasive biomarkers may be considered ‑ biological molecules that can be detected in blood and that provide most relevant picture about the state and development of disease. In fact, all types of cancer cells carry somatic mutations that enable the cells to escape from regulation and to grow and progress. These mutations are only present in the DNA of tumor cells and thus are hallmarks of cancer cells. Genotyping of tumor tissues becomes a common technique in clinical oncology, but it has its limits. Tissue biopsy only yields information about a very small area of tumor at the time of extraction and in some cases it is difficult or impossible to obtain the tissue sample. Furthermore, it is an invasive method that can stress patients. Analysis of circulating tumor DNA from blood – the so ‑ called liquid biopsy – represents one possible solution. Dying tumor cells release fragments of their DNA into the blood stream. From blood, they can be isolated and subjected to analysis using new, sensitive and precise methods that detect genomic changes. These changes are evolving over time because cancer disease is characterized by evolution and ability to select new mutations that bring growth advantages or resistance to treatment. Our inability to capture the heterogeneity during tumor development is one of the major reasons responsible for failure of cancer treatment. Recent technological progress in detection and characterization of circulating DNA could enable tumor evolution monitoring in real time and become a guideline for an accurate and prompt treatment choice.
Key words:
circulating tumor DNA – tumor biomarkers – biopsy – liquid biopsy – blood – mutation
This study was supported by the European Regional Development Fund and the State Budget of the Czech Republic – RECAMO, CZ.1.05./2.1.00/03.0101, by the project MEYS – NPS I – LO1413, GACR 13-00956S, MH CZ – DRO (MMCI, 00209805) and BBMRI_CZ (LM2010004).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
7. 4. 2015
Accepted:
3. 7. 2015
Autoři:
E. Ondroušková; R. Hrstka
Působiště autorů:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Vyšlo v časopise:
Klin Onkol 2015; 28(Supplementum 2): 69-74
doi:
https://doi.org/10.14735/amko20152S69
Souhrn
Nalezení dostatečně citlivé metody pro detekci a monitorování onkologických onemocnění, která by zároveň nepředstavovala zbytečnou zátěž pro pacienta, je dlouholetou výzvou v nádorové diagnostice. Ideálním cílem jsou neinvazivní biomarkery – biologické molekuly, které by bylo možné detekovat z krve, a které by podaly co nejpřesnější obraz o stavu a vývoji onemocnění. V podstatě všechny typy nádorových buněk obsahují somatické mutace, které jim umožňují neregulovaně růst a vyvíjet se. Tyto mutace jsou přítomny pouze v DNA nádorových buněk a představují tedy vhodný biomarker. Genotypizace nádorových tkání se stává v klinické onkologii běžným postupem, má však své limity. Biopsie tkáně podává informaci jen o velmi malé lokalitě nádoru v konkrétním čase odběru a v některých případech je obtížné nebo téměř nemožné vzorek vůbec odebrat. Navíc, jako každá invazivní metoda, představuje zátěž pro pacienty. Řešením by se mohla stát analýza cirkulující nádorové DNA odebrané přímo z krve – tzv. tekuté biopsie. Umírající nádorové buňky uvolňují fragmenty své DNA do krevního oběhu, odkud mohou být izolovány, a díky novým citlivým a přesným metodám podrobeny analýze genomových změn. Tyto změny se navíc v čase vyvíjejí, protože nádorové onemocnění je charakterizováno postupnou evolucí a schopností selektovat nové mutace přinášející růstové výhody či rezistenci k aplikované léčbě. Nezachycení těchto změn je přitom častou příčinou neúspěchu léčby. Současné technologické pokroky v detekci a charakterizaci cirkulující nádorové DNA by mohly umožnit monitorování vývoje nádoru v reálném čase a stát se vodítkem pro včasné a přesné rozhodnutí o nejvhodnější léčbě.
Klíčová slova:
cirkulující nádorová DNA – nádorové biomarkery – biopsie – tekutá biopsie – krev – mutace
Úvod
Biomarkery využívané v současné době při diagnostice a monitorování odpovědi na léčbu jsou většinou proteiny, jejichž hladina v krvi může být snadno detekována a změna exprese je spojena s určitým typem nádorového onemocnění. V poslední době se však díky rozvoji citlivějších technologií detekce stává také DNA cirkulující v krevním oběhu alternativním zdrojem informací o nádorovém onemocnění.
První nález DNA cirkulující v lidské krvi byl popsán již v roce 1948 [1] a její zvýšená hladina v krvi onkologických pacientů potom o 29 let později [2]. V roce 1994 se u pacientů trpících myelodysplastickým syndromem nebo akutní myelogenní leukemií poprvé podařilo prokázat, že tato DNA obsahuje mutace typické pro nádorové buňky, konkrétně mutovaný gen N‑ ras [3]. DNA vyskytující se v krvi je souhrnně označována jako cell‑free DNA (cfDNA), cirkulující nádorová DNA potom konkrétně ctDNA (circulating tumor DNA). V krvi zdravých jedinců byla detekována cfDNA v průměrné koncentraci 13 ng/ ml, zatímco u pacientů s různými typy rakoviny dosahovala průměrné koncentrace 180 ng/ ml [2]. Fragmenty DNA detekovatelné v krvi mohou být velmi krátké (70 – 200 bp) nebo dlouhé až 21 kb, za jejich původce byly proto považovány apoptotické či nekrotické buňky [4]. Některé studie navíc naznačují i možnost, že je cfDNA buňkami aktivně sekretována [5]. Zbytky mrtvých buněk a jejich DNA jsou běžně odstraňovány buňkami imunitního systému, např. makrofágy. Přesto ale v krvi zůstává v mnoha případech dostatečné množství genetického materiálu umožňujícího analýzu, obzvláště u pacientů s rozvinutým nádorovým onemocněním v pozdějších stadiích (obr. 1). V těchto případech může být nádorová DNA dokonce převažujícím typem DNA detekovatelné v krvi. Množství detekovatelné ctDNA se liší také v závislosti na typu nádorového onemocnění. Její podíl může např. představovat přibližně 0,01 – 5 % cfDNA u melanomu [6], méně než 1 % u nemetastazujících kolorektálních nádorů nebo naopak převažujícím typem DNA v plazmě u rozvinutých metastazujících nádorů [7]. Poměr cfDNA/ ctDNA se liší také v průběhu léčby, jak je detailně ukázáno na příkladu rakoviny žaludku [8]. ctDNA se může stát přesným diagnostickým nástrojem, protože při její analýze jsou určeny konkrétní genetické a epigenetické změny. Tyto změny zahrnují mutace v DNA, ztrátu heterozygozity, integraci virových genomů, hypermetylaci tumor ‑ supresorových genů či změny v mitochondriální DNA [9]. Navíc je životnost ctDNA v krevním oběhu mnohem kratší než u proteinů (řádově jen několik hodin), takže podává i přesnější obraz o momentálním stavu a vývoji onemocnění [10].

V tomto přehledovém článku shrneme výhody a nevýhody diagnostiky založené na detekci ctDNA, příklady, kdy byla úspěšně využita při monitorování nebo diagnostice nádorových onemocnění a metodiku, která je k detekci ctDNA používána.
Tkáňová vs. tekutá biopsie
Biopsie a histologické vyšetření tkáně jsou ústředním postupem při diagnostice (nejen) nádorových onemocnění. Její limity ale velmi dobře ukázala nedávná studie heterogenity nádorových tkání, konkrétně nádoru ledvin. Po provedení sekvenace DNA získané odběrem vzorků tkáně z různých oblastí téhož nádoru vědci zjistili, že genetická diverzita je větší, než se dosud předpokládalo. Pouze třetina mutací byla shodná ve všech odebraných vzorcích, jinak se od sebe části nádoru podstatně lišily a ještě větší odlišnosti pak vykazovaly sekundární nádory v jiných částech těla pacienta [11]. Při provedení jediné biopsie je tedy pravděpodobné, že vzorek zdaleka nepostihne všechny podstatné mutace, které mohou ovlivňovat odpověď na léčbu a prognózu onemocnění. Biopsie jsou však invazivní a potenciálně rizikové, mnohdy i obtížně proveditelné v případě nedostupných nebo křehkých tkání, jako jsou např. plíce. Navíc opakované provádění tkáňových biopsií je pro pacienty náročné, a proto jsou lékaři ve své diagnostice a plánování léčby často odkázáni na informace, které rychle zastarávají v kontextu toho, jak se nádor dále vyvíjí.
Nadějnou alternativou se tedy stávají tzv. tekuté biopsie – odběry krve, z níž lze izolovat ctDNA či sekretované proteiny. V klinické praxi je detekce proteinových markerů v krvi běžně používána při diagnostice rakoviny prostaty (prostate ‑ specific antigen – PSA), jater (α ‑ fetoprotein), střeva (carcinoembryonic antigen – CEA), prsu (CA 15.3, cancer antigen) [12] a dalších. V některých případech však diagnóza založená na těchto markerech vede k falešně pozitivním nálezům, protože jejich hladina v krvi může být zvýšena i z jiných důvodů [13]. U většiny nádorů navíc nejsou známy žádné vhodné sekretované proteinové biomarkery, které by bylo možno stanovovat přímo z krevních odběrů.
U většiny pacientů však lze v krvi detekovat ctDNA a pomocí stále přesnějších technik zaměřených na analýzu DNA sestavovat důkladnější obraz mutačních změn v nádorových buňkách, než by umožnila tkáňová biopsie. Protože odběr krve lze provádět v podstatě kdykoliv, otevírá se možnost rychlého testování, zda je zvolená léčba vhodná či zda nedošlo v důsledku terapie k selekci a dalšímu růstu rezistentní populace nádorových buněk. Nevýhodou prozatím zůstává to, že jde o metodu novou a zaváděnou, takže dosud publikované studie jsou provedeny na poměrně malém počtu pacientů a jen u některých typů nádorů. Hladina ctDNA v krvi se liší případ od případu a může být obtížně detekovatelná, obzvlášť u malých nádorů v raných stadiích. Metody pro zpřesnění detekce i malého množství ctDNA se ale stále vyvíjejí a jsou čím dál citlivější, takže lze očekávat, že se tento diagnostický postup časem stane běžným a pro pacienty přínosným.
Využití ctDNA při diagnostice a monitorování nádorových onemocnění
DNA obsahující somatické mutace je vysoce specifická po nádorové buňky, a může se tedy stát optimálním markerem. V solidních nádorech, jako jsou např. kolorektální či prsní tumory, bylo zjištěno až 80 mutovaných genů, které jsou detekovatelné prakticky v každé nádorové buňce, ale nejsou přítomny v DNA zdravých buněk [14].
Velmi obsáhlý vzorek pacientů byl analyzován skupinou dr. Diaze [7]. Technologií založenou na digitální PCR (polymerace chain reaction) zhodnotili využitelnost ctDNA k detekci nádorů u 640 pacientů s různými typy rakoviny. Detekovatelnou hladinu ctDNA zachytili u > 75 % pacientů s rozvinutou rakovinou hlavy a krku, močového měchýře, slinivky, vaječníku, prsu, melanomu, hepatocelulárních buněk, ale u < 50 % pacientů s nádorem v mozku, ledvinách, prostatě nebo thyroidních buňkách. To ukazuje, že množství ctDNA není závislé jen na stupni rozvinutosti nádorového onemocnění, ale i na tom, v jaké tkáni se vyskytuje. Po srovnání hladiny ctDNA a cirkulujících nádorových buněk v této studii se také mj. ukázalo, že tyto dva ukazatele jsou vzájemně nezávislé a že cirkulující nádorové buňky nejsou hlavním zdrojem ctDNA [7].
Modifikace metody zvané BEAMing(beads, emulsion, amplification and magnetics) zvýšila citlivost detekce ctDNA a umožnila studii na 18 pacientech s rakovinou střeva. Hladina ctDNA byla sledována po operaci, kdy poklesla o 99 %, ale v mnoha případech nezmizela zcela. V případech, kdy zůstala ctDNA detekovatelná i po chirurgickém odstranění nádoru, došlo až na jednu výjimku k relapsu onemocnění. Naopak návrat onemocnění nebyl zaznamenán u žádného pacienta, u kterého nebyla ctDNA detekována [15]. Tento výsledek ukázal, že ctDNA může indikovat, zda bude pacient po chirurgickém odstranění nádoru potřebovat i následnou chemoterapii.
Studie srovnávající citlivost proteinových markerů CEA a CA19 - 9 s metodou založenou na analýze ctDNA byla provedena u sedmi pacientů s kolorektálním karcinomem. V DNA izolované z krve bylo detekováno několik nejčastěji mutovaných genů (APC, KRAS, TP53, PIK3CA a BRAF). Hladina ctDNA byla sledována po operaci v několika časových intervalech, vždy úzce korelovala s aktuálním klinickým stavem pacienta a projevila se jako univerzálnější marker pro monitorování klinického vývoje nemoci než běžně používané CEA a CA19 - 9 [16].
Na signální dráhy KRAS, BRAF, EGFR a TP53 je zacíleno mnoho chemoterapeutik [17,18]. Pro predikci léčebné odpovědi je tedy důležité znát a průběžně monitorovat případný vznik mutací v těchto genech a sledovat tak evoluci v genomu nádorových buněk způsobenou selekčním tlakem chemoterapeutik [19]. U kolorektálních nádorů s nemutovaným KRAS se často indikuje léčba cílená na EGFR, ale téměř vždy se během několika měsíců objeví rezistence k této terapii. Analýza ctDNA 24 pacientů ukázala, že během 5 – 6 měsíců od zahájení léčby byla u devíti pacientů, jejichž nádorové onemocnění bylo původně wild-type KRAS, detekována mutace v tomto genu, a tedy že lze metodu monitorování ctDNA využít k predikci odpovědi na léčbu [20]. Podobně, detekce EGFR mutací v ctDNA může být využita k předpovědi vývoje onemocnění a rezistence k chemoterapii u pacientů s rakovinou plic. Ve studii provedené Mackem et al byl analyzován efekt aktivujících mutací v genu pro EGFR detekovaných v ctDNA u nemalobuněčných nádorů plic. U všech pacientů (6 ze 49), u kterých byl tento typ mutace detekován, byl pozorován prodloužený medián OS [21].
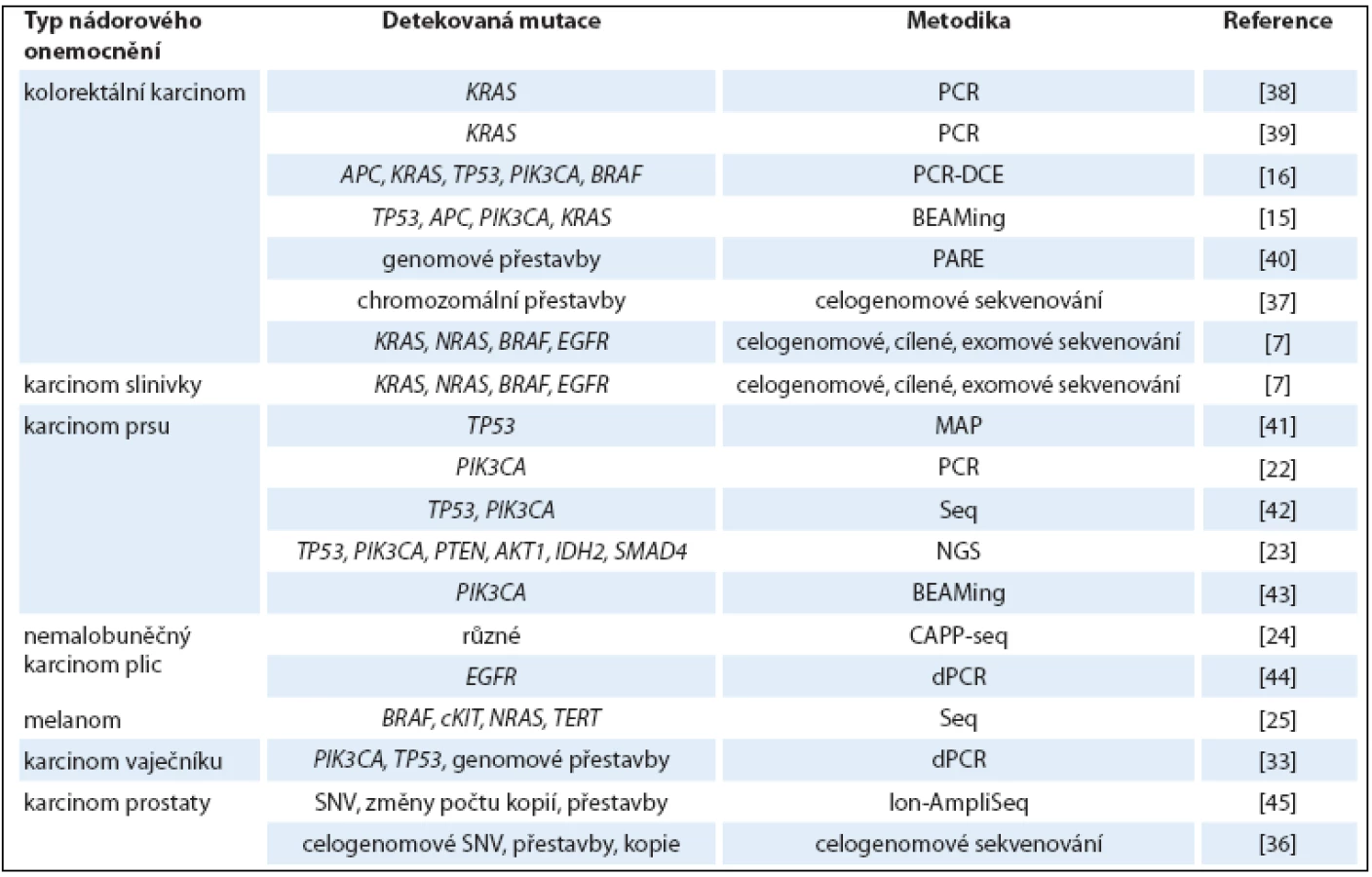
Při analýze ctDNA u pacientek s rakovinou prsu, založené na detekci PIK3CA mutace, byla tato ctDNA v krvi nalezena pouze u rozvinutějších metastatických stadií onemocnění, ale v žádném z 30 vzorků s diagnostikovaným lokalizovaným nádorem. Tento výsledek naznačuje, že ctDNA ‑ PIK3CA detekce je využitelná až u pokročilejších stadií nemoci [22]. Podobně se analýza mutací v genech TP53, PIK3CA, PTEN, AKT1, IDH2 a SMAD4 v ctDNA z plazmy osvědčila jako rovnocenná metoda s tkáňovou biopsií ve studii srovnávající vzorky 17 pacientek s metastatickými karcinomy prsu [23]. Rovněž u nemalobuněčného karcinomu plic se metodika založená na sekvenaci ctDNA osvědčila jako slibná neinvazivní metoda. Hladina ctDNA v plazmě u 13 pacientů korelovala s velikostí nádoru a ukázala se být citlivější při vyhodnocování brzké odpovědi na léčbu v porovnání s konvenčním radiografickým přístupem [24]. U pacientů s metastatickým melanomem (studie zahrnující 12 pacientů) byly v ctDNA detekovány mutace v genech BRAF, cKIT, NRAS a TERT. Hladina ctDNA korelovala s klinickými a radiologickými výsledky a v jednom případě její snížení předcházelo pozdější regresi nádoru [6]. Přehled studií detekujících ctDNA u pacientů s různými typy nádorů je shrnut v tab. 1.
Metodika
Pro širší využití a zavedení testování ctDNA do klinické praxe je potřeba nastavit metodiku uchování a zpracování krevních vzorků, stejně jako zvolit nejvhodnější metodu pro následnou detekci a analýzu ctDNA. DNA může být izolována z plazmy nebo séra, přičemž je potřeba při zpracování vzorků zabránit popraskání membrány krevních buněk a kontaminaci jejich DNA. Plazma je preferována před sérem právě díky nižší pravděpodobnosti kontaminace DNA z bílých krvinek, navíc koncentrace cfDNA v čerstvé plazmě, ač nižší než v séru, přesněji odpovídá koncentraci cfDNA v krevním oběhu [25,26]. cfDNA z ní může být izolována pomocí komerčně dostupných kitů. cfDNA pocházející z nádorových i nenádorových buněk může být kvantifikována pomocí PCR v reálném čase (qPCR) založené na amplifikaci ALU sekvencí nebo jiných markerů (β ‑ globin, β ‑ actin) [27]. Protože však, jak již bylo uvedeno dříve, je DNA pocházející z nádorových buněk většinou ve výrazné menšině, výzvou se stává následná analýza nádorově‑specifických mutací v ctDNA. V tab. 2 jsou uvedeny používané metody, včetně srovnání jejich senzitivity a specificity.

Pro detekci mutací v omezeném počtu genů se jako nejvhodnější jeví různé varianty digitální PCR – na mikročipech či mikrokapkách [28,29] nejlépe v kombinaci s metodou BEAMing, která je založena na zachycení DNA na magnetické kuličky a následné analýze pomocí digitální PCR. ctDNA může být s její pomocí detekována i v případech, kdy ji DNA ze zdravých buněk přečísluje v poměru 10 000 : 1 [30]. Technologie sekvenování nové generace (next-generation sequencing – NGS) jsou aplikovatelné na analýzu DNA v plazmě a umožňují podrobnější detekci mutací v rozsáhlejších oblastech genomu. Jako příklad lze uvést nedávnou studii provedenou na 107 vzorcích plazmy odebraných od pacientů s rakovinou plic. Při analýze DNA z primárního nádoru bylo detekováno 50 mutací v různých genech, z nichž 26 se podařilo detekovat také v krevních vzorcích, se senzitivitou 58 % a specificitou 87 % [31]. Pro detailní analýzu byla NGS technologie využita např. při dlouhodobém sledování pacientky s nádorem prsu, u které bylo analyzováno 300 genů v primárním nádoru, v metastázách a v ctDNA. Sekvenování ctDNA umožnilo sledovat vývoj nádorových buněk při vznikající rezistenci k terapii a bylo využito k identifikaci de novo mutací vznikajících v důsledku aplikované protinádorové terapie [32].
Na sekvenování specifických genomových oblastí v plazmové DNA byly využity varianty cíleného hloubkového sekvenování: TAM ‑ seq (tagged ‑ amplicon deep sequencing) [33], Safe ‑ Seq [34]nebo CAPP ‑ Seq (capture‑based sequencing) [24]. Detekovat lze v ctDNA také chromozomální přestavby, jako jsou translokace nebo ztráta či získání chromozomálních regionů, což představuje vysoce specifickou a senzitivní metodu pro detekci nádorové DNA [35]. V některých případech byla metoda celogenomového sekvenování aplikována přímo na analýzu DNA obsažené v plazmě a poskytla informace o somatických mutacích a změnách v počtu kopií cílových genů v rámci celého genomu [36,37].
S dalším zvyšováním citlivosti metod založených na analýze celého genomu budou technologie NGS hrát zřejmě klíčovou roli v analýze ctDNA a jejím využitím v klinické praxi.
Závěr
Analýzy nádorového onemocnění založené na tkáňové biopsii a zobrazovacích metodách by mohly být v dohledné době vhodně doplňovány také analýzou ctDNA. Tato metoda přináší řadu výhod, např. možnost častého odběru vzorků, detekce mutací v buňkách pocházejících z různých oblastí primárního nádoru i metastáz, monitorování vývoje a predikce odpovědi na léčbu. Před jejím zavedením do klinické praxe však musí být ještě optimalizovány a zavedeny standardizované postupy odběru a uchování krevních vzorků, stejně tak zvoleny nejvhodnější metody pro izolaci ctDNA a analýzu vyskytujících se mutací. Zintenzivnění výzkumu a rostoucí počet publikací v posledních letech naznačuje, že je tato metoda považována za nadějnou a přínosnou, a snad se tedy stane dalším nástrojem zvyšujícím šance pacientů v boji proti nádorovému onemocnění.
Práce byla podpořena Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (RECAMO, CZ.1.05/2.1.00/03.0101), projekty MŠMT – NPU I – LO1413, GAČR 13-00956S, MZ ČR – RVO (MOÚ, 00209805) a BBMRI_CZ (LM2010004).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Eva Ondroušková, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: eva.ondrouskova@mou.cz
Obdrženo/Submitted: 7. 4. 2015
Přijato/Accepted: 3. 7. 2015
Zdroje
1. Mandel P, Metais P. Les acides nucleiques du plasma sanguin chez l‘homme. C R Seances Soc Biol Fil 1948; 142(3 – 4): 241 – 243.
2. Leon SA, Shapiro B, Sklaroff DM et al. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977; 37(3): 646 – 650.
3. Vasioukhin V, Anker P, Maurice P et al. Point mutations of the N ‑ ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br J Haematol 1994; 86(4): 774 – 779.
4. Stroun M, Anker P, Lyautey J et al. Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol 1987; 23(6): 707 – 712.
5. Stroun M, Lyautey J, Lederrey C et al. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta 2001; 313(1 – 2): 139 – 142.
6. Lipson EJ, Velculescu VE, Pritchard TS, et al. Circulating tumor DNA analysis as a real ‑ time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J Immunother Cancer 2014; 2(1): 42. doi: 10.1186/ s40425 ‑ 014 ‑ 0042 ‑ 0.
7. Bettegowda C, Sausen M, Leary RJ et al. Detection of circulating tumor DNA in early ‑ and late‑stage human malignancies. Sci Transl Med 2014; 6(224): 224ra24. doi: 10.1126/ scitranslmed.3007094.
8. Hamakawa T, Kukita Y, Kurokawa Y et al. Monitoring gastric cancer progression with circulating tumour DNA. Br J Cancer 2015; 112(2): 352 – 356. doi: 10.1038/ bjc.2014.609.
9. Gonzalez ‑ Masia JA, Garcia ‑ Olmo D, Garcia ‑ Olmo DC. Circulating nucleic acids in plasma and serum (CNAPS): applications in oncology. Onco Targets Ther 2013; 6 : 819 – 832. doi: 10.2147/ OTT.S44668.
10. Yong E. Cancer biomarkers: written in blood. Nature 2014; 511(7511): 524 – 526. doi: 10.1038/ 511524a.
11. Gerlinger M, Rowan AJ, Horswell S et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366(10): 883 – 892. doi: 10.1056/ NEJMoa1113205.
12. Jacobs EL, Haskell CM. Clinical use of tumor markers in oncology. Curr Probl Cancer 1991; 15(6): 299 – 360.
13. Djavan B, Keffer JH, Molberg K et al. False ‑ positive serum prostate ‑ specific antigen values in a patient with non‑Hodgkin lymphoma of the kidney. Urology 1995; 45(5): 875 – 878.
14. Wood LD, Parsons DW, Jones S et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318(5853): 1108 – 1113.
15. Diehl F, Schmidt K, Choti MA et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008; 14(9): 985 – 990. doi: 10.1038/ nm.1789.
16. Levy M, Benesova L, Lipska L et al. Utility of cell‑free tumour DNA for post‑surgical follow‑up of colorectal cancer patients. Anticancer Res 2012; 32(5): 1621 – 1626.
17. Gadgeel SM, Cote ML, Schwartz AG et al. Parameters for individualizing systemic therapy in non‑small cell lung cancer. Drug Resist Updat 2010; 13(6): 196 – 204. doi: 10.1016/ j.drup.2010.10.001.
18. Rosell R, Vergnenegre A, Liu B et al. Biomarkers in lung oncology. Pulm Pharmacol Ther 2010; 23(6): 508 – 514.
19. Murtaza M, Dawson SJ, Tsui DW et al. Non ‑ invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013; 497(7447): 108 – 112. doi: 10.1038/ nature12065.
20. Diaz LA Jr, Williams RT, Wu J et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012; 486(7404): 537 – 540. doi: 10.1038/ nature11219.
21. Mack PC, Holland WS, Burich RA et al. EGFR mutations detected in plasma are associated with patient outcomes in erlotinib plus docetaxel‑treated non‑small cell lung cancer. J Thorac Oncol 2009; 4(12): 1466 – 1472. doi: 10.1097/ JTO.0b013e3181bbf239.
22. Board RE, Wardley AM, Dixon JM et al. Detection of PIK3CA mutations in circulating free DNA in patients with breast cancer. Breast Cancer Res Treat 2010; 120(2): 461 – 467. doi: 10.1007/ s10549 ‑ 010 ‑ 0747 ‑ 9.
23. Rothe F, Laes JF, Lambrechts D et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol 2014; 25(10): 1959 – 1965. doi: 10.1093/ annonc/ mdu288.
24. Newman AM, Bratman SV, To J et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014; 20(5): 548 – 554. doi: 10.1038/ nm.3519.
25. Ziegler A, Zangemeister ‑ Wittke U, Stahel RA. Circulating DNA: a new diagnostic gold mine? Cancer Treat Rev 2002; 28(5): 255 – 271.
26. Lee TH, Montalvo L, Chrebtow V et al. Quantitation of genomic DNA in plasma and serum samples: higher concentrations of genomic DNA found in serum than in plasma. Transfusion 2001; 41(2): 276 – 282.
27. Rolfo C, Castiglia M, Hong D et al. Liquid biopsies in lung cancer: the new ambrosia of researchers. Biochim Biophys Acta 2014; 1846(2): 539 – 546.
28. Hrstka R, Kolarova T, Michalova E et al. Development of PCR methods and their applications in oncological research and practice. Klin Onkol 2014; 27 (Suppl 1): S69 – S74. doi: 10.14735/ amko20141S69.
29. Wang J, Ramakrishnan R, Tang Z et al. Quantifying EGFR alterations in the lung cancer genome with nanofluidic digital PCR arrays. Clin Chem 2010; 56(4): 623 – 632. doi: 10.1373/ clinchem.2009.134973.
30. Diehl F, Li M, Dressman D et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A 2005; 102(45): 16368 – 16373.
31. Couraud S, Vaca ‑ Paniagua F, Villar S et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never ‑ smokers: a proof ‑ of ‑ concept study from BioCAST/ IFCT ‑ 1002. Clin Cancer Res 2014; 20(17): 4613 – 4624. doi: 10.1158/ 1078 ‑ 0432.CCR ‑ 13 ‑ 3063.
32. De Mattos ‑ Arruda L, Weigelt B, Cortes J et al. Capturing intra ‑ tumor genetic heterogeneity by de novo mutation profiling of circulating cell‑free tumor DNA: a proof ‑ of ‑ principle. Ann Oncol 2014; 25(9): 1729 – 1735. doi: 10.1093/ annonc/ mdu239.
33. Forshew T, Murtaza M, Parkinson C et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012; 4(136): 136ra68. doi: 10.1126/ scitranslmed.3003726.
34. Kinde I, Wu J, Papadopoulos N et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 2011; 108(23): 9530 – 9535. doi: 10.1073/ pnas.1105422108.
35. Shaw JA, Page K, Blighe K et al. Genomic analysis of circulating cell‑free DNA infers breast cancer dormancy. Genome Res 2012; 22(2): 220 – 231. doi: 10.1101/ gr.123497.111.
36. Heitzer E, Ulz P, Belic J et al. Tumor‑associated copy number changes in the circulation of patients with prostate cancer identified through whole ‑ genome sequencing. Genome Med 2013; 5(4): 30. doi: 10.1186/ gm434.
37. Leary RJ, Sausen M, Kinde I et al. Detection of chromosomal alterations in the circulation of cancer patients with whole ‑ genome sequencing. Sci Transl Med 2012; 4(162): 162ra154. doi: 10.1126/ scitranslmed.3004742.
38. Kopreski MS, Benko FA, Borys DJ et al. Somatic mutation screening: identification of individuals harboring K ‑ ras mutations with the use of plasma DNA. J Natl Cancer Inst 2000; 92(11): 918 – 923.
39. Morgan SR, Whiteley J, Donald E et al. Comparison of KRAS mutation assessment in tumor DNA and circulating free DNA in plasma and serum samples. Clin Med Insights Pathol 2012; 5 : 15 – 22. doi: 10.4137/ CPath.S8798.
40. Leary RJ, Kinde I, Diehl F et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med 2010; 2(20): 20ra14. doi: 10.1126/ scitranslmed.3000702.
41. Chen Z, Feng J, Buzin CH et al. Analysis of cancer mutation signatures in blood by a novel ultra ‑ sensitive assay: monitoring of therapy or recurrence in non‑metastatic breast cancer. PLoS One 2009; 4(9): e7220. doi: 10.1371/ journal.pone.0007220.
42. Dawson SJ, Tsui DW, Murtaza M et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013; 368(13): 1199 – 1209. doi: 10.1056/ NEJMoa1213261.
43. Higgins MJ, Jelovac D, Barnathan E et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res 2012; 18(12): 3462 – 3469. doi: 10.1158/ 1078 ‑ 0432.CCR ‑ 11 ‑ 2696.
44. Yung TK, Chan KC, Mok TS et al. Single‑molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non‑small cell lung cancer patients. Clin Cancer Res 2009; 15(6): 2076 – 2084. doi: 10.1158/ 1078 ‑ 0432.CCR ‑ 08 ‑ 2622.
45. Carreira S, Romanel A, Goodall J et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med 2014; 6(254): 254ra125. doi: 10.1126/ scitranslmed.3009448.
46. National Human Genome Research Institute. [homepage on the Internet]. Available from: http:/ / www.genome.gov/ dmd/ img.cfm?node=Photos/Graphics& id=92951.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2015 Číslo Supplementum 2
- Srovnání vlivu tamoxifenu a exemestanu na tloušťku endometria
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Incidence mozkových metastáz a intrakraniální aktivita sotorasibu u pacientů s NSCLC s mutací G12C onkogenu KRAS
- Přibývající důkazy o přínosu léčebného konopí u pacientů s chronickou bolestí
Nejčtenější v tomto čísle
- Adenovírusové vektory v génovej terapii
- Nrf2 – dve tváre regulátora antioxidačného systému
- Rekombinantní protilátky a jejich využití v protinádorové terapii
- Co může přinést studium oligomerizace proteinů v procesu onkogeneze?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy