
Aktivita fosfomanomutázy 2 u pacientů s podezřením na dědičnou poruchu glykosylace
Activity of phosphomannomutase 2 in patients with suspected congenital disorder of glycosylation
Objective:
Aim of study was to establish a method for determining the activity of phosphomannomutase 2 (PMM2) and phosphomannose isomerase (PMI) as a control enzyme in isolated lymphocytes and cultured skin fibroblasts and use it as a differential diagnostic step in a group of 18 patients with suspected CDG syndrome type I.
Design:
Original paper.
Settings:
Department of Pediatrics and Adolescent Medicine, First Faculty of Medicine, Charles University in Prague and General University Hospital in Prague
Material and methods:
Cohort of samples consists of 16 isolated lymphocytes and 8 cultured skin fibroblasts lines from 18 patients from 15 unrelated families with high clinical suspicion for CDG syndrome type I. Group of controls consisted of 59 lymphocytes and 29 fibroblasts cell lines from disease free patients. Activities PMM2 and PMI were measured spectrophotometrically at 37 °C as the reduction of NADP+ to NADPH at 340 nm.
Results:
Statistically significant correlation between activity of PMM2 and PMI and age or gender in lymphocytes or cultured fibroblasts was not found. PMM2 activity in lymphocytes in the control group was from 0.34 to 2.58 nmol/min/mg (2.5% to 97.5% percentile) and in fibroblasts from 0.44 to 9.0 nmol/min/mg. PMM2 activity in lymphocytes in the group of patients was low (0.02-0.18 nmol/min/mg, interquartile range, controls 0.73-1.42 nmol/min/mg, p <0.001). PMM2 activity in fibroblasts from patients was also low (0.2-0.66 nmol/min/mg, controls 1.06-3.17 nmol/min/mg, p<0.001). PMM2 / PMI ratio in patients was significantly decreased in both tissues (p<0.001). The diagnosis of PMM2-CDG was confirmed by finding of mutations in PMM2 gene in all 18 patients.
Conclusion:
Measurement of PMM2 activity in lymphocytes or cultured fibroblasts allows to quickly diagnose PMM2-CDG, the most common congenital disorder of glycosylation
Keywords:
congenital disorders of glycosylation (CDG), phosphomannomutase (PMM2), phosphomannose isomerase (PMI).
Autoři:
H. Hansíková; N. Ondrušková; T. Honzík; K. Veselá; E. Horová; Š. Švecová; M. Tesařová; J. Zeman
Působiště autorů:
Klinika dětského a dorostového lékařství, 1. lékařská fakulta Univerzity Karlovy a Všeobecná fakultní nemocnice v Praze
Vyšlo v časopise:
Klin. Biochem. Metab., 24, 2016, No. 2, p. 67-74
Souhrn
Cíl studie:
Cílem studie bylo zavést metodu pro stanovení aktivity fosfomanomutázy 2 (PMM2) a kontrolního enzymu fosfomanoizomerázy (PMI) v izolovaných lymfocytech a kultivovaných kožních fibroblastech a využít ji jako diferenciální diagnostický krok v souboru 18 pacientů s podezřením na CDG syndrom typu I.
Typ studie:
Původní práce.
Název a sídlo pracoviště:
Klinika dětského a dorostového lékařství, 1. lékařská fakulta Univerzity Karlovy a Všeobecná fakultní nemocnice v Praze.
Materiál a metody:
Analyzovaný soubor představuje 16 vzorků lymfocytů a 8 linií kultivovaných kožních fibroblastů od 18 pacientů z 15 nepříbuzných rodin, u kterých lékaři vyslovili vysoké podezření na CDG syndrom typu I. Kontrolní skupinu tvořilo 59 vzorků lymfocytů a 29 linií fibroblastů od pacientů, u kterých nebylo prokázáno podezření na metabolickou poruchu. Aktivity PMM2 a PMI byly měřeny spektrofotometricky při 37 °C jako redukce NADP+ na NADPH při 340 nm.
Výsledky:
U PMM2 a PMI nebyly v lymfocytech ani kultivovaných fibroblastech nalezeny statisticky významné rozdíly aktivit v závislosti na věku či pohlaví. Aktivita PMM2 v lymfocytech u kontrolního souboru byla 0,34–2,58 nmol/min/mg (2,5% až 97,5% percentil) a ve fibroblastech 0,44–9,0 nmol/min/mg. Aktivita PMM2 v lymfocytech byla v souboru pacientů nízká (0,02–0,18 nmol/min/mg, mezikvartilové rozpětí, kontroly 0,73-1,42, p<0,001). Aktivita PMM2 ve fibroblastech od pacientů byla také nízká (0,2–0,66 nmol/min/mg; kontroly 1,06–3,17, p<0,001). Poměr PMM2/PMI byl u pacientů významně snížený v obou tkáních (p<0,001). U všech 18 pacientů byla diagnóza PMM2-CDG potvrzena i nálezem mutací v PMM2 genu.
Závěr:
Měření aktivity PMM2 v lymfocytech nebo kultivovaných fibroblastech umožňuje rychlé stanovení diagnózy PMM2-CDG, nejčastější dědičné poruchy glykosylace typu I.
Klíčová slova:
dědičné poruchy glykosylace (CDG), fosfomanomutáza (PMM2), fosfomanoizomeráza (PMI).
Úvod
Glykosylace je nejrozšířenější posttranslační modifikace proteinů a zahrnuje kovalentní vazbu cukerných řetězců na amidovou skupinu asparaginu (N-glykoprotein) nebo na hydroxylovou skupinu serinu nebo threoninu peptidového řetězce (O-glykoprotein) [1]. Narušení glykosylačních procesů vede ke skupině závažných onemocnění, které se souhrnně označují jako CDG syndrom (Congenital Disorders of Glycosylation, CDG, dědičné poruchy glykosylace proteinů). Na molekulární úrovni již bylo identifikováno >100 různých CDG, více než polovina z nich se týká poruch glykosylace N-glykoproteinů [2].
Biochemický screening je založen na analýze profilu sialovaných forem transferinu v séru pomocí izoelektrické fokusace (IEF), která umožňuje zachytit pacienty s některými typy poruch na úrovni syntézy (CDG typu I) nebo úpravy N-glykoproteinů (CDG typu II) [3]. Vzhledem k všudypřítomnému výskytu N-glykoproteinů v organismu, vedou poruchy na úrovni jejich biosyntézy k širokému spektru závažných klinických příznaků. Nejčastějším typem CDG syndromu je PMM2-CDG (podle starší nomenklatury CDG Ia) s autosomálně recesivním typem dědičnosti, který tvoří 80 % případů v této skupině onemocnění. V literatuře již bylo popsáno >800 pacientů a výskyt v populaci se odhaduje na cca 1 : 20 000 [4].
Fosfomanomutáza (PMM2) je cytosolární enzym, který katalyzuje reverzibilní přeměnu manózy-6-fosfátu na manózu-1-fosfát (Man-1-P). Snížení enzymové aktivity vede k depleci v zásobách GDP-manózy a dolichol-P-manózy, a tím k hypoglykosylaci řady N-vázaných glykoproteinů. Porucha PMM2 vede k onemocnění, které se nazývá PMM2-CDG. Je způsobena přítomností mutací v PMM2 genu lokalizovaném v oblasti 16p13.3.-p13.2, má 8 exonů a bylo v něm již nalezeno >110 patogenních mutací [5].
Klinicky se PMM2-CDG manifestuje již v kojeneckém věku jako multisystémové onemocnění s heterogenními klinickými příznaky. Mezi univerzální klinické a laboratorní nálezy patří neprospívání, atypické rozložení podkožního tuku v gluteální oblasti, invertované mammily, kraniofaciální dysmorfie, strabismus, hypotonický syndrom, porucha až zástava psychomotorického vývoje, hypoplázie mozečku, hepatopatie a smíšená koagulopatie [6,7]. Prognóza není příznivá, mortalita dětí s nízkou reziduální aktivitou PMM2 dosahuje až 20 % [8]. Esenciální úlohu PMM2 pro vývoj organismu podporuje i zjištění, že inaktivace genu PMM2 u myší je embryonálně letální [9]. Léčba pacientů s PMM2-CDG není známá a ani experimentální terapie pomocí Man-1-P nebyla terapeuticky úspěšná [10].
Dosud bylo stanovení aktivity PMM2 dostupné pouze v několika specializovaných centrech ve světě zaměřených na diagnostiku poruch glykosylace, kde u pacientů s podezřením na CDG typu I je v prvním kroku obecně uznávaným a doporučovaným diagnostickým standardem měření aktivity PMM2 v izolovaných lymfocytech nebo kultivovaných kožních fibroblastech, a to z důvodu, že tento typ CDG je nejčastější.
Cílem studie byla snaha zavést metodu pro stanovení aktivity fosfomanomutázy (PMM2) a kontrolního enzymu fosfomanoizomerázy (PMI) v izolovaných lymfocytech a kultivovaných fibroblastech a využít ji jako diferenciálně-diagnostický krok v našem souboru 18 pacientů s podezřením na CDG syndrom typu I.
Materiál
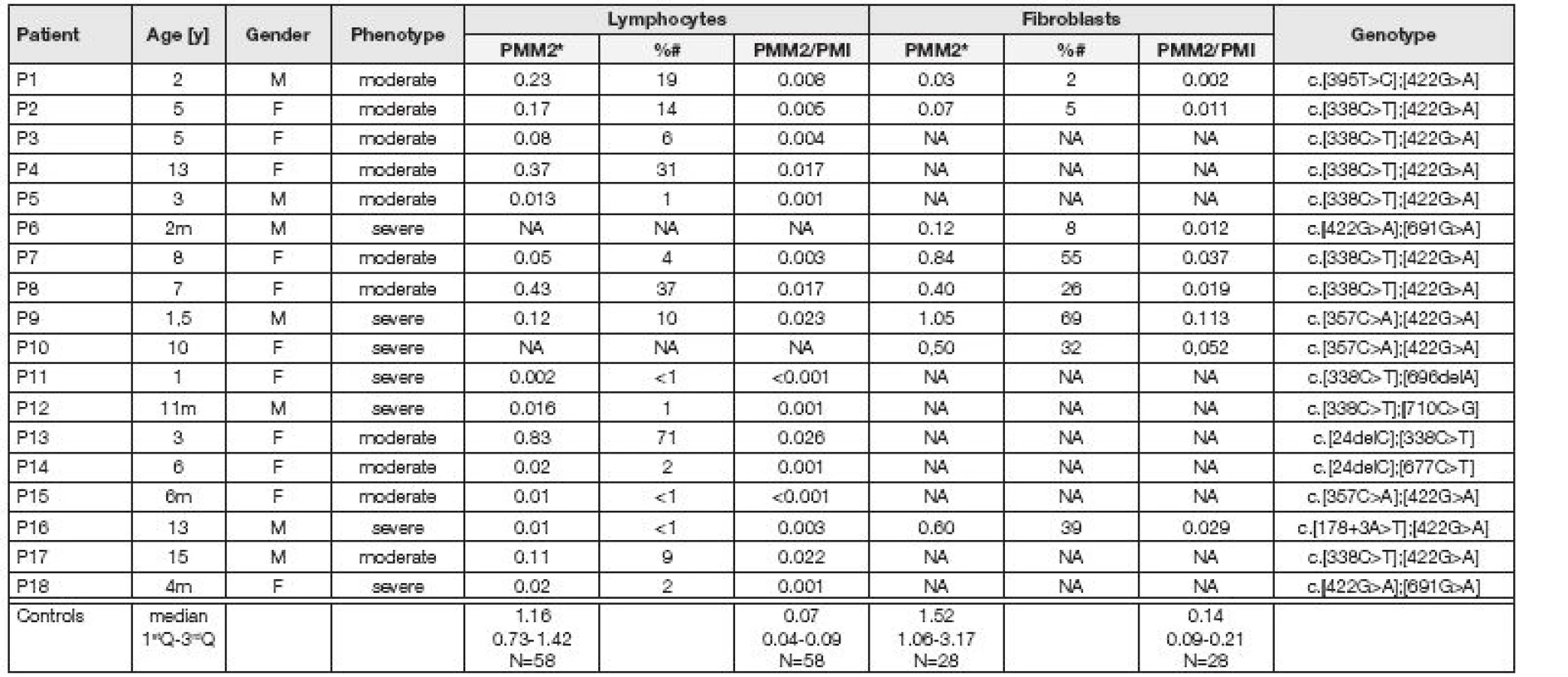
Analyzovaný soubor reprezentovalo 16 vzorků izolovaných lymfocytů a 8 linií kultivovaných kožních fibroblastů od 18 pacientů (0,5-18 let), u kterých bylo na základě klinických příznaků vysloveno podezření na CDG syndrom typu I. U všech pacientů byl pomocí IEF sérového transferinu nalezen patologický profil CDG typu I (Obr 1.). Zároveň bylo analyzováno i 9 vzorků lymfocytů od rodičů vyšetřovaných probandů (obligatorních heterozygotů) ve věku 22-41 let. Kontrolní skupinu tvořilo 59 vzorků lymfocytů a 29 linií kultivovaných fibroblastů od osob ve věku 0,5-48 let, resp. 0,5-18 let bez klinických projevů CDG syndromu.

Chemikálie
Histopaque®1077, Hepes, DTT, KCl, leupeptin a pepstatin, fosfomanoizomeráza, MgCl2, manóza-1-P, manóza-6-P (Sigma), glukóza-6-P-dehydrogenáza, fosfoglukózaizomeráza, NADP (dvojsodná sůl) (Roche), manóza-1,6-di-P byla připravena podle Van Schaftingen [11].
Metody
Lymfocyty byly izolovány ze 7 ml periferní krve s přídavkem EDTA gradientovou centrifugací (Histopaque®1077, Sigma). Kožní fibroblasty byly kultivovány v D-MEM mediu s 10% obsahem fetálního bovinního séra v 5% CO2. Fibroblasty v množství 2x 75 cm2 o konfluenci 80 % byly sklizeny trypsinizací. Peleta obou typů buněk byla zamražena a uchovávána při -80 °C.
Příprava vzorku
Peleta lymfocytů nebo fibroblastů byla rozmražena a homogenizována při 4 °C v 300 µl homogenizačního pufru (20 mmol/l Hepes pH 7,1, 1 mmol/l DTT, 25 mmol/l KCl, leupeptin a pepstatin 10 mg/l) v homogenizátoru sklo-sklo. Poté byl homogenát sonikován (3x8 s, amplituda 20, při 4 °C) a směs byla centrifugována při 1550 g, 8 min, při 4 °C. Supernatant byl uschován při -80 °C přes noc. Druhý den byla rozmražená suspenze centrifugována při 9000 g, 5 min, 4 °C a výsledný supernatant byl použit pro stanovení aktivity enzymu. Na jedno měření bylo používáno 50 µl supernatantu (koncentrace proteinu se pohybovala okolo 3-5 g/l).
Enzymové aktivity
Aktivita PMM2 a PMI byla měřena spektrofotometricky jako přeměna NADP+ na NADPH při 340 nm po dobu 5 min při 37 °C v kyvetách o optické dráze 1 cm v celkovém objemu 0,5 ml. Každé měření bylo provedeno 2x. Aktivita byla vyjádřena jako nmol/min/mg proteinu s použitím molárního extinkčního koeficientu ε340=6,22 (l/mmol/cm). Měření bylo provedeno podle Van Schaftingen (11) s mírnými úpravami uvedenými níže.
Reakční směs pro spektrofotometrické stanovení PMM2 obsahovala: 50 mmol/l Hepes pH 7,1, fosfoglukózaizomeráza (10 g/l), fosfomanoizomeráza (3,5 g/l), glukóza-6-P-dehydrogenáza (10 g/l), 1 mmol/l manóza-1,6-di-P, 0,25 mmol/l NADP, 50 mmol/l MgCl2, 3 mmol/l manóza-1-P. Reakce byla startována přídavkem 50 µl vzorku. Referenční kyveta neobsahovala manózu-1-P.
Reakční směs pro spektrofotometrické stanovení aktivity PMI obsahovala: 50 mmol/l Hepes pH 7,1, fosfoglukózaizomeráza (10 g/l), glukóza-6-P-dehydrogenáza 4 g/l, 15 mmol/l NADP, 50 mmol/l MgCl2, 5 mmol/l manóza-6-P, reakce byla startována přídavkem 50 µl vzorku. Referenční kyveta neobsahovala substrát - manózu-6-P. Aktivita PMI sloužila jako kontrolní enzym k vypočítání poměru PMM2/PMI.
Koncentrace proteinu ve vzorku byla stanovena dle Lowryho [12].
Molekulárně-genetická analýza
Celková DNA byla izolována metodou fenolové extrakce. Všechny exony genu PMM2 (ENSG00000140650, ENST00000268261) a jejich přilehlé intronové oblasti byly analyzovány metodou přímého sekvenování na genetickém analyzátoru ABI 3500xL (Applied Biosystems). Mutace zjištěné u probanda byly vždy potvrzeny i u obou rodičů analýzou jejich DNA.
Statistická analýza
Vzhledem k tomu, že pozorovaná data nemají normální rozdělení, byly použity neparametrické statistické metody. Rozdíly mezi pacienty a kontrolami (resp. mezi heterozygoty a kontrolami) byly testovány Wilcoxonovým dvouvýběrovým testem. Za statisticky významné byly považovány dosažené hladiny testů nižší než 5 %. Analýzy byly provedeny ve statistickém software R verze 3.1.2, R Core Team (2014).
Etika
Všechny odběry a biochemické i molekulární analýzy byly prováděny po informovaném souhlasu rodičů. Studie byla provedena v souladu s Helsinskou deklarací Světové lékařské asociace a byla schválena etickou komisí Všeobecné fakultní nemocnice v Praze.
Výsledky
U PMM2 a PMI nebyly v lymfocytech a v kultivovaných fibroblastech nalezeny statisticky významné rozdíly v aktivitách v závislosti na věku či pohlaví. Rozmezí kontrolních hodnot (2,5 % až 97,5 % kvantil) pro PMM2 v lymfocytech a fibroblastech uvádí obr. 2 a 3. Aktivity kontrolního enzymu PMI v lymfocytech a fibroblastech jsou uvedeny na obr. 4.
Průměrná aktivita PMM2 a poměr PMM2/PMI v lymfocytech a fibroblastech pacientů byly významně snížené (p<0,001) ve srovnání s kontrolním souborem (obr. 2, 3). Medián pro aktivitu PMM2 v lymfocytech vyšetřovaných pacientů odpovídal 6 % mediánu kontrol (p<0,001) (obr. 2, 3). Významně snížená aktivita PMM2 v lymfocytech pod 10 % mediánu kontrolní skupiny byla nalezena u 10 pacientů, středně snížená aktivita mezi 10-30 % u tří pacientů a u tří pacientů byla aktivita PMM2 do hodnoty 71 % mediánu kontrol (Tabulka 1). Nebyl nalezen významný rozdíl mezi skupinou heterozygotů a kontrolním souborem. Medián pro aktivitu PMM2 ve fibroblastech odpovídal 30 % mediánu kontrol (p<0,001). Významně snížená aktivita PMM2 pod 10 % mediánu kontrolní skupiny byla ve fibroblastech nalezena u tří pacientů, středně snížená mezi 10-30 % u jednoho pacienta a u tří byla aktivita PMM2 do hodnoty 69 % mediánu kontrol (Tabulka 1).


Aktivita kontrolního enzymu PMI byla v lymfocytech pacientů vyšší než u kontrolního souboru, zatímco ve fibroblastech nebyly rozdíly statisticky významné (obr. 4).

Posléze byla u všech 18 pacientů diagnóza PMM2-CDG potvrzena i na molekulárně genetické úrovni. Nalezené mutace v PMM2 jsou uvedeny v Tabulce 1. Všichni pacienti jsou složení heterozygoti, přičemž nejčastějšími mutacemi jsou mutace c.422G>A a c.338C>T, které vedou k záměně aminokyseliny R141H a P113L, respektive.
Diskuse
V naší studii jsme v souboru 18 pacientů s klinickým podezřením na CDG syndrom analyzovali aktivitu PMM2, jejíž porucha podporuje diagnózu PMM2-CDG, nejčastějšího typu CDG syndromu. Ačkoliv až u 18 % publikovaných pacientů s PMM2-CDG byla v izolovaných lymfocytech a/nebo kultivovaných fibroblastech nalezena normální aktivita PMM2 [13,14], u většiny našich pacientů jsme prokázali významně sníženou aktivitu PMM2 pod 10 % kontrolních hodnot nebo alespoň středně sníženou aktivitu PMM2 na úroveň 10 až 30 % kontrolních hodnot. Pouze u jednoho pacienta z našeho souboru (P13) dosahovala aktivita PMM2 v lymfocytech 71 % mediánu kontrolních hodnot. Diagnózu PMM2-CDG u pacienta 13 potvrdilo molekulární vyšetření PMM2 genu, které ukázalo přítomnost dvou mutací c.[24delC]+[338C>T]. Delece nukleotidu na jedné alele vede ke vzniku předčasného stop-kodónu, zatímco „missence“ mutace na druhé alele vede k záměně prolinu za lysin na pozici 113, která sice ovlivňuje stabilitu diméru nativního proteinu, ale přímo nezasahuje do aktivního místa PMM2, takže patří mezi mutace s mírnějším dopadem na funkci enzymu [15]. Ve prospěch enzymatické diagnostiky u pacienta 13 svědčí i snížený poměr mezi PMM2 a kontrolním enzymem PMI.
Ve fibroblastech jsme závažné snížení aktivity PMM2 pod 10 % zaznamenali pouze u tří z osmi námi testovaných pacientů. Také literární údaje uvádějí poměrně vysokou residuální aktivitu PMM2 ve fibroblastech pacientů, což při enzymatickém vyšetření zvyšuje riziko falešně negativního výsledku [16].
Nesoulad mezi residuální aktivitou PMM2 v lymfocytech a fibroblastech ukazuje na tkáňově specifické rozdíly. Studie na mutantních proteinech prokázaly narušenou kinetiku enzymu a sníženou stabilitu mutantního proteinu ve srovnání s kontrolním vzorkem [17]. Residuální aktivita je vyšší ve fibroblastech také díky tomu, že fibroblast je rychle se dělící buňka s aktivní proteosyntézou ve srovnání s lymfocyty. Navíc tento fakt může být doprovázen i možností selektivní výhody podmínek kultivace u fibroblastů [16].
V souladu s literárními údaji jsme u našich pacientů s PMM2-CDG nezjistili významnější korelaci mezi jejich fenotypem, genotypem a residuální aktivitou PMM2 v obou tkáních. Většina pacientů s PMM2-CDG jsou složení heterozygoti, přičemž nejfrekventovanější mutace R141H, která se vyskytuje cca u 40 % pacientů, ovlivňuje vazbu Man-1-P do aktivního místa enzymu. Její dopad na aktivitu PMM2 je natolik zásadní, že dosud nebyla nalezena v homozygotní formě [15]. Deset z 16 pacientů našeho souboru má mutaci P113L, která ovlivňuje stabilitu diméru nativního enzymu a patří k mutacím s mírnějším dopadem [15].
Vzhledem k tomu, že je již známo vice než 50 různých dědičných poruch glykosylace na úrovni syntézy N-glykoproteinů a že již byli popsáni i PMM2-CDG pacienti s normálním profilem transferinu při IEF [18], je stanovení aktivity PMM2 v izolovaných lymfocytech nebo kultivovaných fibroblastech i přes řadu svých omezení významným přínosem při diferenciální diagnostice poruch glykosylace.
Závěr
Byla zavedena metoda měření aktivity PMM2 v izolovaných lymfocytech a kultivovaných fibroblastech a bylo stanoveno rozmezí hodnot u kontrol pro tyto dva typy tkání. Metoda je úspěšně využívána v diferenciální diagnostice při podezření na PMM2-CDG.
Zkratky
PMM2 fosfomanomutáza
PMI fosfomanoizomeráza
IEF izoelektrická fokusace
Man-1-P manóza-1-fosfát
Práce byla podporována IGA MZ NT12166-5/2011.
Do redakce došlo 25. 1. 2016
Adresa pro korespondenci:
RNDr. Hana Hansíková, CSc.
1. LF UK a VFN v Praze
Laboratoř pro studium mitochondriálních poruch
Klinika dětského a dorostového lékařství
Ke Karlovu 2
128 08 Praha 2
email: hana.hansikova@lf1.cuni.cz
Zdroje
1. Schachter, H., Freeze, H. H. Glycosylation diseases: quo vadis? Biochim Biophys Acta. 2009, 1792 (9): p. 925-30
2. Hennet, T., Cabalzar, J. Congenital disorders of glycosylation: a concise chart of glycocalyx dysfunction. Trends Biochem Sci. 2015, 40 (7): p. 377-84.
3. Aebi, M., Helenius, A., Schenk, B. et al. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG. First International Workshop on CDGS. Glycoconj J. 1999, 16 (11): p. 669-71.
4. Haeuptle, M. A., Hennet, T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009, 30 (12): p. 1628-41.
5. Matthijs, G., Schollen, E., Pardon, E. et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nature Genet. 1997, 16: p. 88-92.
6. Honzík, T., Malonová, E., Hansíková, H. et al. Congenital disorder of type Ia protein glycosylation: clinical, biochemical and molecular characteristics in 2 siblings with cerebellar hypoplasia. Cas. Lek. Cesk., 2003, 142 (5): p. 276-9.
7. Honzík, T., Magner, M., Krijt, J. et al. Clinical picture of S-adenosyl-homocysteine hydrolase deficiency resembles phosphomannomutase 2 deficiency. Mol. Genet. Metab., 2012, 107 (3): p. 611-3.
8. Matthijs, G., Schollen, E., Bjursell, C. et al. Mutations in PMM2 that cause congenital disorders of glycosyla-tion, type Ia (CDG-Ia). Hum Mutat. 2000, 16 (5): p. 386-94.
9. Thiel, C., Lübke, T., Matthijs, G., von Figura, K., Körner, C. Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol. Cell. Biol., 2006, 26 (15): p. 5615-20
10. Higashidani, A., Bode, L., Nishikawa, A., Freeze, H. H. Exogenous mannose does not raise steady state mannose-6-phosphate pools of normal or N-glycosy-lation-deficient human fibroblasts. Mol. Genet. Metab., 2009, 96 (4): p. 268-72
11. Van Schaftingen, E., Jaeken, J. Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett. 1995, 377: p. 318-320
12. Lowry, O. H., Rosebrough, N. J., Farr, A. L., Randall, R. J. Protein measurement with the folin phenol reagent. J Biol. Chem., 1951, 193: p. 265-275.
13. Jaeken, J., Artigas, J., Barone, R. et al. Phosphomannomutase deficiency is the main cause of carbohydrate-deficient glycoprotein syndrome with type I isoelectrofocusing pattern of serum sialotransferrins. J Inherit Metab. Dis., 1997, 20 (3): p. 447-9.
14. Westphal, V., Peterson, S., Patterson, M. et al. Functional significance of PMM2 mutations in mildly affected patients with congenital disorders of glycosylation Ia. Genet. Med., 2001, 3 (6): p. 393-8.
15. Vega, A. I., Pérez-Cerdá, C., Abia, D. al. Expression analysis revealing destabilizing mutations in phosphomannomutase 2 deficiency (PMM2-CDG): expression analysis of PMM2-CDG mutations. J Inherit. Metab. Dis., 2011, 34 (4): p. 929-39.
16. Grünewald, S., Schollen, E., Van Schaftingen, E., Jaeken, J., Matthijs, G. High residual activity of PMM2 in patients’ fibroblasts: possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am. J Hum. Genet., 2001, 68 (2): p. 347-54.
17. Kjaergaard, S., Skovby, F., Schwartz, M. Carbohydrate-deficient glycoprotein syndrome type 1A.: expression and characterisation of wild type and mutant PMM2 in E. coli. Eur J Hum. Genet., 1999, 7 (8): p. 884-8
18. Fletcher, J. M., Matthijs, G., Jaeken, J., Van Schaftingen, E., Nelson, P. V. Carbohydrate-deficient glycoprotein syndrome: beyond the screen. J Inherit. Metab. Dis., 2000, 23 (4): p. 396-8.
Štítky
Biochemie Nukleární medicína Nutriční terapeutČlánek vyšel v časopise
Klinická biochemie a metabolismus

2016 Číslo 2
- Moderní přístupy zvyšující efektivitu antibiotické léčby v nemocniční praxi
- Zpracované masné výrobky a červené maso jako riziko rozvoje kolorektálního karcinomu u žen? Důkazy z prospektivní analýzy
- GLP-1RA a PCOS: Je to „jenom“ o hmotnosti?
- Efektivita léčby a možné indikace liraglutidu v gynekologii
- Farmakologická léčba obezity u pacientek se syndromem polycystických ovarií – systematický přehled a klinická doporučení
Nejčtenější v tomto čísle
- Kvalita, kontrola a validace glukometrů a CGM systémů. Přehled stavu.
- Postanalytická fáze a interpretace laboratorního testu (post-postanalytická fáze)
- Abstrakta přednášek
- Od laboratoří k čipům. Od čipů k teranostice, nanočásticím, mikrofluidice. Nové cesty, staré iluze?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy