
Fylogenetická a molekulární analýza virů chřipky A/H1N1pdm izolovaných v epidemické sezoně 2012/2013 od pacientů hospitalizovaných s příznaky ILI
Phylogenetic and molecular analysis of A/H1N1pdm influenza viruses isolated in the epidemic season 2012/2013 from hospitalised patients with symptoms of influenza-like illness
Aim:
To perform phylogenetic and molecular analysis of A/H1N1pdm influenza viruses isolated in the epidemic season 2012/2013 from hospitalised patients with symptoms of influenza-like illness (ILI).
Material and methods:
The study set included 34 strains of the A/H1N1pdm influenza virus isolated in the Czech Republic in the epidemic season 2012/2013. The strains were analysed by partial or whole-genome sequencing. The genome segments were compared at the nucleotide and amino acid levels, absolute and percentage sequence identity were determined, and phylogenetic relations were identified. The last steps were the comparison of the H1 molecule with that of the most recent vaccine strain and identification of the genotypic structure and molecular markers linked to the pathogenicity and antiviral resistance.
Results:
Phylogenetic analysis of the H1 molecule suggested that all 34 A/H1N1pdm isolates from the 2012/2013 season in the Czech Republic should be assigned to H1 group 6 divided into sublineages 6A and 6B. The comparison of the known antigenic regions of the H1 molecule with those in the most recent vaccine strain revealed two stable changes in antigenic regions Sb and Ca1. Furthermore, sporadic mutations were identified in antigenic regions Ca2, Cb, and Sb. Genotyping revealed co-circulation of two related but clearly distiguishable genotypes of A/H1N1pdm. All isolates showed sensitivity to oseltamivir. One strain consisted of two N1 sub-populations, one oseltamivir sensitive and the other oseltamivir resistant, in nearly equimolar proportions.
Conclusion:
All A/H1N1pdm isolates from the epidemic season 2012/2013 in the Czech Republic formed a phenotypically uniform group. At the nucleotide level, the divergence was relatively more pronounced and H1 sublineages and discrete genotypes were possible to identify. H1 molecules were highly identical to those of the vaccine strain A/California/7/2009 (H1N1) which showed that the current vaccine was protective enough. All strains were sensitive to oseltamivir; however, the selection of oseltamivir resistant N1 subpopulations was observed.
Keywords:
influenza – H1N1 – phylogenetic analysis – genotype – resistance
Autoři:
A. Nagy 1,5; H. Jiřincová 1; M. Havlíčková 1; O. Džupová 2,3; K. Herrmannová 2,4; M. Trojánek 2,4; J. Kynčl 1,3
![]() ; Z. Blechová 2,4; V. Marešová 2,4
; Z. Blechová 2,4; V. Marešová 2,4
Působiště autorů:
Centrum epidemiologie a mikrobiologie, Státní zdravotní ústav, Praha
1; Klinika infekčních nemocí, Nemocnice Na Bulovce, Praha
2; 3. lékařská fakulta, Univerzita Karlova v Praze
3; 2. lékařská fakulta, Univerzita Karlova v Praze
4; Státní veterinární ústav, Praha
5
Vyšlo v časopise:
Epidemiol. Mikrobiol. Imunol. 63, 2014, č. 2, s. 83-87
Kategorie:
Souhrnná sdělení, původní práce, kazuistiky
Souhrn
Cíl práce:
Provést fylogenetickou a molekulární analýzu virů chřipky A/H1N1pdm izolovaných v epidemické sezoně 2012/2013 od pacientů hospitalizovaných s příznaky ILI (influenza-like illnes).
Materiál a metodiky:
Analyzovaný soubor představoval 34 kmenů viru pandemické chřipky A/H1N1pdm v epidemické sezoně 2012/2013 v ČR. Kmeny byly podrobeny parciálnímu nebo celogenomovému sekvenování. Jednotlivé genomové segmenty se porovnaly na nukleotidové i aminokyselinové úrovni, určila se absolutní i procentní sekvenční identita a identifikovaly se fylogenetické vztahy. V posledním bodu se analyzovaly rozdíly v molekule H1 oproti aktuálnímu vakcinačnímu kmenu a identifikovala se genotypová struktura a přítomnost molekulárních markerů majících vztah k patogenitě a rezistenci na antivirotika.
Výsledky:
Fylogenetická analýza molekuly H1 naznačila příslušnost všech 34 A/H1N1pdm izolátů reprezentujících sezonu 2012/2013 v ČR do H1 skupiny 6 s členěním do dvou H1 sublinií 6A a 6B. Mapování rozdílů do známých antigenních oblastí molekuly H1 ukázalo dvě stabilní změny oproti aktuálnímu vakcinačnímu kmenu, a to v antigenních oblastech Sb a Ca1. Dále byly identifikovány sporadické mutace spadající do antigenní oblasti Ca2, Cb a Sb. Genotypizace odhalila ko-cirkulaci dvou příbuzných, ale jasně rozlišitelných genotypů A/H1N1pdm. Všechny izoláty se vyznačovaly citlivostí na oseltamivir. U jednoho kmene byly identifikovány dvě N1 subpopulace oseltamivir senzitivní a rezistentní v přibližně ekvimolárním poměru.
Závěr:
Všechny A/H1N1pdm izoláty reprezentující epidemickou sezonu 2012/2013 v ČR představovaly fenotypově jednotnou skupinu. Na nukleotidové úrovni byla divergence relativně výraznější s možností identifikace H1 sublinií i diskrétních genotypů. Molekuly H1 se vyznačovaly vysokou identitou s vakcinačním kmenem A/California/7/2009 (H1N1), což dokazovalo dostatečnou protektivitu aktuální vakcíny. Všechny kmeny se vyznačovaly citlivostí na oseltamivir, nicméně v některých případech docházelo k selekci oseltamivir-rezistentních N1 subpopulací.
Klíčová slova:
chřipka – H1N1 – fylogenetická analýza – genotyp – rezistence
Úvod
Chřipka představuje akutní febrilní virové onemocnění, jež atakuje především horní cesty dýchací – s maximem výskytu v zimních měsících roku. U většiny nemocných mívá infekce středně těžký průběh, obvykle postačí symptomatická léčba a po 5–7 dnech dochází k ústupu příznaků. Rekonvalescenci však může doprovázet únavový syndrom, často i několik týdnů. Nicméně, u části nemocných se chřipka může projevit jako závažná infekce, která se manifestuje postižením dolních cest dýchacích (primární chřipková pneumonie) a nezřídka i projevy mimoplicními. Závažnost průběhu je obtížně odhadnutelná, často se může těžká infekce objevit i u velmi mladého pacienta bez jakýchkoliv rizikových faktorů. Chřipková sezóna 2012/2013 se, na rozdíl od sezony 2011/2012, vyznačovala delším trváním i výskytem těžkých forem infekce [1]. Dominujícím subtypem byla chřipka A/H1N1pdm, subtyp A/H3N2 a typ B ko-cirkulovaly v menší míře ve druhé polovině epidemické vlny. Cílem naší studie byla charakteristika genetické variability virů chřipky subtypu A/H1N1pdm, u pacientů kteří byli hospitalizováni s příznaky ILI v sezoně 2012/2013.
MATERIÁL A METODY
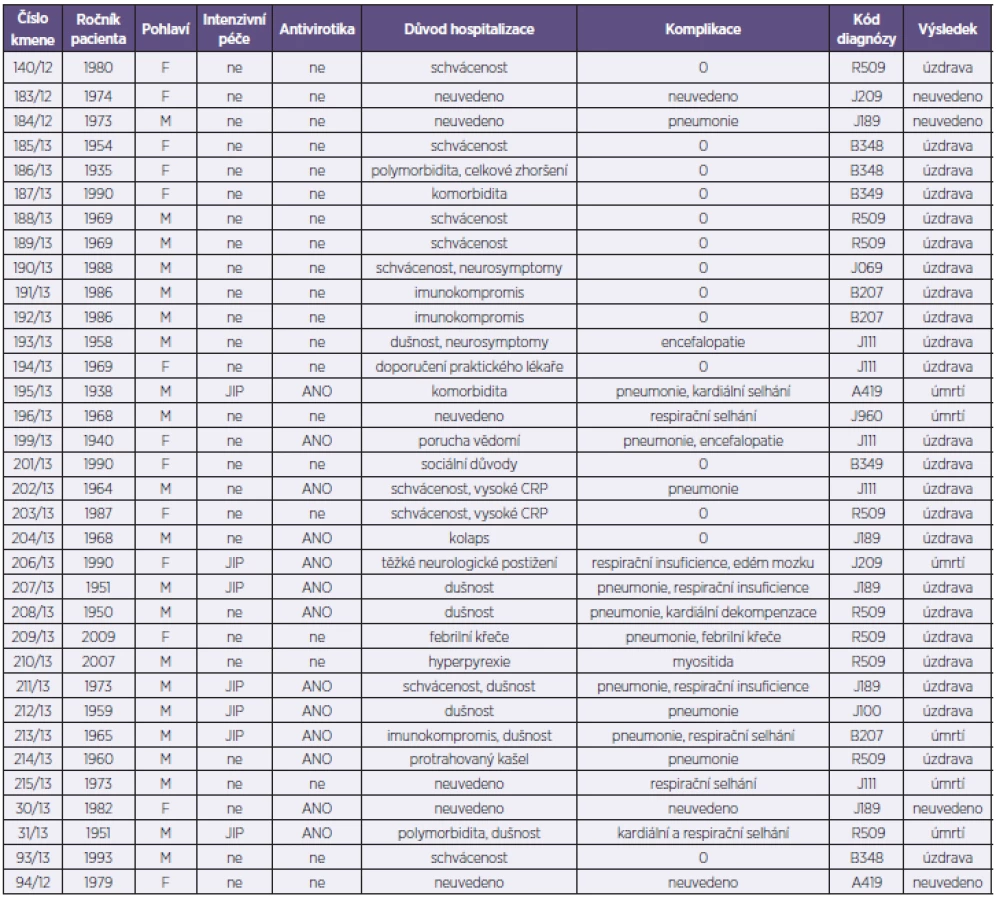
Analyzovaný soubor představoval 34 kmenů viru pandemické chřipky A/H1N1pdm. Z uvedeného počtu bylo od 26 pacientů hospitalizovaných s příznaky ILI v Nemocnici na Bulovce v období od 1. 10. 2012 do 30. 4. 2013 získáno 28 kmenů (2 pacienti byli z důvodů prolongované hospitalizace odebráni dvakrát), 6 kmenů pocházelo od pacientů indikovaných k vyšetření mimo Nemocnici na Bulovce v rámci standardní diagnostické činnosti NRL a z toho 2 kmeny byly získány post mortem od pacientů s perakutním průběhem chřipky vedoucím k exitu (tab. 1). Při příjmu byl u nemocných rutinně odebrán stěr z nosohltanu a z obou nosních průduchů, který byl následně použit pro účely molekulární detekce a typizace a rovněž pro izolační pokus na tkáňových kulturách MDCK. Molekulární detekce a typizace byla provedena s využitím metody RT-qPCR (Center for Disease Control, Atlanta, USA). Kmeny A/H1N1pdm byly izolovány v buněčném systému MDCK SIAT-1[2]. Získané izoláty byly podrobeny parciálnímu nebo celogenovému sekvenování a fylogenetické analýze. Sekvenování proběhlo na přístroji GA 3130 (Life Technologies) za použití selektovaných oligonukleotidů z naší knihovny primerů pokrývající všechny segmenty viru chřipky A (sekvence použitých primerů jsou dostupné na základě vyžádání). Editace, sestava a kvalitativní analýza sekvencí se uskutečnila pomocí programů SeqScape (Life Technologies) a BioEdit [3] a získané sekvence se následně srovnaly s využitím MAFFT analýzy [4]. Dále se stanovil stupeň sekvenční identity všech segmentů na nukleotidové i aminokyselinové úrovni ve formě tzv. „sequence difference count“ (SDC) matrixu [3]. Fylogenetická analýza pro každý segment byla provedena s využitím Neighbour-Joining nebo Maximum Likelihood aplikací programu PHILIP [5] – obr. 1. Dendrogramy se graficky upravily s využitím TreeExplorer aplikace programu MEGA 4 [6]. Reprezentativní sekvence byly zadány do databáze GenBank.
![Fylogenetická analýza molekuly hemaglutininu H1 Strom byl sestrojen metodou Maximum Likelihood (100replikací) na základě reprezentativních H1 sekvencí z roku 2011 získaných z databáze GISAID [7] a vztažen k vakcinačnímu kmenu A/California/7/2009(H1N1). Pro přehlednost jsou analyzované viry uvedeny pod kmenovým číslem a zvýrazněné tučně.
Fig. 1. Phylogenetic analysis of the hemagglutinin molecule H1 The tree was generated using the maximum likelihood method (100 replications) on the basis of representative H1 sequences 2011 obtained from the GISAID database [7] and related to the vaccine strain A/California/7/2009(H1N1). Viruses are shown under strain numbers in bold.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/a645a2ad42b66b003acf24f1cb138be4.jpg)
VÝSLEDKY
V naší studii bylo analyzováno 34 izolátů viru A/H1N1pdm pocházejících od 32 pacientů ve věku 4–78 let, průměr i medián věku byl 42 let. Důvodem přijetí k hospitalizaci byl závažnější obraz infekce se schváceností, dušností, protrahovaným kašlem a horečkou nebo komorbidity. Třináct pacientů bylo léčeno oseltamivirem, šest pacientů bylo přijato na JIP a u patnácti pacientů došlo k rozvoji pneumonie a respiračního selhávání. Šest pacientů infekci podlehlo, z toho ve dvou případech se jednalo o perakutní průběh infekce.
Fylogenetická analýza molekuly H1, provedena na základě reprezentativních sekvencí databáze GISAID [7] naznačila příslušnost všech 34 A/H1N1pdm izolátů reprezentujících sezonu 2012/2013 v ČR do H1 skupiny 6 [8]. Studium fylogenetických vztahů dále naznačilo, že v rámci skupiny 6 jsou naše kmeny separované do dvou jasně rozlišitelných sublinií. První sublinii, s označením 6A, tvořil jediný kmen s číslem 140, kdežto zbylých 33 kmenů bylo uniformně začleněných do druhé sublinie 6B. Pozorované fylogenetické vztahy H1 segmentů se na nukleotidové úrovni projevily relativně výrazně. Konstrukce SDC matrixu ukázala rozdíly mezi subliniemi 6A a 6B ve 23–29 pozicích na 1635 nukleotidů (1,41–1,77 %), zatímco v rámci sublinie 6B byl stupeň nukleotidové identity relativně vysoký: 0–11/1635 (≤ 0,67 %). Na aminokyselinové úrovni byly rozdíly méně výrazné, a to 7–12/536 (1,31–2,24%) mezi subliniemi 6A a 6B a 0–7/536 (≤ 1,31 %) v rámci sublinie B (tab. 2). Porovnání H1 segmentu studovaných kmenů s vakcinačním kmenem A/California/7/2009(H1N1) odhalilo 12/536 (2,24 %) aminokyselinových rozdílů u sublinie 6A a 11–13/536 (2,05–2,43 %) u sub-linie 6B (tab. 2). Mapování rozdílů do známých antigenních oblastí ukázalo dvě stabilní změny oproti aktuálnímu vakcinačnímu kmenu, a to v antigenních oblastech (H3 číslování): Sb (S188T) a Ca1 (S206T). Mutace v těchto antigenních pozicích jsou obecně známé a byly uniformně přítomny u všech analyzovaných kmenů A/H1N1pdm. Vedle stabilních rozdílů se dále identifikovaly sporadické mutace spadající do antigenní oblasti Ca2 (6A a 2 kmeny sublinie 6B) a Sb a Cb (po jednom kmeni sublinie 6B). Stanovení sekvenční variability u zbývajících genomových segmentů ve formě fylogenetických vztahů a SDC matrixu (neuvedeno) dále naznačilo, že se dichotomie analyzovaných A/H1N1pdm kmenů, pozorovaná na úrovni molekuly H1, zachovává. V rámci H1 sublinie 6B byla pozorována vysoká sekvenční identita s hodnotami nukleotidové a aminokyselinové variability v rozmezí 0,3–1,12 %, respektive 0,40–1,37 % s nejvýraznějšími rozdíly u segmentů kódujících protein NS1 a N1. Naopak, A/H1N1pdm kmen 140 se od zbylých kmenů odlišoval ve všech segmentech relativně výrazně. Na úrovni nukleotidů a aminokyselin byly v porovnání se sublinií B rozdíly v rozmezí 1,13–2,42 % a 0,53–3,09 %, a to hlavně v proteinech N1, M2 a NS1 (viz tab. 2).

Analýza přítomnosti a distribuce molekulárních markerů majících vztah k patogenitě a rezistenci na antivirotika odhalila, že všechny kmeny až na kmen 207 se vyznačují citlivostí na inhibitory neuraminidáz. U kmene 207 byly identifikovány dvě koexistující N1 varianty lišící se v pozici 275, která je nejvýznamnější v rezistenci na inhibitory neuraminidáz. To naznačuje, že u kmene 207 byly přítomny dvě N1 subpopulace, a to senzitivní H275 a rezistentní H275Y v přibližně ekvimolárním poměru. Další molekulární znaky byly shledány jako nevýznamné.
Vedle indikátorů rezistencí a patogenity se všechny analyzované kmeny vyznačovaly uniformní přítomností typických A/H1N1pdm znaků, a to M2 mutace S31N a I43T a uzavřeným čtecím rámcem pro protein PB1f2. Mutace S31N v protonovém kanálu M2, indikující rezistenci na adamantanové blokátory, je charakteristickým znakem pandemické chřipky a kořeny této mutace sahají až k tzv. „Eurasian avian-like“ H1N1 virům jako jedním z prekurzorů A/H1N1pdm [9]. Čtecí rámec pro protein PB1-F2 je přerušen třemi stop kodony, proto se tento protein v analyzovaných kmenech neexprimuje. Dalším znakem pandemické chřipky je M2 mutace I43T. Ačkoliv není tato mutace spojována s patogenním fenotypem, představuje znak, kterým se A/H1N1pdm odlišuje od jiných subtypů a linií chřipky A jako sezonní H1N1, H5N1 apod. [9]. Z toho pohledu stojí za pozornost, že kmen 140 (jediný kmen H1 sublinie 6A) se v této pozici odlišuje, a to M2 aminokyselinovou mutací T43A.
DISKUSE A ZÁVĚR
Výsledky našich analýz prokázaly, že A/H1N1pdm kmeny reprezentující chřipkovou sezonu 2012/2013 v ČR tvořily antigenně jednotnou skupinu. Na základě klasifikace ECDC patřil hlavní povrchový antigen H1 do skupiny 6, která spolu se skupinami 5 a 7 tvořila nejčastěji detekované H1 varianty v Evropě v předcházející sezoně 2011/2012 [8]. V rámci skupiny 6 bylo možné mezi analyzovanými kmeny identifikovat dvě sublinie. Aminokyselinové pozice ve všech epitopech molekuly H1 se vyznačovaly vysokou identitou s vakcinačním kmenem A/California/7/2009 (H1N1), což naznačuje dostatečnou protektivitu aktuální vakcíny. To dokazují i séra pacientů testovaná za stejné epidemické období (data neuvedena).
Parciální nebo kompletní sekvenování jednotlivých genomových segmentů odhalilo, že se rozdíly mezi H1 subliniemi projevily na úrovni celého genomu, což umožnilo rozdělit A/H1N1pdm kmeny do dvou příbuzných genotypů. První genotyp tvořil jediný kmen 140, zatímco všechny zbývající genomy spadaly do genotypu 2. Kmen 140 byl na nukleotidové úrovni jasně rozlišitelný, a to u všech segmentů s variabilitou 2–4krát vyšší, nežli byla pozorována v rámci genotypu 2. To indikuje odlišný evoluční původ. Kmen 140 byl izolován z výtěru dospělé pacientky již v polovině listopadu 2012, proto lze předpokládat i jistou odlišnost od A/H1N1pdm kmenů, které následně predominovaly v roce 2013. Pacientka neuváděla pozitivní cestovní anamnézu ani jiné epidemiologické souvislosti. Na aminokyselinové úrovni byly rozdíly mezi kmeny méně výrazné. Z uvedeného plyne, že A/H1N1pdm kmeny reprezentující chřipkovou sezonu 2012/2013 v ČR patří i fenotypově do jednotné skupiny.
Analýza přítomnosti a distribuce mutací podmiňujících rezistenci na antivirotika identifikovala, že všechny studované kmeny si zachovaly citlivost na inhibitory neuraminidáz. Z tohoto pohledu byl nejzajímavější kmen 207, který se vyznačoval senzitivní i rezistentní variantou neuraminidázy N1. Kmen 207 byl získán od pacienta s bilaterální pneumonií, který byl indikován k virologickému vyšetření až pátý den léčby oseltamivirem, což mohlo vytvořit dostatečný prostor pro selekci rezistentní N1 subpopulace. Pacient se uzdravil.
Dopad a charakter chřipkových epidemií a průběh infekcí v dané epidemické sezoně není v mnoha ohledech předvídatelný a četné souvislosti doposud nejsou plně probádány. Průběh infekce chřipkovým virem může být u člověka velmi variabilní – od téměř asymptomatických průběhů až po kritické, život ohrožující stavy. Vždy záleží na vzájemné interakci viru a jeho popřípadě specifických mutací s imunitním systémem hostitele. I z tohoto důvodu se izoláty de facto výrazně nelišily u nekomplikovaných a fatálních průběhů infekce. Introdukce nového subtypu A/H1N1pdm v roce 2009 s sebou přinesla prakticky úplné vytěsnění původní varianty A/H1N1 a v současnosti virus A/H1N1pdm ko-cirkuluje v lidské populaci spolu s A/H3N2. Oba subtypy vyvolávají sezonní epidemie se střídavou dominancí. Z tohoto pohledu je zajímavé, že H3N2 i H1N1pdm viry v lidské populaci evolují jako diskrétní paralelní linie bez stabilního H3N2-H1N1pdm „intersubtype reassortment“. Nicméně, genetický reassortment A/H1N1pdm virů s prasečími viry chřipky A jsou popisovány neustále, a to i na evropském kontinentu [10]. Diverzifikace A/H1N1pdm kmenů jejich křížením s prasečími viry a zpětná introdukce nových variant do lidské populace proto představuje zvýšené zdravotní riziko. Kromě toho A/H1N1pdm má relativně vysokou afinitu k epiteliálním buňkám dolních cest dýchacích, což představuje jeden z důvodů těžších průběhů chřipky tak, jak byly registrovány během uplynulé sezony [1].
Vzhledem ke klinické i epidemiologické závažnosti chřipky i genetické nestabilitě chřipkových virů je sledování molekulárněbiologických vlastností tohoto mnohotvárného patogenu naprostou nezbytností.
Tato práce vznikla za podpory grantu IGA MZ ČR 12493-3: Virologická a genetická charakteristika viru chřipky A ve vztahu ke klinické závažnosti infekce a projektu Ministerstva zdravotnictví ČR číslo 34/13/NAP. Děkujeme všem členům nemocničního i laboratorního personálu podílejícím se na odběru a zpracování vzorků. Děkujeme také všem přispívatelům do databází GISAID a IVSD.
Do redakce došlo dne 20. 12. 2013.
Adresa pro korespondenci:
RNDr. Alexander Nagy, Ph.D.
Státní veterinární ústav Praha
Sídlištní 136/24
165 03 Praha 6
e-mail: alexandernagy17@hotmail.com
Zdroje
1. Kyncl J, Havlickova M, Nagy A, Jirincova H, Piskova I. Early and unexpectedly severe start of influenza epidemic in the Czech Republic during influenza season 2012–13. Euro Surveill, 2013;18(6):pii 20396.
2. Manual for the laboratory diagnosis and virological surveillance of influenza[online]. Geneva: World Health Organization, 2011[cit. 2012-12-18]. ISBN: 9789241548090. Dostupné na www: http://whqlibdoc.who.int/publications/2011/9789241548090_eng.pdf
3. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser, 1999;41 : 95–98.
4. Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res; 2002;30(14):3059–3066.
5. Felsenstein J. PHYLIP (Phylogeny Inference Package) Version 3.6. Distributed by the Author. Department of Genome Sciences[online]. Seattle: University of Washington, 2004[cit. 2012-12-18]. Dostupné na www: http://evolution.genetics.washington.edu/phylip.html.
6. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol, 2007;24(8):1596–1599.
7. EPIFLU™ database[online]. GISAID. The Global Initiative on Sharing All Influenza Data, 2011[cit. 2014-12-18]. Dostupné na www: http://platform.gisaid.org/epi3/frontend#4efb5b.
8. Influenza virus characterisation. Summary Europe, February 2012. Technical document[online]. Stockholm: ECDC, 2012[cit. 2012-12-18]. Dostupné na www: http://www.ecdc.europa.eu/en/publications/publications/1203_ted_cnrl_report_feb2011.pdf
9. Deyde VM, Sheu TG, Trujillo AA, Okomo-Adhiambo M, Garten R, Klimov AI, Gubareva LV. Detection of molecular markers of drug resistance in 2009 pandemic influenza A(H1N1) viruses by pyrosequencing. Antimicrob Agents Chemother, 2010;54(3):1102–1110.
10. Howard WA, Essen SC, Strugnell BW, Russell C, Barrass L, Reid SM, Brown IH. Reassortant pandemic (H1N1) 2009 virus in pigs, United Kingdom. Emerg Infect Dis, 2011;17(6):1049–1052.
Štítky
Hygiena a epidemiologie Infekční lékařství MikrobiologieČlánek vyšel v časopise
Epidemiologie, mikrobiologie, imunologie

2014 Číslo 2
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
Nejčtenější v tomto čísle
- Výskyt Candida dubliniensis v klinickém materiálu a možnosti její identifikace
- Bodové prevalenčné sledovanie nozokomiálnych nákaz na Slovensku – súčasť projektu Európskej únie
- Q-horečka jako profesionální onemocnění vedoucí k invaliditě – kazuistika
- Diagnostika Clostridium difficile infekcí – porovnávací studie dvou imunoenzymatických metod s konfirmací pomocí PCR a kultivace s následnou ribotypizací kmene*
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy