
Současný pohled na vrozené syndromy selhání kostní dřeně (vyžádaný článek)
Current perspectives on inherited bone marrow syndromes
Inherited bone marrow failure syndromes are a heterogeneous group of diseases characterised by bone marrow failure with cytopenia in peripheral blood, associated somatic anomalies and increased tendency to malignancies. The degree of bone marrow damage can lead to various blood count changes from isolated cytopenia to bicytopenia or severe pancytopenia. The diseases in this group are caused by mutations in genes coding for proteins involved in important cellular processes such as DNA repair, ribosomal biogenesis, telomere maintenance and cell cycle regulation. Rapid development of new molecular-genetic techniques, especially next-generation sequencing, have led to the elucidation of several causative genetic defects. Besides the understanding of the pathogenesis of the different diseases, these new discoveries have significantly contributed to a better understanding of haematopoiesis regulation as well as the pathophysiology of malignancies. The new pieces of knowledge have enabled to create new disease groups such as ribosomopathies or telomeropathies. Revealing the genetic defect in individual diseases is extremely important for appropriate approach to treatment strategies and genetic counselling.
The aim of the paper is to present the overview of current knowledge about this group of diseases.
Key words:
bone marrow failure, pancytopenia, DNA repair, ribosomal biogenesis, telomere length, hematopoietic stem cell transplantation, gene therapy
Autoři:
D. Pospíšilová
Působiště autorů:
Dětská klinika LF UP a FN, Olomouc
přednosta prof. MUDr. V. Mihál, CSc.
Vyšlo v časopise:
Čes-slov Pediat 2016; 71 (4): 216-228.
Kategorie:
Přehledový článek
Souhrn
Syndromy selhání kostní dřeně představují heterogenní skupinu onemocnění, která jsou charakterizovaná selháním kostní dřeně s cytopenií v periferní krvi, vrozenými anomáliemi orgánů a zvýšeným rizikem vzniku maligního onemocnění. Stupeň postižení kostní dřeně se může projevit různými změnami v krevním obraze od izolované cytopenie přes bicytopenii k těžké pancytopenii. Příčinou onemocnění jsou mutace genů kódujících proteiny, které se účastní důležitých buněčných pochodů: reparace poškozené DNA, biogeneze ribozomů, udržování délky telomer a regulace buněčného cyklu. Díky rozvoji metod molekulární biologie, především celogenomového sekvenování, byla v průběhu posledních 10 let odhalena řada vyvolávajících genetických defektů, což kromě pochopení patogeneze jednotlivých nemocí významně přispělo k lepšímu pochopení regulace hematopoezy i patofyziologie maligních onemocnění. Nové poznatky se staly základem definice nových skupin onemocnění, jako jsou ribozomopatie nebo telomeropatie. Odhalení genetického defektu jednotlivých nemocí je velmi důležité pro správnou strategii péče o pacienty a genetické poradenství.
Cílem práce je shrnout přehled současných poznatků o této skupině onemocnění.
Klíčová slova:
selhání kostní dřeně, pancytopenie, reparace DNA, biogeneze ribozomů, délka telomer, transplantace kmenových buněk, genová léčba
ÚVOD
Vrozené syndromy selhání kostní dřeně (inherited bone marrow failure syndromes, IBMFS) patří mezi vzácné poruchy krvetvorby.
Jsou charakterizovány:
- geneticky podmíněným selháním funkce jedné nebo více hematopoetických linií (erytrocytární, granulocytární nebo trombocytární);
- vrozenými anomáliemi nebo funkčními poruchami postihujícími různé tkáně a orgány: kůži, skelet, srdce, plíce, ledviny, centrální nervový systém, které jsou často doprovázené i malým vzrůstem;
- zvýšeným rizikem vzniku maligních onemocnění: nejčastěji akutní myeloidní leukémie (AML), myelodysplastického syndromu (MDS) nebo solidních nádorů [1].
Společným rysem těchto onemocnění je výrazná variabilita jak hematologických, tak i somatických změn. Závažnější formy onemocnění se zřetelnými vrozenými anomáliemi, časnou manifestací cytopenií a rozvojem selhání kostní dřeně jsou obvykle diagnostikovány již v kojeneckém, batolecím nebo předškolním věku. Lehčí formy s nenápadnými anomáliemi a pozdní manifestací cytopenií mohou na druhé straně dlouho unikat pozornosti a jsou potom diagnostikovány až v dospělosti nebo dokonce až při vypuknutí maligního onemocnění.
Znalost příznaků těchto vzácných nemocí je proto velmi důležitá pro jejich včasnou diagnostiku a správnou volbu léčebného postupu.
Do skupiny vrozených syndromů setkání kostní dřeně jsou řazena následující onemocnění:
- Fanconiho anémie (FA)
- Dyskeratosis congenita (DC)
- Syndrom hypoplastických chrupavek a vlasů (Cartilage-hair hypoplasia – CHH)
- Diamondova-Blackfanova anémie (DBA)
- Shwachmanův-Diamondův syndrom (SDS)
- Těžká vrozená neutropenie (SCN)
- Retikulární dysgeneze (RD)
- Amegakaryocytární trombocytopenie (CAMT)
- Trombocytopenie s chyběním radia (TAR)
- Pearsonův syndrom (PS)
Někteří autoři řadí do této skupiny i další onemocnění charakterizovaná cytopenií a v některých případech i vrozenými anomáliemi, např. Kongenitální dyserytropoetickou anémii (CDA), Familiární trombocytopenii s rizikem rozvoje AML, MonoMAC syndrom a některá velmi vzácná onemocnění doprovázená cytopenií. Otázka zařazení těchto onemocnění mezi IBMS je však dosud předmětem diskuse.
ROZDĚLENÍ
Uvedenou skupinu onemocnění dělíme obvykle podle rozsahu postižených hematopoetických linií na izolované cytopenie a pancytopenie. U izolovaných cytopenií (DBA, SCN, TAR) obvykle nedochází k rozvoji pancytopenie, u ostatních onemocnění se postupně rozvíjí pancytopenie s aplazií kostní dřeně (FA, DC, SDS, CAMT).
Schematický přehled vrozených syndromů selhání kostní dřeně se základními klinickými, laboratorními příznaky a kumulativním rizikem vzniku maligního onemocnění je uveden v tabulce 1.

EPIDEMIOLOGIE
Všechna tato onemocnění jsou vzácná, incidence některých z nich není přesně známa. V řadě případů je regionálně odlišná a závislá na lokální frekvenci nosičství mutací příslušných genů. V některých uzavřených populačních skupinách je vyšší frekvence výskytu v důsledku tzv. „founder efektu“ (ztráta genetické variability, která nastává při vytvoření uzavřené nové populace s malým počtem jedinců). Typ dědičnosti je u jednotlivých chorob variabilní a závisí na charakteru vyvolávajícího genetického defektu.
KLINICKÝ OBRAZ
Při klinickém vyšetření nacházíme jednak příznaky odpovídající přítomné cytopenii (známky anemického syndromu, krvácení do kůže a sliznic, kožní infekce) a jednak různé kombinace somatických anomálií. Tyto jsou relativně charakteristické pro každý syndrom, ale často se překrývají. Mohou na onemocnění upozornit ještě před rozvojem hematologických příznaků (obr. 1). Někdy jsou však velmi nenápadné a mohou uniknout pozornosti méně zkušeného klinika. Nepřítomnost somatických anomálií však onemocnění nevylučuje.

Přehled anomálií popsaných u pacientů se syndromy selhání kostní dřeně je uveden v tabulce 2.

LABORATORNÍ VYŠETŘENÍ
Při laboratorním vyšetření nacházíme podle typu onemocnění různě vyjádřené cytopenie v periferní krvi, od izolované cytopenie až po pancytopenii a hypoplazii nebo aplazii jedné či více hematopoetických linií v kostní dřeni. Typická je makrocytóza erytrocytů, známky „stresové erytropoezy“, např. zvýšení HbF s jeho nerovnoměrnou distribucí v erytrocytech, při poruchách erytropoezy a trombopoezy potom vysoké hladiny erytropoetinu a trombopoetinu. Důležitým rysem některých onemocnění je chromozomální instabilita, která se dá prokázat in vitro zvýšením počtu spontánních chromozomálních zlomů nebo jejich zvýšením po klastogenním stresu (mitomycin, diepoxybutan). Pro DC, v menší míře i pro jiné typy IBMFS je typické zkrácení délky telomer.
PATOFYZIOLOGIE
Významné pokroky v rozvoji technik molekulárně-genetických analýz, především sekvenování nové generace (NGS), umožnily identifikovat řadu kauzálních mutací nových genů, které jsou příčinou onemocnění a mohou vést nejen k selhání kostní dřeně, ale i k dalším klinickým projevům. Významně přispěly k objasnění dů-ležitých aspektů patofyziologie onemocnění této skupiny.
MOLEKULÁRNÍ PODSTATA JEDNOTLIVÝCH TYPŮ ONEMOCNĚNÍ
Některá z těchto onemocnění byla definována na molekulární úrovni teprve v posledních letech díky rychlému rozvoji metod molekulární genetiky. Na jejich vzniku se podílejí mutace takzvaných „provozních genů“ (housekeeping genes), které kódují proteiny účastnící se čtyř důležitých regulačních buněčných procesů: reparace DNA (FA), udržování délky telomer (DC), biogeneze ribozomů (DBA, SDS, RD, DC) a regulace buněčného cyklu [1]. Většinou se tedy nejedná o nitrobuněčné procesy specifické pouze pro hematopoezu a poruchy těchto důležitých dějů mají proto dopad i na řadu jiných buněčných systémů.
Velmi zajímavým a překvapivým poznatkem posledních let bylo zjištění, že u čtyř z této skupiny onemocnění (DBA, DC, SDS, CHH) byly nalezeny mutace v proteinech, které se přímo nebo nepřímo podílejí na sestavování ribozomů. Tato onemocnění jsou dnes řazena do nově definované skupiny onemocnění zvaných „ribozomopatie“ [2]. Všechny proteiny v buňce jsou syntetizovány pomocí translačního aparátu, jehož nejdůležitější složkou jsou ribozomy. Mutace v proteinech ovlivňujících biogenezi ribozomů vede v konečném důsledku v buňkách k tzv. ribozomálnímu stresu s následnou aktivací p53 proteinu [2, 3], zvýšené apoptóze buněk a snížení úrovně translace. Vedle aktivace apoptózy může být důsledkem těchto buněčných dějů i snížení množství produkce klíčových proteinů nezbytných pro diferenciaci a proliferaci buněk. Předpokládá se, že různé tkáně jsou různě citlivé na sníženou úroveň translace. Porucha funkce ribozomů se nejvýrazněji uplatňuje v buněčných populacích s nejrychlejším obratem buněčného dělení, ke kterým hematopoeza patří. Lze očekávat, že u této skupiny chorob budou identifikovány poruchy v dalších proteinech translačního aparátu, které mohou hrát roli i při vzniku maligního bujení.
DIAGNOSTIKA A LÉČBA
Diferenciálně-diagnostická úvaha o vrozených syndromech selhání kostní dřeně je nutná u všech dlouhotrvajících cytopenií a také při diagnostice MDS a AML [4], obzvláště při současné přítomnosti vrozených anomálií a pozitivní rodinné anamnéze výskytu IBMFS. Včasné stanovení diagnózy a určení genetického defektu je nutné především pro stanovení adekvátního plánu léčebného postupu včetně transplantací kmenových buněk (HSCT), plánu dlouhodobého sledování pacienta a stanovení prognózy onemocnění. Má velký význam i pro genetické poradenství a vyhledávání dalších postižených členů rodiny.
Péče o pacienty s IBMFS v dnešní době již není pouze výsadou pediatrie. S výrazným rozvojem poznatků v oblasti podpůrné péče a především s dynamickým rozvojem nových přístupů v oblasti transplantací kmenových buněk (indikace, načasování, zdroj kmenových buněk, výběr přípravných režimů) se osud pacientů se závažnými vrozenými poruchami krvetvorby podstatně zlepšil. Pacienti s tímto onemocněním se dnes častěji dožívají dospělého věku. Proto se s touto problematikou stále více setkávají hematologové a další specialisté v oboru genetiky, onkologie, gynekologie, plicního lékařství, gastroenterologie, kteří ošetřují dospělé pacienty.
Členové rodin postižených pacientů by měli podstoupit základní hematologické a genetické vyšetření. Vyloučení přítomnosti mírné formy onemocnění u ostatních rodinných příslušníků je velmi důležité nejen pro stanovení léčebného plánu, ale také při výběru dárce před transplantací kostní dřeně.
CHARAKTERISTIKA JEDNOTLIVÝCH TYPŮ ONEMOCNĚNÍ
Fanconiho anémie (FA)
Charakteristika
Fanconiho anémie je klinicky i geneticky výrazně heterogenní onemocnění charakterizované postupně progredujícím selháním kostní dřeně, přítomností různých kombinací somatických anomálií a zvýšeným výskytem maligních onemocnění: MDS, AML a solidních nádorů rezistentních na léčbu [5]. Byla poprvé popsána v roce 1927 švýcarským pediatrem Guidem Fanconim, molekulární podstata onemocnění byla postupně odhalena v devadesátých letech.
Epidemiologie a dědičnost
Incidence onemocnění je 1–5 na 1 milion živě narozených, přičemž jsou popsány výrazné regionální rozdíly. Frekvence nosičů genu je v populaci USA odhadována na 1 : 181, vyšší je popisována např. v židovské populaci Ashkenazi Jews (1 : 93) [6]. Onemocnění je autozomálně recesivně dědičné kromě jednoho z genetických defektů (FANCB), u kterého je dědičnost gonozomální vázaná na chromozom X.
Klinické a laboratorní nálezy
Na onemocnění může již v útlém věku upozornit přítomnost pestré palety vrozených anomálií. Jsou popisovány u 80 % pacientů. Patří k nim: malý vzrůst spojený často s intrauterinní růstovou retardací, hyperpigmentace kůže, malformace palce, skeletu, ledvin, srdce, poruchy sluchu a hypogonadismus [7]. Jejich přehled je uveden v tabulce 2. Selhání KD se klinicky manifestuje obvykle kolem sedmého roku věku trombocytopenií, postupně se rozvíjí pancytopenie. Asi ve 4 % případů se aplazie kostní dřeně rozvíjí již během kojeneckého věku, vzácně se naopak může projevit až v dospělosti, někdy až současně s rozvojem maligního onemocnění. U 50 % pacientů, kteří se prezentují trombocytopenií, dochází k rozvoji pancytopenie v průběhu následujících 4 let, kumulativní incidence rozvoje selhání kostní dřeně je 50–90 % ve věku 40 let.
Nejnovější literární zdroje uvádějí, že u 25–40 % jedinců s FA jsou vrozené anomálie nenápadné, nebo dokonce nejsou přítomny vůbec. Totéž platí pro známky selhání kostní dřeně, které mohou být pouze diskrétní a dlouho unikat pozornosti [1].
U pacientů s FA je 50x vyšší riziko vzniku maligních onemocnění ve srovnání se zdravou populací: kumulativní incidence MDS, AML a solidních nádorů ve věku 40 let je 30, 10 a 30 % [8]. Nejčastějšími solidními tumory jsou spinocelulární karcinomy hlavy, krku, jazyka, jícnu a vulvy.
Molekulární podstata onemocnění
Charakteristickou vlastností buněk pacientů s FA je chromozomální nestabilita, kterou je možno in vitro dále akcentovat přidáním látek navozujících klastogenní stres (mitomycin C, diepoxybutan). Působení těchto látek indukuje vznik chromozomálních zlomů a fúzí s charakteristickými radiálními formami. Popisuje se i zvýšená tendence k apoptóze a zvýšená náchylnost k toxickému působení volných kyslíkových radikálů. Pestrému a variabilnímu klinickému obrazu odpovídá i výrazná heterogenita vyvolávajících genetických defektů, z nichž většina byla postupně analyzována až v posledních letech. Jednotlivé geny kódují proteiny, které jsou složkami systému reparace DNA. Geny, jejichž mutace byly popsány u pacientů s FA, se nazývají FANC geny – tzv. FANC komplementační skupiny. Jejich genové produkty regulují tři důležité procesy v průběhu oprav meziřetězcových křížových vazeb DNA: nukleolytické incize, translézní opravu DNA a homologní rekombinaci. Buňky pacientů s FA mají v důsledku poruchy funkce uvedených genů nízkou toleranci vůči poškození DNA jak in vitro, tak in vivo.
Od prvního popisu molekulárně-genetického defektu u FA v roce 1992 bylo dodnes identifikováno přes 1000 různých mutací 15 genů (FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, PALB2 (FANCN), RAD51C (FANCO), BRCA2, BRIP1 a SLX4 (FANCP). 60–65 % mutací tvoří FANCA mutace, z ostatních jsou nejčastější FANCB a FANCC. Asi 84 % pacientů je řazeno do subtypu A, C nebo G [5].
Osm z FA proteinů se sdružuje v tzv. jaderný komplex a společně s FAN1 proteinem jsou nezbytné pro mono-ubikvitinaci FANCD2 proteinu. Porucha funkce každého z proteinů tohoto komplexu přeruší FANCD2 monoubikvitinaci, klíčový krok, který ovlivňuje translokaci jaderného komplexu k chromatinu a jeho asociaci s ostatními jadernými proteiny účastnícími se buněčné odpovědi na poškození DNA (obr. 2).

Diagnostika a léčba
U všech pacientů s cytopenií, typickými somatickými anomáliemi s podezřením na FA je indikováno provedení testů chromozomální fragility z lymfocytů periferní krve s použitím diepoxybutanu (DEB) nebo mitomycinu C (MMC). V případech chromozomálního mozaicismu však tyto testy mohou prokázat pouze malé procento buněk s chromozomálními zlomy, průměrný počet zlomů potom může výsledně být normální. Proto u pacientů s podezřením na FA a negativním DEB testem se doporučuje doplnit vyšetření chromozomální nestability z fibroblastů. Vzhledem k možnostem sekvenování nové generace je v dnešní době molekulárně-genetická diagnostika k jednoznačnému průkazu diagnózy lépe přístupná a snáze proveditelná.
Při plánování léčebné strategie je nezbytný komplexní multidisciplinární přístup, na kterém se kromě hematologů podílejí specialisté z oboru genetiky, onkologie, neurologie, kožního lékařství. Nedílnou součástí týmu by měl být i endokrinolog, protože u pacientů s FA se setkáváme s poruchami růstu, glukózového a lipidového metabolismu, funkcí štítné žlázy, gonadálních funkcí a kostní denzity. Pacienti jsou hypersenzitivní k radiaci, kyslíkovým radikálům, a zejména k vlivu chemických mutagenů, proto musí být důsledně chráněni před jejich vlivem v běžném životě a zejména pak v případě nutnosti léčby maligních onemocnění.
Zcela nezbytné je pečlivé monitorování hematologických nálezů a periodické vyšetření kostní dřeně k vyloučení rozvoje MDS. K nepříznivému vývoji hematologického onemocnění může dojít velmi rychle. Proto je vhodné připravit rámcový plán transplantace kostní dřeně (HSCT).
Vzhledem k radio - a chemosenzitivitě je nezbytné použít přípravné režimy s redukovanou intenzitou. Na funkci kostní dřeně může mít u některých pacientů příznivý efekt léčba androgeny, na druhé straně však může mít nežádoucí účinky včetně zvýšeného rizika hepatocelulárního karcinomu. K časnému odhalení karcinomů v oblasti hlavy, krku a genitálu je nezbytné periodické sledování ve specializovaných ambulancích, většinou 1x za půl roku.
Jsou vyvíjeny nové techniky prevence a léčby FA, např. HSCT od sourozence po výběru geneticky zdravého embrya, genová léčba, genomové editování s použitím genetické rekombinace nebo konstruovaných nukleáz. Tyto přístupy jsou však zatím v experimentální fázi.
Dyskeratosis congenita (DC)
Charakteristika
Do kategorie onemocnění nazývaných souborně Dyskeratosis congenita jsou řazeny multisystémové fenotypově heterogenní syndromy s různým genetickým podkladem. Onemocnění jsou charakterizována typickou klinickou trias: přítomností slizničních leukoplakií, dystrofií nehtů a retikulárními změnami kůže, různě rychlým rozvojem selhání kostní dřeně a zvýšeným rizikem rozvoje maligního onemocnění [9]. Původně byla DC považována za primárně kožní onemocnění. Vzácněji jsou u pacientů přítomny i jiné anomálie: urogenitální, gastrointestinální, skeletální, anomálie CNS a/nebo malý vzrůst (tab. 2).
Díky pokrokům v odhalování genetické příčiny onemocnění jsou dnes do skupiny DC řazena další multisystémová onemocnění: Hoyeraalův-Hreidarssonův syndrom a Reveszův syndrom [2], část pacientů s aplastickou anémií, myelodysplazií, leukémií a idiopatickou plicní fibrózou. Toto široké spektrum nemocí od klasické DC k aplastické anémii vznikající na podkladě defektních telomer je někdy označováno jako „telomeropatie“.
Epidemiologie a dědičnost
Incidence onemocnění není přesně známa. Vzhledem k různé molekulární patofyziologii je dědičnost různá a závisí na typu vyvolávajícího defektu: autozomálně dominantní, autozomálně recesivní i gonozomální s vazbou na chromozom X.
Molekulární patofyziologie
U DC byly dosud popsány mutace 9 genů: DKC1, TERC, TERT, TINF2, NOP10, NHP2, WRAP53, CTC1, RTEL1. Všechny jmenované geny mají souvislost s udržováním délky telomer. Společné pro tuto skupinu onemocnění je zkrácení délky telomer. Telomery jsou zodpovědné za stabilizaci konců chromozomů, zabraňují jejich zkrácení během replikace odlišením chromozomálních konců od indukovaných zlomů. Jsou udržovány telomerázou, jejíž součástí je RNA komponenta (TERC), reverzní transkriptáza (TERT) a jiné proteiny jako dyskerin.
Telomeráza je silně exprimována ve tkáních obsahujících buňky s vysokým obratem replikace (hematopoetické buňky, pohlavní buňky a orgánové kmenové buňky). Pokud je hladina telomerázy snížena, telomery se s každým buněčným dělením progresivně zkracují. Výsledkem je zástava buněčného dělení, stárnutí a posléze smrt buňky jako mechanismus prevence chromozomálních přestaveb. Klinicky se u transgenních zvířat projeví jako šedivění ochlupení, alopecie, genetická nestabilita.
Pacienti s autozomálně dominantní formou mají mutace RNA komponenty telomerázy (TERC) nebo telomerázové reverzní transkriptázy (TERT). Klinický fenotyp je tedy důsledkem haploinsuficience TERC nebo TERT (10). Gen zodpovědný za vznik gonozomálně dědičného onemocnění, tzv. DKC1 gen – dyskerin je lokalizován na X chromozomu. Je exprimován ve všech tkáních, patří do skupiny ribonukleáz, které zahrnují i telomerázovou RNA. Dyskerin navíc hraje roli v biogenezi ribozomů (maturace primárního transkriptu genů pro ribozomální RNA). Ribozomální dysfunkce pravděpodobně ovlivňuje klinickou závažnost choroby v kontextu s dysfunkcí telomerázy. V roce 2008 byly u autozomálně dědičného subtypu DC identifikovány mutace genů kódujících komponenty tzv. shelterinového komplexu. Tento komplex určuje strukturu telomerického konce, účastní se tvorby t-kliček a kontroluje syntézu telomerické DNA pomocí telomerázy. Při nedostatečné protektivní funkci shelterinu dochází ke ztrátě ochrany telomer a konce chromozomů jsou nedostatečně procesovány systémem oprav DNA.
Přehled proteinů, v jejichž kódujících genech byly nalezeny mutace u DC, je uveden v obrázku 3.
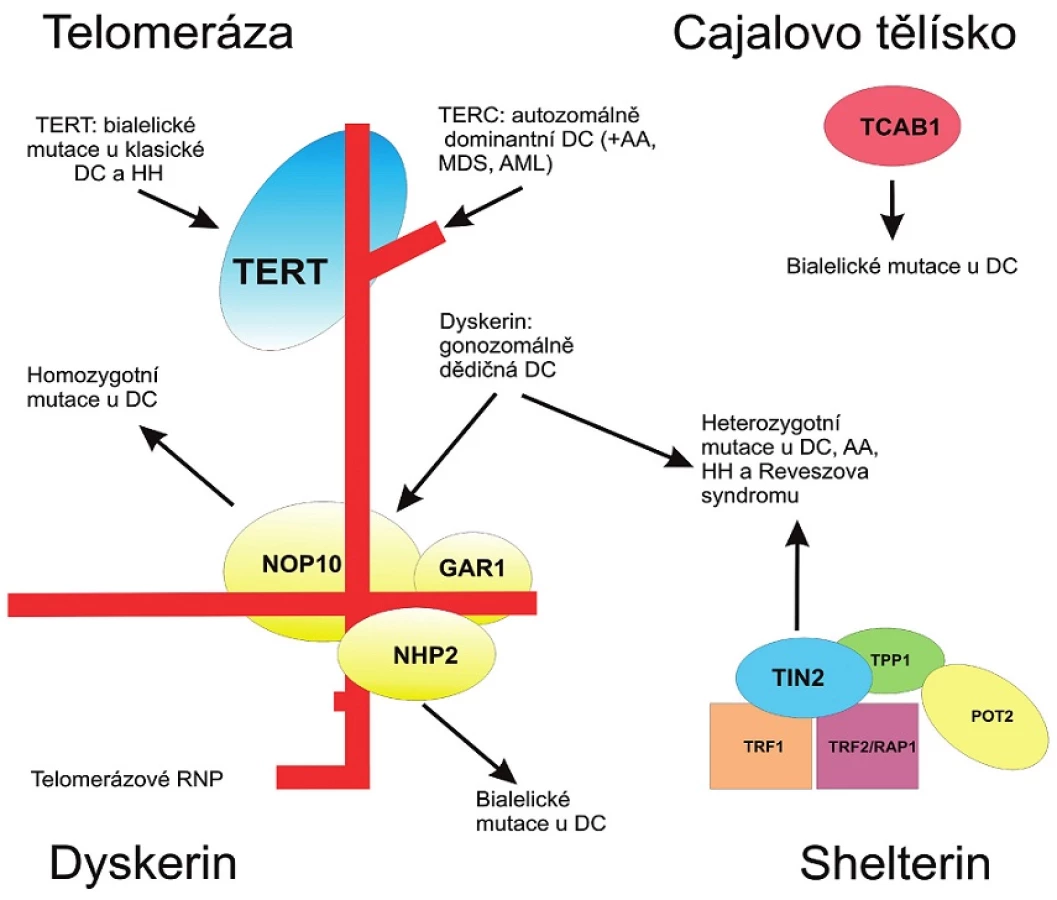
Klinické a laboratorní nálezy
Kromě typických anomálií, ke kterým patří retikulární pigmentace kůže, dystrofie nehtů a slizniční leukoplakie, může být v některých případech přítomna řada dalších anomálií. Opoždění vývoje, malý vzrůst, avaskulární nekrózy stehenní a ramenní kosti, stenózy jícnu, uretry, slzných kanálků a hypogonadismus. Byla popsána i řada dalších anomálií, jejich přehled je shrnut v tabulce 2.
Klasický klinický obraz s mukokutánními příznaky a pancytopenií se asi v polovině případů rozvíjí již od dětského věku. V prvním deceniu se nejprve objevují kožní příznaky, selhání kostní dřeně se postupně může manifestovat až u 80 % pacientů ve druhém a třetím deceniu. Dalším typickým klinickým příznakem onemocnění je plicní fibróza, která se rozvíjí v důsledku dysfunkce plicních kmenových buněk. Může být prvním příznakem onemocnění. Komplikací může být i rozvoj jaterní cirhózy. V čase manifestace jak hematologických, tak i ostatních příznaků však existuje výrazná variabilita, dokonce i mezi postiženými členy v jednotlivých rodinách. Do skupiny DC je dnes řazen: Hoyeraalův-Hreidarssonův syndrom (HHS) charakterizovaný růstovou retardací, mikrocefalií, cerebelární hypoplazií, aplastickou anémií a imunodeficitem, a Reveszův syndrom, který je charakterizován oboustrannou exsudativní retinopatií, hypoplazií kostní dřeně, dystrofií nehtů, jemnými vlasy, cerebelární hypoplazií a růstovou retardací. U pacientů byla prokázána mutace genu komplexu shelterinu: TINF2, ale i mutace DKC1 genu.
U DC byla dosud popsána následující maligní onemocnění: MDS, AML, non-Hodgkinský lymfom, karcinomy oblasti hlavy, krku a gastrointestinálního traktu, karcinom děložního čípku a bazaliom. Riziko rozvoje maligního onemocnění je 20–30 % ve věku 50 let [7]. Nejčastěji se manifestuje v mladém dospělém věku. Rozvoj MDS, AML nebo solidních nádorů může být první manifestací DC u pacientů s lehčí formou onemocnění.
Diagnostika a léčba
Na onemocnění je nutno myslet při cytopenii a přítomnosti dalších popsaných klinických příznaků. Pro diagnostiku je možno využít vyšetření délky telomer v buňkách periferní krve, jejichž zkrácení patří k základním znakům nemoci. Měření délky telomer je senzitivním, ale ne dostatečně specifickým diagnostickým testem u DC, protože zkrácení pod 1 percentil může být přítomno až u 30 % pacientů s IBMFS způsobených jinou příčinou. Tento test rovněž není vhodný k vyhledávání nosičů genových mutací u DC, protože zkrácení telomer nemusí být u pacientů bez známek BMF patrné.
U mutací TERC a TERT byl popsán fenomén genetické anticipace, který označuje postupné časnější manifestace onemocnění v následujících generacích. U mutací TINF2 proteinu, který je součástí tzv. „shelterinového komplexu“ chránícího telomery, je popisována časná manifestace onemocnění.
Formy DC s autozomálně recesivní dědičností jsou způsobené bialelickými mutacemi NOP10, NHP2, WRAP53, CTC1 – komponenty CTS komplexu zajišťujícího udržování telomer a RTEL1. Vzhledem k tomu, že u části pacientů s klasickým klinickým obrazem DC nebyla nalezena mutace žádného z výše uvedených genů, očekává se odhalení mutací dalších nových genů.
Léčba pacientů s DC se řídí podobnými zásadami jako léčba FA. Při plánování HSCT je nutno mít na paměti, že peritransplantační mortalita pacientů s DC je vyšší než u jiných BMFS a je proto nutné volit speciální přípravné režimy, protože volba adekvátního režimu může mít vliv na přežití pacienta. Standardní myeloablativní režimy jsou spojeny s četnými nežádoucími efekty, jako jsou plicní komplikace nebo venookluzivní nemoc. Nemyeloablativní režimy s použitím fludarabinu jsou spojeny s menším počtem komplikací a nižší toxicitou. První výsledky s použitím těchto režimů u DC jsou zatím povzbudivé.
Pro pacienty je důležitá pečlivá prevence rozvoje plicní fibrózy: dodržování přísného nekuřáctví, omezení podávání léků negativně ovlivňujících plicní funkce, ochrana plic před ozářením. U pacientů bez vhodného dárce je možné ke zlepšení funkce kostní dřeně použít podání androgenů.
V budoucnu se očekává použití nových léčebných možností. Bylo například prokázáno, že exogenní TERC může korigovat telomerázový defekt, upravit délku telomer a zlepšit buněčný růst lymfocytů s mutací DKC1 a TERC [11]. Je nutné prokázat, že zvýšená exprese TERC s použitím různých přístupů by se mohla stát novou léčebnou možností pro pacienty s DC.
Syndrom hypoplastických chrupavek a vlasů (Cartilage hair hypoplasia – CHH)
Charakteristika
Syndrom hypoplastických chrupavek a vlasů také známý jako McKusickův typ metafyzální chondrodysplazie, je vzácná, vysoce pleiotropní genetická porucha. Klinicky se projevuje jako forma nanismu s krátkými dolními končetinami, způsobeným skeletální dysplazií, variabilním stupněm imunodeficitu a v některých případech zvýšenou predispozicí pro nádorová bujení.
Epidemiologie, dědičnost
CHH je autozomálně recesivně dědičné onemocnění, v některých případech je popsána i dědičnost formou uniparentální dizomie, kdy jedinec zdědí dvě kopie chromozomu od jednoho rodiče místo jedné kopie od každého z rodičů.
Pacienti s CHH syndromem obvykle trpí imunodeficitem, který postihuje specifickou buněčnou imunitu. Ve studii 108 finských pacientů trpících CHH byla detekována různá úroveň lymfopenie, snížená hypersenzitivní odpověď IV. typu a neschopnost odpovědi na stimulaci fytohemaglutininem in vitro. To vede k náchylnosti a ve vážnějších případech i k podlehnutí infekcím v časném dětství. U některých pacientů s CHH byla pozorována kombinovaná forma imunodeficitu.
Molekulární patofyziologie
Byla pozorována asociace mezi mutací lokusu, ve kterém leží gen pro ncRNA komponentu RNázy MRP, RMRP. Endoribonukleáza RNáza MRP je komplex RNA molekuly a několika proteinů a účastní se na štěpení mitochondriálních primerů zodpovědných za replikaci mitochondriální DNA, její další identifikovanou funkcí je zpracování pre-rRNA v jadérku. Gen leží v lokusu na krátkém rameni lidského 9. chromozomu [12].
U CHH bylo rovněž popsáno zvýšené riziko maligních onemocnění (Hodgkinův lymfom, non-Hodgkinský lymfom, CLL, spinocelulární karcinom, bazaliom) [13].
CHH je dnes řazena do skupiny ribozomopatií [2].
Léčba
U závažné anémie (6 % pacientů) se podávají transfuze erytrocytární masy. Benefit může mít v některých případech i podání kortikoidů, jejich podávání je však diskutabilní vzhledem k současně přítomnému imunodeficitu a malému vzrůstu. Ačkoli u značné části pacientů se selhání kostní dřeně zmírňuje, u některých perzistuje. U pacientů s těžkou kombinovanou imunodeficiencí může mít dlouhodobý pozitivní efekt HSCT.
Diamondova-Blackfanova anémie (DBA)
Charakteristika
Diamondova-Blackfanova anémie je vzácná vrozená aplazie erytropoezy, charakterizovaná normochromní makrocytární anémií, těžkou retikulocytopenií, normocelulární kostní dření se selektivním nedostatkem erytroidních prekurzorů, normálním nebo lehce nižším počtem leukocytů a normálním nebo lehce vyšším počtem trombocytů. U 40–50 % pacientů jsou přítomny přídatné anomálie. Poprvé byla popsána jako klinická jednotka v roce 1938 Diamondem a Blackfanem.
Diagnóza je v 90 % stanovena do konce prvního roku života, většinou mezi 2. a 4. měsícem věku. Vzácně bývá onemocnění zjištěno později během dětského věku a výjimečně i v dospělosti [14]. U 50–60 % pacientů se nacházejí vrozené anomálie (tab. 2). Vzácně se může postupně rozvinout selhání kostní dřeně. Riziko rozvoje MDS a maligního onemocnění je relativně nízké, je udáváno v rozmezí 4–6 % [15], kumulativní incidence vzniku maligního onemocnění do věku 46 let je nověji uváděna až 22 % [1].
Epidemiologie a dědičnost
Incidence je udávána v rozmezí 5–8 případů na 1 milion živě narozených. DBA se vyskytuje u mužského a ženského pohlaví v poměru 1,1 : 1. Většinu pacientů tvoří příslušníci indoevropské populace, nemoc však byla popsána i u afrických černochů, Japonců a Arabů. U 20–40 % případů se jedná o hereditární onemocnění s autozomálně dominantním typem dědičnosti. Ve zbývajících 80 % jde o sporadické případy, v jejichž etiologii se mohou uplatnit nové mutace nebo dosud neobjasněný etiologický činitel.
Klinické a laboratorní nálezy
Prvními klinickými příznaky DBA jsou bledost, dušnost, u kojenců je často popisováno neprospívání. U 30–50 % pacientů s DBA jsou přítomny vrozené anomálie, postihující převážně oblast lbi a obličeje (kraniofaciální dysmorfie, mikrocefalie, rozštěp patra, hypertelorismus a gotické patro). K dalším anomáliím patří vývojové vady palce, horní končetiny (tříčlánkový palec, duplikace palce nebo hypoplazie palce či radia), srdce, ledvin, urogenitálního traktu a kostí.
U 30–50 % pacientů může být pozorován malý vzrůst. Na růstové retardaci u starších pacientů se může podílet dlouhodobá léčba kortikosteroidy a přetížení organismu železem.
Při laboratorním vyšetření je dominujícím nálezem makrocytární anémie s retikulocytopenií. Může být zvýšená hladina HbF jako známka stresové erytropoezy. Důležitým znakem je zvýšení hladiny erytrocytární adenosindeaminázy, které může být jedním z prvních diagnostických vodítek. I přes specificitu tohoto nálezu pro DBA nebyla dosud příčina zvýšení e-ADA vysvětlena. V kostní dřeni je v typických případech výrazně snížen počet erytroidních prekurzorových buněk, často na 1–2 %.
Podle analýzy údajů z registru DBA v USA je riziko rozvoje solidních nádorů nebo leukémie u DBA 5,4x vyšší než v populaci. Byla popsána především zvýšená incidence myelodysplastického syndromu, akutní myeloidní lekémie, karcinomu plic, tlustého střeva, bazaliomu, osteosarkomu a gynekologických karcinomů u žen. Podle novějších statistik je incidence solidních nádorů vyšší než výskyt leukémií. Malý počet popsaných případů a značná diverzita typů maligních onemocnění však zatím nedovolují definitivní závěry.
Molekulární podstata onemocnění
V roce 1999 byla poprvé odhalena genetická podstata DBA: heterozygotní mutace genu kódujícího ribozomální protein RPS19, součást malé ribozomální podjednotky. Mutace genu kódujícího RPS19 byly v roce 2000 prokázány u 25 % pacientů s DBA, v dalších letech byly nalezeny mutace genů kódujících další RP: RPS7, RPS10, RPS15, RPS24, RPS26, RPS27A, RPS17, RPL5, RPL11, RPL27 a RPL35a, a mutace jediného genu, který nekóduje RP: GATA1, důležitého transkripčního faktoru pro erytropoezu [3]. RPS19 je komponentou malé 40S ribozomální podjednotky a je lokalizován v nukleolu, hlavním místě transkripce biogeneze ribozomů. Role některých RP není dosud u vyšších eukaryont přesně známa. Delece jedné alely RP vede k poruše růstu a snížené tvorbě 40S ribozomální podjednotky, což vede k tzv. „ribozomálnímu stresu“. Buňka reaguje aktivací p53 proteinu a indukci apoptózy [3]. Dalším důsledkem je snížení translace a tím i proteosyntézy [16] s následným ovlivněním procesů s velkým nárokem na přísun proteinů – tedy s rychlým obratem produkce buněk, jako je hematopoeza, obnova kožních buněk a buněk GIT.
DBA vzniká v důsledku haploinsuficience ribozomálních proteinů. Aktivace p53 proteinu i snížení translace bylo prokázáno na buněčných i zvířecích modelech DBA. Jsou dále intenzivně studovány extraribozomální funkce RP a jejich možná úloha při vzniku somatických anomálií.
Odhalení poruchy biogeneze ribozomů jako příčiny hematologického onemocnění patří mezi nejzajímavější objevy moderní hematologie a otevřelo mnoho otázek týkajících se souvislosti poruch ribozomální biogeneze se somatickými anomáliemi a zvýšenou incidencí maligního onemocnění.
Diagnostika a léčba
Pro diagnózu DBA svědčí makrocytární anémie s retikulocytopenií při normálním počtu trombocytů a leukocytů. V době stanovování diagnózy se může objevit neutropenie a/nebo trombocytopenie, u některých pacientů naopak trombocytóza. Pro diagnostiku je stěžejní vyšetření kostní dřeně, kde je klasickým nálezem nízký počet erytroidních prekurzorových buněk v jinak normocelulární kostní dřeni. U pacientů, kteří nejsou závislí na transfuzní terapii, je přínosné vyšetření erytrocytární adenosindeaminázy (eADA), jejíž hodnota je zvýšená. Normální hladina eADA nevylučuje onemocnění. Jedinou metodou, která jednoznačně potvrzuje diagnózu, je nález mutací genů pro ribozomální proteiny. Kromě mutací genů pro RP dosud neexistuje žádný jednoznačně specifický marker k průkazu DBA.
Při diagnostice je vždy nutné vyloučit některá jiná onemocnění, o kterých lze diferenciálně diagnosticky uvažovat. Především se jedná o: Fanconiho anémii, erytroblastopenii vzniklou po infekci parvovirem B19, tranzientní erytroblastopenii malých dětí (TEC) a další získané erytroblastopenie.
Léčebně lze onemocnění ovlivnit podáním kortikoidů, na které až 60 % pacientů odpoví akcelerací erytropoezy. Kortikoidy se podávají po dosažení 1. roku života, nejprve po dobu 3 týdnů v dávce 2 mg/kg/den, při dobré odpovědi potom dlouhodobě v obdenní dávce nepřesahující 0,5 mg/kg. U zbývajících pacientů je nutno podávat dlouhodobě transfuze erytrocytární masy současně s chelátory železa k prevenci orgánového postižení při přetížení železem. Je nutno sledovat pečlivě růst pacienta a možné nežádoucí účinky přetížení železem i chelatační léčby. Nedílnou součástí týmu pečujícího o pacienty s DBA je i endokrinolog. U části pacientů může nastat remise onemocnění (spontánně nebo při léčbě kortikoidy), která nastupuje obvykle do 8.–10. roku věku, velmi vzácně i později. Pokud lze nalézt v rodině pacienta příbuzenského dárce, je u pacientů trvale závislých na podávání transfuzí indikována transplantace kmenových buněk. HSCT od nepříbuzenských dárců má horší výsledky, i když v posledních letech došlo k jejich významnému zlepšení [17].
Shwachmanův-Diamondův syndrom (SDS)
Charakteristika
Shwachmanův-Diamondův syndrom je vzácné onemocnění popsané poprvé v šedesátých letech . Je charakterizováno chronickou leukopenií, poruchou zevně sekretorické funkce pankreatu, anomáliemi skeletu, rozvojem pancytopenie a zvýšením rizika rozvoje MDS/AML. Riziko rozvoje MDS a AML je udáváno různě v rozmezí 0–36 % [1, 18].
Etiologie a dědičnost
Incidence onemocnění není přesně známa. Dědičnost je autozomálně recesivní. U 90 % pacientů byly popsány bialelické mutace genu pro Shwachmanův-Bodianův-Diamondův protein (SBDS).
Klinické a laboratorní nálezy
Neutropenie se manifestuje často již v kojeneckém věku, u části pacientů později v předškolním věku. U části pacientů jsou popisovány metafyzární dysostózy, anomálie žeber a hrudního koše a osteoporóza. U většiny pacientů bývá popisováno neprospívání a porucha růstu. Při biochemickém vyšetření jsou obvykle snížené hodnoty pankreatické amylázy a nižší hladina elastázy ve stolici. Sonografické vyšetření břicha může upozornit na změny echogenity pankreatu. U části pacientů se postupně rozvíjí selhání kostní dřeně [18].
Při vyšetření karyotypu je u pacientů s SDS nejčastěji nacházen izochromozom i(7). Způsobuje duplikaci SBDS lokusu a většinou se nachází na alele s určitou reziduální funkcí SBDS. Tato chromozomální abnormalita zatím nebyla prokázána u pacientů s DC, u kterých se vyvinula AML, pravděpodobně tedy nepřispívá k transformaci do AML [3].
Molekulární patologie
SBDS protein je lokalizován v cytoplazmě a je transportován do nukleolu, hlavního místa biogeneze ribozomů v závislosti na buněčném cyklu. Podílí se na sestavování ribozomu, má důležitou roli v napojování malé a velké ribozomální podjednotky. K jeho dalším funkcím patří: chemotaxe a destabilizace mitotického vřeténka. Role ribozomální dysfunkce v etiologii SDS byla již definitivně potvrzena, Shwachmanův-Diamondův syndrom je dnes proto řazen rovněž do skupiny ribozomopatií [2].
Diagnostika a léčba
Diagnóza je obvykle stanovena v časném dětství, malá část pacientů s dominující mírnou pankreatickou insuficiencí je diagnostikována až v dospělosti.
Na diagnózu SDS je nutno pomýšlet u dětí s neutropenií, pankreatickou insuficiencí a neprospíváním. Snížené hladiny sérového trypsinogenu, pankreatické amylázy, snížené hodnoty elastázy ve stolici a ultrazvukové vyšetření pankreatu jsou rovněž důležitými diagnostickými znaky. Vždy je nutno vyloučit cystickou fibrózu. Diagnózu jednoznačně potvrdí molekulárně-genetické vyšetření s nálezem bialelické mutace genu pro SBDS protein.
V kojeneckém a batolecím věku, pro který je u pacientů charakteristické neprospívání a porucha růstu, je vhodná substituce pankreatických enzymů. Je zajímavé, že porucha zevně exkretorické funkce pankreatu se může postupně zmírňovat a v některých případech je možno substituční léčbu vysadit. U některých pacientů se onemocnění může projevovat dlouhodobě mírnou neutropenií a diagnóza může být stanovena až v adolescentním nebo vzácně v mladém dospělém věku.
Ve velkém souboru pacientů z francouzského SDS registru bylo popsáno 20leté kumulativní riziko rozvoje těžké cytopenie 24 %, přičemž věk manifestace prvních symptomů pod 3 měsíce byl spojen se signifikantně vyšším rizikem závažných hematologických komplikací [19].
U pacientů s rychlým rozvojem pancytopenie je indikováno provedení HSC.
Těžká vrozená neutropenie (SCN)
Charakteristika
Je vzácné recesivně dědičné onemocnění, které je charakterizované těžkou vrozenou neutropenií s absolutním počtem neutrofilů (ANC) <0,2 x 109/L a časnou manifestací závažných bakteriálních infekcí. První případ onemocnění byl popsán švédským lékařem R. Kostmannem v roce 1956 ve velké rodině s vysokou mírou konsanguinity.
Název Kostmannův syndrom je v současnosti nahrazován termínem „těžká vrozená neutropenie“. U pacienta poprvé popsaného Kostmannem byla dodatečně popsána mutace HOX1 (viz tab. 2). Pro recesivně dědičnou formu vrozené agranulocytózy způsobenou mutacemi genu pro HOX1 je proto někdy používán stále název Kostmannova agranulocytóza.
Epidemiologie a dědičnost
Incidence onemocnění je udávána 1–2 případy na milion obyvatel. Dědičnost je variabilní, byla popsána autozomálně recesivní i dominantní dědičnost, vzácněji i gonozomální dědičnost s vazbou na chromozom X v závislosti na typu genetického defektu.
Klinické a laboratorní nálezy
Onemocnění se většinou projevuje již v prvních měsících života těžkými hnisavými záněty: omfalitidou, recidivujícími záněty středouší, záněty horních cest dýchacích, pneumoniemi, abscesy kůže a bakteriální sepsí.
V periferní krvi je těžká agranulocytóza s ANC <0,2 x 109/L. V kostní dřeni nacházíme typicky blok ve vyzrávání na úrovni promyelocyt – myelocyt. Myelocyty, metamyelocyty, tyče a segmenty jsou nalézány jen ojediněle. Zástava vyzrávání bílé řady je vysvětlována urychlenou apoptózou promyelocytů a následných stadií vývoje granulocytu.
Molekulární podstata onemocnění
Příčinou onemocnění jsou nejčastěji u 30–60 % případů mutace genu pro elastázu neutrofilů (ELA2), proteázu syntetizovanou v promyelocytech, skladovanou v granulech neutrofilů v aktivní formě a uvolňovanou v místě zánětu. Mutace genu ELA2 je nacházena u většiny pacientů s Kostmannovým syndromem a u všech pacientů s cyklickou neutropenií, což je dokladem blízké příbuznosti obou onemocnění [1]. Dalšími kandidátními geny, jejichž mutace byly popsány u vrozené těžké neutropenie, jsou: HAX1, G6PC3 [21], GFII mutace a mutace genu pro G-CSF (CSF3R) [22] a WASP. V případě mutací genu pro WAS protein se jedná o „gain of function“ mutace, tedy mutace odlišné od genetických změn způsobujících Wiskottův-Aldrichův syndrom.
Dosud nebylo jednoznačně objasněno, jakým způsobem mutace genu pro ELANE způsobují neutropenii. Je zajímavé, že dosud popsané mutace uchovávají čtecí rámec nebo jsou lokalizovány v oblasti C-terminu proteinu. Patogenetický mechanismus tedy vede k syntéze abnormálního proteinu spíše než k haploinsuficienci. Jeden z modelů soudí, že porucha sbalování – „misfolding“ proteinu aktivuje aberantně složený/nezabalený protein, což indukuje apoptózu neutrofilních prekurzorů. K patogenezi může přispívat i aberantní/narušený intracelulární transport mutovaného proteinu. Je diskutován možný vliv stresu endoplazmatického retikula, lysozomálního transportu, mitochondriální dysfunkce a poruchy glykosylace [20].
U pacientů s SCN je popisována zvýšená tendence k akvizici změn chromozomu 7.
Rozvoji MDS/AML u pacientů s mutacemi ELANE, HAX11 a aktivační mutací genu pro WAS většinou předchází klonální expanze buněk nesoucích nesmyslnou mutaci genu CSF3R [22]. Tyto mutace predikují leukemickou transformaci. U MDS/AML u pacientů s mutací ELANE byly rovněž popsány abnormality chromozomu 7 a mutace RUNX, RAS a KIT genů.
Diagnostika a léčba
Diagnóza je obvykle stanovena již v prvním roce života na podkladě agranulocytózy s klinickými příznaky charakteru těžkých purulentních infekcí a na podkladě vyšetření kostní dřeně, kde nacházíme typický blok vyzrávání v granulocytární řadě na úrovni promyelocytu. Definitivně ji potvrzuje výsledek molekulárně-genetické analýzy.
Zahájení léčby pomocí G-CSF významně zlepšilo přežití a morbiditu pacientů s těžkou vrozenou neutropenií. Dále však pokračuje diskuse o možné souvislosti dlouhodobého podávání G-CSF s rozvojem MDS/AML. Transformace do MDS/AML je nejvyšší u pacientů se špatnou odpovědí na léčbu vyžadujících vysoké dávky G-CSF. Odpověď na otázku, jestli má podávání G-CSF přímý vliv na rozvoj MDS/AML nebo zda se jedná spíše o vliv základního onemocnění, zůstává zatím kontroverzní. I přes uvedené pochybnosti je však u pacientů s SCN i nadále doporučeno dlouhodobé podávání G-CSF v dávce 5–10 µg/kg.
Transplantace kostní dřeně je indikována u pacientů, kteří neodpovídají na léčbu G-CSF nebo u kterých je nutno použít vysokých dávek. Doporučuje se u pacientů, pro které je k dispozici příbuzenský dárce.
V souboru transplantovaných pacientů z francouzského registru s SCN bylo 3leté celkové přežití 82 % a mortalita 17 %. Kumulativní incidence chronické GvHD byla 20 %. Autoři zdůrazňují velmi důkladný výběr kandidátů HSCT vzhledem k výši peritransplantační mortality [23]. Výsledky HSCT u pacientů, u kterých se vyvinula maligní transformace (MDS, AML), nejsou příznivé.
Retikulární dysgeneze (RD)
Charakteristika
Retikulární dysgeneze je extrémně vzácné autozomálně recesivně dědičné onemocnění, které je nejtěžší formou těžkého vrozeného kombinovaného imunodeficitu. Je charakterizována chyběním granulocytů a téměř kompletním deficitem lymfocytů v periferní krvi, hypoplazií thymu a sekundárních lymfoidních orgánů, chyběním přirozené imunity, adaptivních humorálních a buněčných odpovědí a senzorineurickou hluchotou. Důsledkem jsou závažné infekce, především fatální septikémie v průběhu několika dní po porodu. V kostní dřeni se nachází typický blok vyzrávání na úrovni promyelocytu, vyzrávání erytroidních a megakaryocytárních buněk je normální. Nejedná se tedy o defekt kmenové buňky, ale o unikátní aberaci myeloidní-lymfoidní linie, genetickým defektem způsobujícím onemocnění jsou mutace genu kódujícího adenylát kinázy 2 (AK2), mitochondriální protein důležitý pro regulaci intracelulárních hladin adenozin-difosfátu a udržování potenciálu mitochondriální membrány, u které byla prokázána specifická role při vyzrávání granulocytů. RD je prvním případem imunodeficitu spjatým kauzálně s energetickým metabolismem. Je nyní řazena spíše mezi mitochondriální onemocnění a těžké imunodeficity [8].
Amegakaryocytární trombocytopenie
Charakteristika
Je velmi vzácné onemocnění charakterizované závažnou trombocytopenií projevující se většinou již v novorozeneckém a kojeneckém věku a progresivním rozvojem selhání kostní dřeně v prvních letech života u většiny pacientů. U části pacientů jsou popisovány vrozené anomálie (srdeční vady, poruchy růstu, psychomotorická retardace) a zvýšené riziko vzniku leukémie (AML, ALL).
Epidemiologie a dědičnost
Onemocnění se vyskytuje velmi vzácně, jeho přesná incidence není známa. Dědičnost je autozomálně recesivní.
Molekulární patogeneze
Příčinou onemocnění jsou mutace genu kódujícího receptor pro hlavní růstový faktor pro trombopoezu: trombopoetinový receptor – C-MPL [24].
Klinické a laboratorní nálezy
Onemocnění se manifestuje většinou již v novorozeneckém věku těžkou trombocytopenií s krvácivými projevy. Velikost a tvar trombocytů je normální. U novorozenců je nutno odlišit Wiskottův-Aldrichův syndrom a imunitní trombocytopenie. V kostní dřeni je výrazně snížený počet megakaryocytů. Hladina endogenního trombopoetinu (TPO) je vysoká, ale trombocyty ani krevní progenitory na něj nereagují v důsledku kauzálních mutací C-MPL. Závažnost onemocnění závisí na typu mutace a jejím výsledném vlivu na funkci proteinu. U většiny pacientů se rozvíjí selhání kostní dřeně. Vzácně byly popsány případy s pozdější manifestací onemocnění a lehčím průběhem, který závisí na typu mutace [24].
Diagnostika a léčba
Jako prevence krvácení je při nízkém počtu trombocytů nutno podávat transfuze trombocytů. Jedinou možností vyléčení je transplantace kmenových buněk. Kongenitální amegakaryocytární trombocytopenie jako onemocnění s monogenní dědičností je do budoucna vhodným kandidátem na genovou terapii.
TAR syndrom
Charakteristika
Je vzácné vrozené onemocnění charakterizované anomálií předloktí způsobenou oboustrannou dysplazií radia, těžkou trombocytopenií a u části pacientů dalšími anomáliemi skeletu, kardiovaskulárního a gastrointestinálního systému. Do skupiny TAR syndromu je někdy řazena i trombocytopenie s radioulnární synostózou, která je způsobena mutacemi HOXA11 genu.
Molekulární podstata, dědičnost
Genetický podklad onemocnění byl poprvé popsán teprve v roce 2012, kdy byla u pacienta s TAR nalezena bialelická mutace genu RBM8A. RBMA kóduje podjednotku Y13 v tzv. exon-junction komplexu (EJC), která je důležitá pro RNA procesování a je exprimovaná ve všech hematopoetických tkáních [25]. TAR syndrom je tedy důsledkem kombinované dědičnosti nízké frekvence nekódujícího SNP a raritní nulové alely v genu RBM8A. Jedná se o první popsaný defekt EJC komplexu asociovaný s onemocněním u člověka. TAR syndrom je v kontextu abnormálního fenotypu asociován s proximální a distální 1q21.1 mikrodelecí a mikroduplikací s inkompletní penetrancí a variabilní expresivitou.
Diagnostika a léčba
Diagnóza je ve většině případů stanovena na podkladě klinických příznaků: krvácivých projevů a přítomnosti anomálií horních končetin v prvních dnech po narození. V léčbě onemocnění jsou v prvním roce života používány trombocytární náplavy, později se jejich spotřeba výrazně snižuje. Důležitou roli hraje především péče ortopedů a plastických chirurgů při řešení anomálií předloktí.
Pearsonův syndrom (PS)
Charakteristika
Pearsonův syndrom je velmi vzácná sideroblastická anémie s exokrinní pankreatickou dysfunkcí, poruchou funkce jater a defekty tubů ledvin. Onemocnění se manifestuje často již v novorozeneckém věku a bylo popsáno jako jedna z příčin fetálního hydropsu. Pro PS je typická perzistující makrocytární anémie v periferní krvi s charakteristickou vakuolizací v prekurzorových buňkách kostní dřeně. Vzácně byly u pacientů s PS popsány asociované anomálie jako retinopatie, ataxie, svalová slabost spojená s neprospíváním nebo poruchou růstu. V 90. letech byla odhalena molekulární podstata onemocnění: delece a duplikace úseků mitochondriální DNA. Důsledkem mitochondriálního defektu je dysfunkce enzymů oxidativní respirační kaskády, která vede ke vzniku typické acidózy. U pacientů, kteří přežijí novorozenecké období, může dojít ke zlepšení anémie se zvýšením hladiny hemoglobinu.
Jedinou možností léčby je podávání transfuze erytrocytů. Léčba růstovými faktory neprokázala efekt. Mortalita onemocnění je vysoká v důsledku acidózy, sepsí nebo selhání jater a ledvin. Maligní onemocnění nebyla v souvislosti s PS popsána.
ZÁVĚR
Vrozené syndromy selhání kostní dřeně jsou narůstající skupinou onemocnění, jejíž přesná patogeneze je postupně odhalována s rozvojem metod molekulární genetiky, především sekvenování nové generace. Odhalení podstaty jednotlivých syndromů vedlo nejen k lepšímu porozumění řadě buněčných pochodů, ale dokonce k vytvoření nových skupin onemocnění v humánní medicíně, jako jsou například „ribozomopatie“ nebo „telomeropatie“.
Tato onemocnění s výraznou variabilitou klinických příznaků u stejných genetických defektů jsou příkladem vlivu epigenetických změn na fenotyp onemocnění a jsou modelem pro studium inkompletní penetrance a variabilní expresivity genetické informace včetně modifikujících genů a spontánní genové konverze, která vede k mozaicismu somatických buněk a je příkladem přirozeného modelu genové terapie. U některých pacientů jsou hematologické nálezy a vrozené anomálie velmi nenápadné a diagnóza tak může být stanovena v dospělosti, někdy až při diagnóze maligního onemocnění (MDS/AML) nebo typických solidních nádorů, případně nemaligních onemocnění jako plicní fibróza, jaterní cirhóza a striktura uretry.
Při rychlosti vývoje nových diagnostických možností a rozšiřování poznatků o jednotlivých onemocněních lze předpokládat, že pacienti s atypickou prezentací již známých syndromů, případně s nově zařazenými vrozenými syndromy selhání kostní dřeně, budou především v mladém dospělém věku diagnostikováni stále častěji.
Nové poznatky získané při molekulární a biochemické analýze genů a proteinů, které hrají roli v patogenezi vrozených syndromů selhání kostní dřeně, poskytují unikátní pohled na jejich funkci v průběhu hematopoezy. Další výzkum může poskytnout cenné informace v oblasti regulace hematopoezy, kancerogeneze a přinést nové možnosti cílené léčby těchto závažných onemocnění.
Prof. MUDr. Dagmar Pospíšilová, Ph.D.
Dětská klinika LF UP a FN
I. P. Pavlova 6
775 20 Olomouc
e-mail: Dagmar.Pospisilova@fnol.cz
Zdroje
1. Wilson DB, Link DC, Mason PJ, Bessler M. Inherited bone marrow failure syndromes in adolescents and young adults. Ann Med 2014; 46 (6): 353–363.
2. Chirnomas SD, Kupfer GM. The inherited bone marrow failure syndromes. Pediatr Clin North Am 2013; 60 (6): 1291–1310.
3. Parikh S, Bessler M. Recent insights into inherited bone marrow failure syndromes. Curr Opin Pediatr 2012; 24 (1): 23–32.
4. Babushok DV, Bessler M. Genetic predisposition syndromes: when should they be considered in the work-up of MDS? Best Pract Res Clin Haematol 2015; 28 (1): 55–68.
5. Bogliolo M, Surrallés J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev 2015; 33 : 32–40.
6. Rosenberg PS, Tamary H, Alter BP. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am J Med Genetics A 2011; 155A: 1877–1883.
7. Bessler M, Mason PJ, Link DC, Wilson DB. Inherited Bone Marrow Failure Syndromes. In: Orkin SH, et al. Hematology of Infancy and Childhood. 7th ed. Philadelphia: Saunders Elsevier, 2009 : 307–395.
8. Alter BP, Giri N, Savage SA, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 2010; 150 (2): 179–188.
9. Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program 2011; 2011 : 480–486.
10. Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Developm 2011; 25 : 11–16.
11. Kirwan M, Beswick R, Vulliamy T, et al. Exogenous TERC alone can enhance proliferative potential, telomerase activity and telomere length in lymphocytes from dyskeratosis congenita patients. Br J Haematol 2009; 144 (5): 771–781.
12. Ridanpaa M, van Eenennaam H, Pelin K, et al. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell 2001; 104 (2): 195–203.
13. Taskinen M, Ranki A, Pukkala E, et al. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet A 2008; 146A: 2370–2375.
14. Vlachos A, Ball S, Dahl N, et al. Participants of Sixth Annual Daniella Maria Arturi International Consensus Conference. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol 2008; 142 (6): 859–876.
15. Vlachos A, Rosenberg PS, Atsidaftos E, et al. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood 2012; 119 : 3815–3819.
16. Cmejlova J, Dolezalova L, Pospisilova D, et al. Translational efficiency in patients with Diamond-Blackfan anemia. Haematologica 2006; 91 (11): 1456–1464.
17. Fagioli F, Quarello P, Zecca M, et al. Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: a report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol 2014; 165 (5): 673–681.
18. Dror Y, Donadieu J, Koglmeier J, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci 2011; 1242 : 40–55.
19. Donadieu J, Fenneteau O, Beaupain B, et al.; Associated investigators of the French Severe Chronic Neutropenia Registry. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica 2012; 97 (9): 1312–1319.
20. Grenda DS, Murakami M, Ghatak J, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood 2007; 110 : 4179–4187.
21. Hayee B, Antonopoulos A, Murphy EJ, et al. G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for neutrophil dysfunction. Glycobiology 2011; 21 : 914–924.
22. Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: Results of a long-term survey. Blood 2007; 109 : 93–99.
23. Fioredda F, Iacobelli S, van Biezen A, et al. Stem cell transplantation in severe congenital neutropenia: an analysis from the European Society for Blood and Marrow Transplantation. Blood 2015 Jul 16. pii: blood-2015-02-628859.
24. Ballmaier M, Germeshausen M. Congenital amegakaryocytic thrombocytopenia: clinical presentation, diagnosis, and treatment. Semin Thromb Hemost 2011 Sep; 37 (6):673–681.
25. Albers CA, Paul DS, Schulze H, et al. Compound inheritance of a low-frequency regulatory SNPand a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nature Genetics 2012; 44 (S1–2): 435–439.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2016 Číslo 4
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- OZP pečuje o zdraví svých pojištěnců již 30 let. V čem je lepší a jaké výhody nabízí?
- Cytomegalovirové infekce u novorozenců a dětí
- Cytomegaloviróza a spalničky u dětí
Nejčtenější v tomto čísle
- Bolest na hrudi
- Hemoptýza
- Jak hodnotit vysoce senzitivní troponin T u novorozenců?
- Současný pohled na vrozené syndromy selhání kostní dřeně (vyžádaný článek)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy