
Progresivní familiární intrahepatální cholestáza
Progressive Familial Intrahepatic Cholestasis
The term progressive familial intrahepatic cholestasis (PFIC) represents complex designation for a heterogeneous group of diseases developing due to mutations in genes coding canalicular proteins associated with the bile production. According to specific mutation we can distinguish 3 types of PFIC (PFIC1–3). PFIC1 and PFIC2 usually appear during the first months of the life; PFIC3 then appears much later - during the whole childhood and rarely even in adults. Phenotypic manifestations of PFIC are diverse, but the main symptoms are pruritus and jaundice.
Diagnostics is based on precise evaluation of clinical course of the disease, biochemical liver tests (normal levels of gammaglutamyltransferase in PFIC1 and PFIC2 are unique), and histological, immunohistochemical and electron microscopic examination of the liver tissue. Biliary tract imaging together with biochemical bile analysis is sometimes indicated in PFIC3. Molecular genetic examination of all three types of PFIC is also available. Conservative treatment, which consists of standard cholestasis management including ursodeoxycholic acid application, usually fails. The natural history of the disease may be influenced by biliary diversion, but most of the patients progresses to liver failure and requires liver transplantation.
Key words:
progressive familial intrahepatic cholestasis
Autoři:
P. Dědek
Působiště autorů:
Dětská klinika Fakultní nemocnice a Lékařská fakulta, Hradec Králové
přednosta prof. MUDr. M. Bayer, DrSc.
Vyšlo v časopise:
Čes-slov Pediat 2010; 65 (4): 197-205.
Kategorie:
Postgraduální vzdělávání
Souhrn
Progresivní familiární intrahepatální cholestáza (PFIC) je souhrnné označení pro heterogenní skupinu autozomálně recesivně dědičných onemocnění dětského věku podmíněných mutacemi v genech kódujících kanalikulární proteiny odpovědné za tvorbu žluče. Podle typu mutace rozeznáváme 3 formy PFIC (PFIC1–3). PFIC1 a PFIC2 se manifestují obvykle v novorozeneckém a kojeneckém věku, PFIC3 pak v průběhu celého dětského věku, vzácně až u dospělých. Fenotypové projevy PFIC jsou velmi variabilní. Hlavními klinickými příznaky jsou pruritus a ikterus.
Unikátním laboratorním rysem je normální hodnota gamaglutamyltransferázy u PFIC1 a PFIC2. Diagnostika PFIC je založena na zhodnocení klinického průběhu, laboratorního vyšetření, histologického, imunohistochemického a elektronmikroskopického vyšetření jaterní tkáně. U PFIC3 je někdy indikováno zobrazovací vyšetření žlučových cest s odběrem žluče k biochemické analýze. Diagnostické možnosti všech forem PFIC rozšiřuje molekulárně genetické vyšetření. Konzervativní léčba, která spočívá kromě obvyklých zásad léčby cholestázy v podávání kyseliny ursodeoxycholové, je obvykle neúčinná. Přirozený průběh onemocnění může ovlivnit provedení biliární diverze, většina nemocných je však indikována k transplantaci jater.
Klíčová slova:
progresivní familiární intrahepatální cholestáza
Úvod
Progresivní familiární intrahepatální cholestáza (PFIC) označuje vzácnou skupinu cholestatických chorob s autozomálně recesivní dědičností a progresivním průběhem s rozvojem portální hypertenze a/nebo jaterního selhání v první či druhé dekádě života. Incidence onemocnění není přesně známá, odhaduje se na 1 : 50 000–1 : 100 000 [1, 12]. Na celkovém počtu cholestáz novorozeneckého a kojeneckého období se PFIC podílí asi 1–10 % [2, 3].
Poprvé bylo onemocnění popsáno v roce 1965 jako Bylerova choroba u potomků ortodoxních Amishů v rodině Jacoba Bylera a Nancy Kaufmannové. Následovaly kazuistiky s podobnou symptomatologií, ale bez příbuzenského vztahu k této rodině, označované jako Bylerův syndrom. Tato skupina chorob připomínala v časných stadiích vývoje již dříve definovanou benigní rekurentní intrahepatální cholestázu (BRIC). Progresivní charakter onemocnění a familiární výskyt vedl k souhrnnému názvu progresivní familiární intrahepatální cholestáza. V 80. a 90. letech minulého století byla na základě rozdílných histologických a elektronmikroskopických nálezů jaterní tkáně vyslovena hypotéza, že se jedná o heterogenní skupinu onemocnění [4, 5]. Další vývoj tuto domněnku potvrdil.
S rozvojem metod molekulární genetiky byly identifikovány geny a jejich produkty lokalizované převážně v kanalikulární membráně jaterní buňky, které se podílejí na transportu žlučových kyselin a fosfolipidů z hepatocytů do žluče. Současná klasifikace PFIC je založena na typu defektního genu [5, 6, 7, 8, 9]. Charakteristiku jednotlivých forem PFIC uvádí tabulka 1.

Progresivní forma FIC1 onemocnění
Progresivní forma FIC1 (PFIC1, Bylerova choroba, FIC = Familial Intrahepatic Cholestasis) je onemocnění podmíněné mutací genu ATP8B1 lokalizovaného na chromozomu 18 (18q2-22) [1, 6, 7, 8, 9, 10]. Produkt genu (FIC1 protein) patří mezi tzv. přenašeče P-typu ATP-ázy účastnící se transportu aminofosfolipidů, jako je fosfatidylserin a fosfatidyletanolamin. Nejedná se o typický přenašeč, protože pouze „švihá“ aminofosfolipidy („aminofosfolipid flipáza“) z vnějšího listu buněčné membrány na vnitřní a zajišťuje tak žádoucí asymetrii buněčné membrány. FIC1 gen je exprimován nejen v kanalikulární membráně jaterní buňky a apikální membráně cholangiocytů, ale i v jiných orgánech, např.v enterocytech a acinárních buňkách slinivky břišní. Extrahepatální exprese genu je dokonce výraznější a vysvětluje oproti ostatním formám PFIC systémový charakter onemocnění.
Existuje řada mutací FIC1 genu. Výsledky studií zabývající se vztahem mezi jednotlivými typy mutací a závažností onemocnění nejsou jednoznačné [6]. Mutace D554N způsobuje jednu z variant progresivní formy FIC1 onemocnění u Inuitů v Grónsku a Kanadě, tzv. grónskou familiární cholestázu [7]. Patofyziologické mechanismy vysvětlující důsledek porušené funkce FIC1 proteinu nejsou zcela objasněny. Předpokládá se účast tzv. jaderného farnesoid X receptoru, který je klíčovým regulátorem homeostázy žlučových kyselin, protože zasahuje do jejich metabolismu a transportu nejen v játrech, ale i ve střevě [6, 7, 8]. Deficit FIC1 proteinu vede k downregulaci farnesoid X receptoru a následně ke snížené expresi BSEP proteinu (viz dále), zvýšené syntéze žlučových kyselin a zvýšené expresi apikálního přenašeče žlučových kyselin v tenkém střevě. Výsledkem je toxická koncentrace žlučových kyselin v séru a v jaterní buňce. Na patogenezi onemocnění se pravděpodobně podílí také porucha funkce CFTR (cystic fibrosis transmembrane conductance regulator) genu.
Symptomatologie a klinický průběh progresivní formy FIC1 onemocnění je značně variabilní [6, 7, 8, 9]. V klinickém obraze je iniciálním příznakem intenzivní pruritus, který se objevuje obvykle již v novorozeneckém věku a příliš nekoreluje s hladinou bilirubinu. Mezi další symptomy či komplikace patří: ikterus, hepatomegalie, splenomegalie, neprospívání, příznaky z nedostatku vitaminů rozpustných v tucích, cholelitiáza, cholecystitida, respirační potíže („astma-like“ symptomatologie), porucha růstu, chronický průjem, recidivující akutní pankreatitida, epistaxe nesouvisející s trombocytopenií či poruchou hemokoagulace, centrální hluchota a u dospívajících opožděná puberta.
Laboratorně je onemocnění charakterizováno normální hodnotou gamaglutamyltransferázy (GMT), která je odrazem nízké koncentrace žlučových kyselin ve žluči a chyběním jejich detergentního účinku na membránu kanalikulů a buněk žlučové výstelky obsahující molekuly GMT [7, 8, 9]. Konjugovaná hyperbilirubinémie je kolísavá, aktivita aminotransferáz se pohybuje od normálních hodnot až po 10násobek horní hranice normy, je zvýšená aktivita alkalické fosfatázy, hladina cholesterolu je normální nebo jen lehce zvýšená, je vysoká hladina žlučových kyselin v séru, někdy je falešně pozitivní výsledek potního testu.
Při histologickém vyšetření jaterní tkáně je popisována mírná kanalikulární cholestáza, žlučové válce, pseudoacinární rozety a balonová degenerace hepatocytů. I když centrilobulárně mohou být přítomny změny ve smyslu obrovskobuněčné hepatitidy, typický histologický obraz neonatální hepatitidy je vzácný. S progresí onemocnění dochází k poruše architektoniky jaterní tkáně až do stadia cirhózy [6, 8]. Imunohistochemickým vyšetřením ze zamraženého vzorku jater zjišťujeme přítomnost FIC1 proteinu. Specifické je elektronmikroskopické vyšetření, při kterém vždy prokazujeme hrubě granulární žluč v kanalikulech, tzv.”Bylerovu žluč” (obr. 1). Méně charakteristická je redukce mikroklků kanalikulární membrány a ztluštění stěny perikanalikulárních filament.

V diagnostice FIC1 onemocnění, ale i dalších typů PFIC, je významná přímá DNA analýza s přímým průkazem obou patologických mutací.
K rozvoji chronického jaterního selhání dochází v první či druhé dekádě života [6]. Konzervativní léčba může zmírnit některé příznaky, jako je svědění, ale přirozený vývoj onemocnění obvykle neovlivní. Užívá se kyselina ursodeoxycholová (UDCA), cholestyramin či rifampicin. Naopak chirurgické metody zasahující do enterohepatální cirkulace žlučových kyselin a provedené ještě před rozvojem cirhózy jsou asi u poloviny nemocných provázeny výrazným zlepšením klinického stavu, laboratorních ukazatelů cholestázy, upravuje se i histologický a elektronmikroskopický nález [9]. K indikaci chirurgické léčby může přispět efekt endoskopické nazobiliární drenáže. Parciální externí biliární diverze je charakterizována anastomózou mezi fundem žlučníku a krátkým jejunálním segmentem, který je vyveden na kůži jako koncové stoma. Další možností je tzv. metoda “ileal exclusion“, při které je vyřazeno asi 15–25 % délky ilea anastomózou mezi proximálním ileem a cékem. Výhodou této metody je nepřítomnost stomie. V průběhu několikaletého sledování však dochází u části pacientů k rekurenci potíží, proto je více preferována předešlá metoda. V pokročilých stadiích je indikována transplantace jater, ta však neřeší extrahepatální projevy onemocnění, které nadále přetrvávají nebo se dokonce zhoršují (např. průjem). V transplantovaných játrech byl také pozorován rozvoj steatohepatitidy až do obrazu cirhózy s nutností retransplantace [1].
Koncem 90. let bylo prokázáno, že mutace v obou genech ATP8B1 (FIC1) jsou také zodpovědné za některé případy benigní rekurentní intrahepatální cholestázy [6, 8, 10]. Tato tzv. benigní forma FIC1 onemocnění (benigní rekurentní intrahepatální cholestáza typ 1 = BRIC1) je charakterizována rekurentními epizodami cholestázy. Onemocnění se manifestuje od kojeneckého věku až po dospělost, frekvence a délka atak je velmi variabilní. V popředí potíží je výrazný pruritus zásadně ovlivňující kvalitu života nemocných. Dále bývá přítomna únava, nechutenství, ikterus, acholické a průjmovité stolice. Spouštěcí faktor epizod není většinou odhalen, někdy může být provokujícím momentem infekce, jindy těhotenství či užívání hormonální antikoncepce. Výsledky laboratorního vyšetření v době ataky jsou srovnatelné s vyšetřením u progresivní formy FIC1 onemocnění. Histologický nález prokazuje fokální kanalikulární cholestázu. Benigní charakter onemocnění je založen na pozorování, že v průběhu choroby nedochází k významné progresi fibrózy. Mezi atakami je nemocný klinicky zcela bez potíží, laboratorní odchylky i histologický nález se normalizují. Neexistuje spolehlivá léčba a prevence cholestatických epizod. V současné době je u pacientů s atakou cholestázy doporučováno provedení krátkodobé endoskopické nazobiliární drenáže, po které následuje dlouhodobá klinická i laboratorní remise [11].
Vztah mezi progresivní a benigní variantou FIC1 onemocnění není vždy jednoznačný, existují i intermediární formy [6, 10]. Progresivní a benigní varianta tak představuje oba konce spojitého spektra fenotypových projevů FIC1 onemocnění, které není možné vždy vysvětlit typem patologické mutace. Identické mutace se dokonce podílejí na obou formách onemocnění. Byly také publikovány kazuistiky s benigní formou v kojeneckém a batolecím věku s přechodem do progresivní formy v dospělosti. Průběh onemocnění bude pravděpodobně ovlivněn dalšími dosud neidentifikovanými geny či vlivy prostředí.
Progresivní forma BSEP onemocnění
Progresivní forma BSEP onemocnění (PFIC2, BSEP = Bile Salt Export Pump) je způsobená mutacemi genu ABCB11 lokalizovaném na chromozomu 2 (2q24-31) [1, 6, 7, 8, 12]. Produktem genu je hlavní přenašeč žlučových kyselin (BSEP protein), který je součástí širší skupiny ATP dependentních přenašečů přenášející žlučové kyseliny z jaterní buňky do žluče proti extrémnímu koncentračnímu gradientu. BSEP protein je lokalizován především v kanalikulární membráně. Důsledkem porušené funkce je značně vysoká koncentrace žlučových kyselin v jaterní buňce. Je popsáno velké množství mutací genu ABCB11 [12]. V Evropě má nejvyšší frekvenci výskytu mutace E297G (30 % případů).
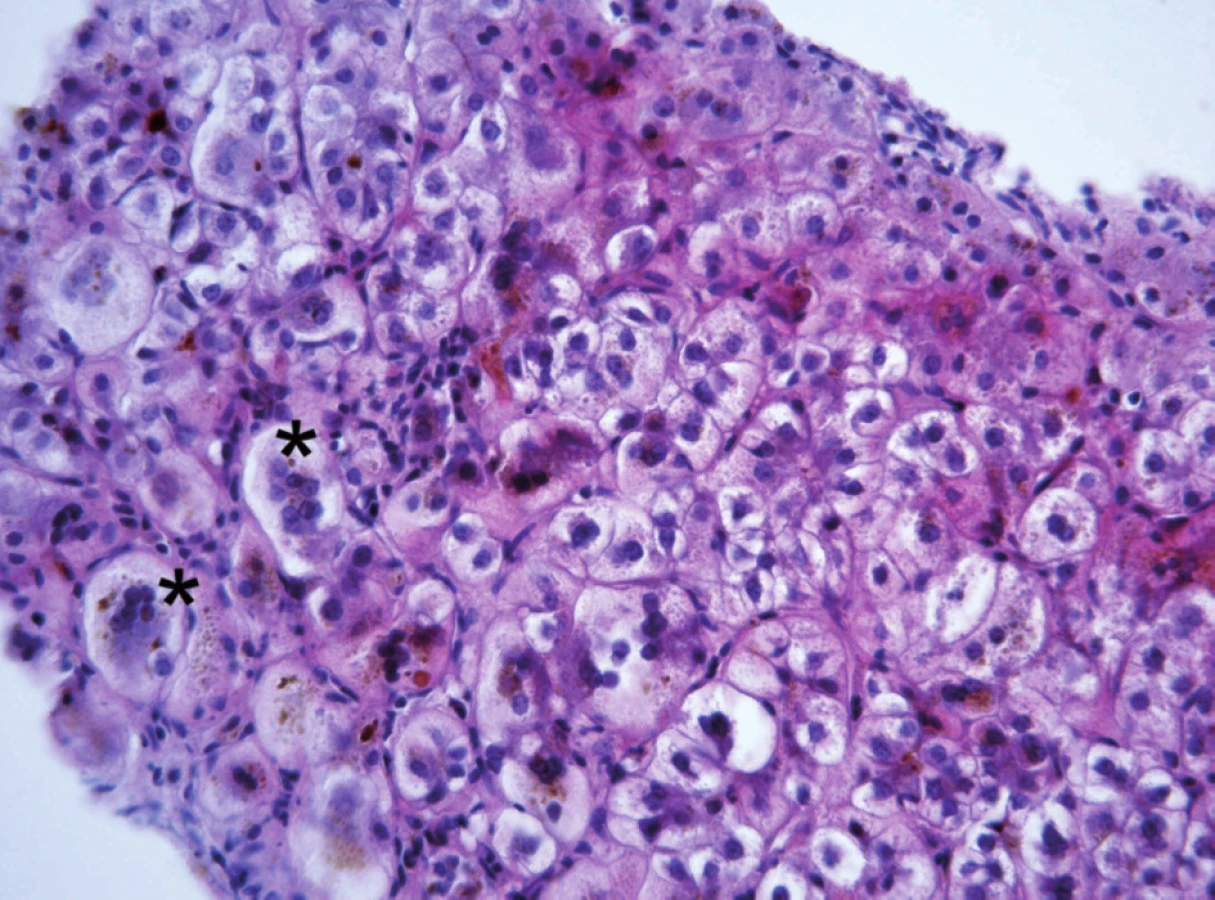
Progresivní forma BSEP onemocnění se klinicky i laboratorně podobá progresivní formě FIC1 onemocnění včetně normální hodnoty GMT. Na rozdíl od ní však chybí extrahepatální příznaky, je rychlejší progrese do cirhózy a jaterního selhání a je zvýšené riziko malignity [6, 7, 9]. V histologickém vyšetření je přítomen obraz obrovskobuněčné hepatitidy a s postupující fibrotizací porucha architektoniky jaterní tkáně (obr. 2). Diagnosticky významná je absence BSEP proteinu při imunohistochemickém vyšetření z parafinových bločků jaterní tkáně fixované formalinem (obr. 3). Vzácně u mírnějších forem onemocnění přítomnost BSEP proteinu nevylučuje jeho funkční poruchu. Elektronmikroskopický nález amorfní žluče s filamentózními strukturami postrádá rysy Bylerovy žluče a je tak méně specifický (obr. 4).


Konzervativní léčba je obvykle neúčinná. Efekt UDCA je pozorován jen asi v 10 % případů [6, 8]. U nemocných s “mírnějšími” mutacemi (D482G, E297G) vede ke zlepšení stavu parciální externí biliární diverze provedená v časných fázích vývoje. V případě již pokročilé jaterní léze, při selhání dosavadní konzervativní či chirurgické terapie nebo při nezvladatelném pruritu je během dětství (výjimečně již v kojeneckém věku nebo až u mladých dospělých) indikována transplantace jater. Vzhledem k velmi příznivým potransplantačním výsledkům doporučují některá centra při selhání konzervativní terapie jako metodu volby přímo transplantaci jater a na parciální biliární diverzi pohlížejí jako na “bridging” terapii [13, 14]. Zařazení na čekací listinu také významně ovlivňuje maligní potenciál choroby. Progresivní forma BSEP onemocnění je totiž spojena se zvýšeným rizikem rozvoje hepatocelulárního karcinomu [12]. Byly také popsány kazuistiky dětí s výskytem cholangiokarcinomu a hepatoblastomu [15]. Pro zvýšené riziko malignity jsou proto již od kojeneckého období doporučovány pravidelné kontroly jater pomocí UZ vyšetření a hladin α-fetoproteinu v séru.
V domácím písemnictví referují o klinicko-patologickém průběhu PFIC u 2 sourozenců Bartoš a spol. [16] Diagnostika progresivní formy BSEP onemocnění (PFIC2) založená na molekulární úrovni a imunohistochemickém vyšetření jaterní tkáně je poprvé publikována autory Kotalová a spol. [17].
Mutace v obou genech ABCB11 jsou také příčinou některých benigních rekurentních intrahepatálních cholestáz [6, 7]. Hovoříme o tzv. benigní formě BSEP onemocnění (BRIC2). Klinický průběh a laboratorní vyšetření BRIC1 a 2 nerozliší. Pouze u BRIC2 je popisován častější výskyt cholecystolitiázy. V současnosti se neví, je-li možný přechod benigní formy do progresivní podobně jako u FIC1 onemocnění, ale předpokládá se [12].
Progresivní forma MDR3 onemocnění
Progresivní forma MDR3 onemocnění (PFIC3, MDR = Multiple Drug Resistance) je podmíněna mutacemi genu ABCB4 lokalizovaném na chromozomu 7 (7q21) [1, 6, 7, 8, 9, 18]. Produkt genu, tzv. MDR3 glykoprotein, je exprimován v kanalikulární mebráně. Jde o tzv. fosfatidyl-flopázu či fosfatidylcholin-translokázu, která přemísťuje fosfatidylcholin z vnitřní části kanalikulární membrány na vnější. Zde pak dochází za pomoci žlučových kyselin k uvolňování fosfatidylcholinu do žluče. Tvorbou smíšených micel obsahujících fosfolipidy, žlučové kyseliny a cholesterol je zajištěna adekvátní solubilita žluče a ochrana cholangiocytů před detergentními účinky žlučových kyselin. Také u této formy PFIC byla identifikována celá řada mutací. Na rozdíl od předchozích forem je popisována korelace mezi jednotlivými genotypy a fenotypovými projevy onemocnění včetně odpovědi na léčbu.
Onemocnění se manifestuje pod obrazem cholestatického ikteru od kojeneckého věku až po 2.–3. decenium [6, 7, 8, 18]. V kojeneckém období se projevuje asi u poloviny nemocných. Zbylá část je diagnostikována v pozdějším věku většinou již ve stadiu kompenzované či dekompenzované “kryptogenní” cirhózy jater. Na pozdní manifestaci se pravděpodobně podílí zbytková aktivita MDR3 proteinu.
Cholestatický laboratorní profil se od předchozích forem PFIC odlišuje zvýšenou hodnotou GMT.
Histopatolog v časných stadiích onemocnění popisuje chronickou zánětlivou infiltraci v lehce rozšířených portálních polích se zřetelnou duktulární proliferací, v kojeneckém věku obvykle doprovázenou obrovskobuněčnou transformací [6, 18]. V pozdějším věku je přítomný obraz biliární cirhózy. Diagnosticky přínosný je imunohistochemický průkaz MDR3 proteinu z parafinových bločků jaterní tkáně fixované formalinem s absencí proteinu asi u jedné třetiny nemocných [6]. Častější pozitivní nález nevylučuje funkční deficit [12]. Diagnostické možnosti rozšiřuje stanovení fosfatidylcholinu ve žluči odebrané při endoskopickém vyšetření žlučových cest [1]. Elektronmikroskopický nález jaterní tkáně je nespecifický a k diagnostice nepříspívá.
Efekt léčby UDCA je pozorován asi u poloviny nemocných, kteří vykazují zbytkovou aktivitu MDR3 proteinu [6, 18]. V pokročilých stadiích onemocnění je indikována transplantace jater. Zvýšená incidence malignit nebyla pozorována.
Mutace obou genů ABCB4 jsou vedle progresivní formy MDR3 onemocnění zodpovědné za další fenotypové projevy vyskytující se až u dospělých. Byly publikovány kazuistiky s cholesterolovou intrahepatální cholelitiázou či cholecystolitiázou (“low phospholipid associated cholelithiasis“) projevující se chronickou cholestázou, biliárními kolikami, recidivujícími pankreatitidami nebo rekurencemi potíží po cholecystektomii s dobrým efektem léčby UDCA [6, 9]. Podíl mutací v ABCB4 na etiologii dětské cholesterolové cholelitiázy prokázán nebyl [19].
Zajímavá je skutečnost, že k rozvoji cholestatického stavu je za některých situací dostačující přítomnost pouze jedné z patologických mutací [6, 9]. Zásadním faktorem spolupodílejícím se na rozvoji cholestázy jsou estrogeny. Mutace pouze jednoho genu ABCB4 jsou zodpovědné za některé intrahepatální cholestázy v těhotenství či cholestázy indukované kontraceptivy obsahujících estrogeny. Bylo prokázáno, že tyto poznatky se vztahují i na nosiče mutací genu ATP8B1 a ABCB11 [1, 6, 17]. Jelikož jsou matky dětí s PFIC obligátními nosičkami těchto mutací, pátráme v rodinné anamnéze po příznacích cholestázy během těhotentsví či po podání kontraceptiv.
Fenotypové projevy mutací v genech ATP8B1, ABCB11 a ABCB4 s jednotlivými nozologickými jednotkami ukazuje tabulka 2.

Diagnostika a diferenciální diagnostika PFIC
U cholestatického ikteru v novorozeneckém a kojeneckém věku vylučujeme nejdříve častější příčiny, jako jsou biliární atrézie, deficit α1-antitrypsinu, syndrom arteriohepatální dysplazie (Alagilleův syndrom), fibrocystická onemocnění jater, infekční hepatitida, cysta choledochu, cystická fibróza či některá onemocnění ze skupiny vrozených vad metabolismu (galaktosémie, hereditární intolerance fruktózy, tyrosinémie). Diferenciální diagnostika cholestatického ikteru v tomto věkovém období přesahuje rámec článku. Přehled příčin cholestáz s charakteristikou jednotlivých onemocnění uvádí Nevoralova monografie [20].
Při podezření na PFIC pátráme v rodinné anamnéze po údaji o cholestáze u matek, po příbuzenských sňatcích či cholestáze u sourozenců. Někdy může napomoci věk v době manifestace, přítomnost výrazného pruritu či některých extrahepatálních symptomů, ale klinický obraz je nespecifický. V laboratorním vyšetření mají nemocní s PFIC1 a PFIC2 normální hodnotu GMT v séru. Aktivita aminotransferáz je relativně nejvyšší u pacientů s PFIC2. Je vždy zvýšená hladina primárních žlučových kyselin v séru. Je potřeba zdůraznit, že před jakýmkoli vyšetřením žlučových kyselin je nezbytné léčbu UDCA přerušit na dobu alespoň 10–14 dnů [1, 16]. UZ vyšetření jater a žlučových cest může prokázat cholecystolitiázu. Nezastupitelnou úlohu plní jaterní biopsie.
Kromě charakteristických známek popsaných histopatologem při histologickém vyšetření umožňuje imunohistochemický průkaz jednotlivých membránových proteinů pomocí cílených protilátek (FIC1 ze zamraženého vzorku, BSEP a MDR3 z parafinových bločků fixovaných formalinem). Absence proteinu nebo jen mírná pozitivita při imunohistochemickém vyšetření svědčí pro konkrétní formu PFIC. Normální výsledek nevylučuje funkční poruchu proteinu, obzvláště pokud máme podezření na PFIC3. Významné je elektronmikroskopické vyšetření jaterní tkáně, které diferencuje mezi PFIC1 a PFIC2. Jaterní tkáň k elektronmikroskopickému vyšetření je nezbytné bezprostředně po odběru umístit do odpovídajícícho fixačního činidla, dodatečné vyšetření z parafinových bločků je nespolehlivé. U cholestázy nejasné etiologie se zvýšenou hodnotou GMT a histologickým obrazem biliární obstrukce se provádí endoskopické vyšetření žlučových cest především k vyloučení sklerózující cholangoitidy. Zobrazovací vyšetření žlučových cest při podezření na PFIC3 doplňujeme o biochemickou analýzu žluče, protože u tohoto onemocnění je nízký podíl fosfolipidů (1–15 % z celkových lipidů, norma 19–24 %) a je několikanásobně zvýšený poměr žlučové kyseliny/fosfolipidy a cholesterol/fosfolipidy [1].
U všech typů PFIC zpřesňuje diagnostiku přímá mutační analýza umožňující správnou diagnostiku až v 90 % případů [12]. V současné době existuje velké množství mutací genu s rozdílným zastoupením v odlišných geografických oblastech. Mutace jsou identifikovány v homozygotní či heterozygotní kombinaci („složení heterozygoti“). Jelikož DNA analýza je metodologicky i finančně náročná, pořadí analýzy genů determinují výsledky dosavadních vyšetření (hodnota GMT, histologické, imunohistochemické a elektronmikroskopické vyšetření jaterní tkáně). Mutační analýzu je možné využít také v prenatální diagnostice [21].
Pod obrazem PFIC s normální hodnotou GMT se manifestuje většina poruch syntézy a konjugace žlučových kyselin. Mezi nejčastější patří deficit 3β-hydroxy-C27-steroid dehydrogenázy/isomerázy a deficit 3-oxosteroid 5β-reduktázy [5, 9]. Na rozdíl od PFIC zde chybí pruritus. Diagnostika je založena na stanovení žlučových kyselin, intermediálních produktů a jejich metabolitů v séru a/nebo moči. Zahajuje se screeningovým vyšetřením hladiny primárních žlučových kyselin v séru, která je nízká nebo normální. K přímému průkazu intermediálních metabolitů či atypických žlučových kyselin, které si zachovávají typickou strukturu substrátu před konkrétním enzymatickým blokem, se využívá metody LSIMS (“liquid secondary ionization mass spectrometry“). Každá porucha syntézy i konjugace žlučových kyselin je charakterizována unikátním hmotnostním spektrem metabolitů žlučových kyselin v moči. Potvrzení nálezu při LSIMS a stanovení definitivní diagnózy poskytuje plynová chromatografie s hmotnostní spektrometrií. Jelikož byly u některých poruch syntézy a konjugace žlučových kyselin identifikovány geny a jejich mutace, přispívají k diagnostice molekulárně genetické metody. Familiární hypercholémie je charakterizována poruchou konjugace primárních žlučových kyselin s následnou vysokou hladinou nekonjugovaných žlučových kyselin v séru (deficit BAAT = „bile acid coenzyme A:amino acid N-acyltransferáza“) [7]. Na cholestáze se podílí také defekt mezibuněčných spojení („tight junction“) mezi jaterními buňkami v oblasti kanalikulů zvyšujících paracelulární permeabilitu s průnikem žluče zpět do plazmy [7, 22].
Výskyt dalších vzácných vrozených cholestatických stavů s normální či zvýšenou hodnotou GMT je v našich podmínkách pro vazbu na určitá etnika či světové oblasti málo pravděpodobný, ale nelze je zcela vyloučit [1, 7, 22, 23]. K cholestáze s normální hodnotou GMT vede např. tzv. abnormální exprese villinu (“villin functional defect”) se ztrátou strukturální integrity mikroklků v kanalikulech. Syndrom arthrogrypózy, renální dysfunkce a cholestázy (ARC syndrom) je způsoben mutací genu vedoucí k porušenému transportu a lokalizaci proteinů apikální membrány buněk jater a ledvin. Do kategorie onemocnění s vysokou hodnotou GMT patří např. neonatální sklerózující cholangoitida, syndrom neonatální ichtyózy – alopecie – sklerózující cholangoitidy, cirhóza severoamerických indiánů či syndrom lymfedému a cholestázy (Aagenaesův syndrom).
Závěr
Detailní studium jednotlivých typů PFIC a dalších familiárních cholestatických syndromů významně přispělo k pochopení fyziologie a patofyziologie tvorby žluče. Jak již bylo opakovaně konstatováno, PFIC je vzácné heterogenní cholestatické onemocnění jater dětského věku se závažnou prognózou. Většina nemocných je indikována v průběhu onemocnění k transplantaci jater.
I když výsledky po transplantaci jater jsou v porovnání s jinými diagnózami velmi dobré, očekáváme další zlepšení prognózy pokračujícím výzkumem ve vztahu genotyp-fenotyp-odpověď na léčbu, v odhalování patogeneze malignizace na molekulární úrovni či zavedením moderní farmakoterapie zasahující cíleně do patofyziologie tvorby žluče.
MUDr. Petr Dědek, PhD.
Dětská klinika
Fakultní nemocnice a Lékařská fakulta
Sokolská 581
500 05 Hradec Králové
e-mail: dedek@fnhk.cz
Zdroje
1. Davit-Spraul A, Gonzales E, Baussan CH, et al. Progressive familial intrahepatic cholestasis. Orphanet. J. Rare Dis. 2009; 4: I.
2. Balisteri WF, Bezerra JA. Whatever happend to „neonatal hepatitis“? Clin. Liver Dis. 2006; 10 : 27–53.
3. Rafeey M, Golzar A, Javadzadeh A. Cholestatic syndromes of infancy. Pak. J. Biol. Sci. 2008; 11 : 1764–1767.
4. Bull LN, Carlton VE, Stricker NL, et al. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): evidence for heterogeneity. Hepatology 1997; 26 : 155–164.
5. Knisely AS. Progressive familial intrahepatic cholestasis: a personal perspective. Pediatr. Dev. Pathol. 2000; 3 : 113–125.
6. Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J. Pediatr. Gastroenterol. Nutr. 2008; 46 : 241–252.
7. Carlton VE, Pawlikowska L, Bull LN. Molecular basis of intrahepatic cholestasis. Ann. Med. 2004; 36 : 606–617.
8. Harris MJ, Le Couter DG, Arias IM. Progressive familial intrahepatic cholestasis: genetic disorders of biliary transporters. J. Gastroenterol. Hepatol. 2005; 20 : 807–817.
9. Tomer G, Schneider BL. Disorders of bile formation and biliary transport. Gastroenterol. Clin. N. Am. 2003; 32 : 839–855.
10. Van Mill SW, Klomp LW, Bull LN, et al. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin. Liver Dis. 2001; 21 : 535–544.
11. Stapelbroek JM, van Erpecum KJ, Klomp LW, et al. Nasobiliary drainage induces long-lasting remission in benign recurrent intrahepatic cholestasis. Hepatology 2006; 43 : 51–53.
12. Strautnieks SS, Byrne JA, Pawlikowska L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008; 134 : 1203–1214.
13. Englert C, Grabhorn E, Richter A, et al. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation 2007; 84 : 1361–1363.
14. Wanty C, Joomye R, Van Hoorebeek N, et al. Fifteen years single center experience in the management of progressive familial intrahepatic cholestasis of infancy. Acta Gastroenterol. Belg. 2004; 67 : 313–319.
15. Richter A, Grabhorn E, Schulz A, et al. Hepatoblastoma in a child with progressive familial intrahepatic cholestasis. Pediatr. Transplant. 2005; 9 : 805–808.
16. Bartoš V, Lauko L, Szépeová R, et al. Bylerov syndróm. Čes.-slov. Pediat. 2006; 61 : 32–35.
17. Kotalová R, Cebecauerová D, Knisely AS, et al. Progressive familial intrahepatic cholestasis – manifestations and diagnosis in infancy. Čes.-slov. Pediat. 2006; 61 : 200–206.
18. Jacquemin E, De Vree JM, Cresteil D, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001; 120 : 1448–1458.
19. Bronský J, Hřebíček M, Jirásek T, et al. Úloha variací genu ABCB4 v etiologii idiopatické cholelitiázy dětského věku. Čes.-slov. Pediat. 2009; 64 : 337–343.
20. Nevoral J. Onemocnění jater v dětském věku. Praha: Scientia Medica, 1994.
21. Jung C, Driancourt C, Baussan C, et al. Prenatal molecular diagnosis of inherited cholestatic diseases. J. Pediatr. Gastroenterol. Nutr. 2007; 44 : 453–458.
22. Knisely AS. Progressive familial intrahepatic cholestasis: an update. Pediatr. Dev. Pathol. 2004; 7 : 309–314.
23. Kelly DA. Diseases of the Liver and Biliary System in Children. 2nd ed. Oxford: Blackwell Publishing, 2004.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2010 Číslo 4
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- OZP pečuje o zdraví svých pojištěnců již 30 let. V čem je lepší a jaké výhody nabízí?
- Cytomegalovirové infekce u novorozenců a dětí
- Cytomegaloviróza a spalničky u dětí
Nejčtenější v tomto čísle
- Progresivní familiární intrahepatální cholestáza
- Syndrom třeseného dítěte
- Modelování vývoje tělesné délky a výšky dětí s pomocí údajů o výšce rodičů
- Nedožitých 50 rokov RNDr. Libora Kozáka, CSc.
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy