
Muskuloskeletální postižení u pacientů s lyzozomálním střádavým onemocněním
Musculoskeletal involvement in patients with lysosomal storage disease
Lysosomal storage diseases (LSD) represent a heterogeneous group of > 60 severe hereditary metabolic disorders due to impaired function of one or more lysosomal enzymes or disorders of protein processing and transport across the lysosomal membrane. The diagnosis of LSD is based on clinical suspicion and the results of enzymatic and molecular analyses.
Of the 836 LSD patients diagnosed in the last 30 years, mucopolysaccharidoses (n = 150), Gaucher disease (n = 57), and Fabry disease (74 hemizygous men and 116 heterozygous women) were the most common. Initial symptoms of LSD that were initially diagnostically non-specific included swelling and limited mobility of large and/or small joints, knee and hip pain, joint disfiguration and bone deformity, hepatomegaly and/or splenomegaly, recurrent upper respiratory tract infections, obstructive and/or restrictive pulmonary disorder, decreased renal function, anemia and, in some types of LSD, growth disorders, craniofacial dysmorphia and cognitive impairment. Patients went through a number of specialist outpatient clinics including rheumatology, orthopedics, ENT, neurology, nephrology or hematology, which in most cases led to late diagnosis. Although individual types of LSD are rare, many are treatable or at least therapeutically modifiable. Enzyme replacement therapy (ERT), substrate reducing therapy (SRT), and hematopoietic stem cell transplantation are useful in therapy, which can significantly improve disease prognosis or at least stabilize its progression.
Conclusion: The efficacy of treatment in patients with lysosomal storage diseases is significantly dependent on early diagnosis, in which rheumatology specialists should participate as well.
Keywords:
lysosomal storage diseases – mucopolysaccharidoses – Gaucher disease – Fabry disease – musculoskeletal involvement
Autoři:
J. Májovská 1; V. Malinová 1; G. Dostálová 2; Lenka Murgašová 1,3
![]() ; H. Poupětová 1; J. Zeman 1; M. Magner 1,4
; H. Poupětová 1; J. Zeman 1; M. Magner 1,4
Působiště autorů:
Klinika dětského a dorostového lékařství 1. LF UK a VFN, Praha
1; Centrum pro Fabryho chorobu II. interní kliniky kardiologie a angiologie 1. LF UK a VFN, Praha
2; Klinika ušní, nosní a krční 2. LF UK a FN Motol, Praha
3; Pediatrická klinika 1. LF UK a Thomayerovy nemocnice, Praha
4
Vyšlo v časopise:
Čes. Revmatol., 27, 2019, No. 4, p. 204-210.
Kategorie:
Přehledový článek
Souhrn
Lyzozomální střádavá onemocnění (lysosomal storage diseases – LSD) představují heterogenní skupinu > 60 závažných dědičných poruch metabolismu způsobených poruchou funkce jednoho nebo více lyzozomálních enzymů nebo poruchami procesování a transportu proteinů přes lyzozomální membránu. Diagnostika LSD je založena na klinickém podezření a výsledcích enzymatických a molekulárních analýz.
Z 836 pacientů s LSD, které jsme diagnostikovali v posledních 30 letech, byly nejčastější mukopolysacharidózy (n = 150), Gaucherova nemoc (n = 57) a Fabryho nemoc (74 hemizygotních mužů a 116 heterozygotních žen). Mezi počáteční příznaky LSD, které byly zpočátku diagnosticky nespecifické, patřily otoky a omezená hybnost velkých a/nebo malých kloubů, bolesti kolen a kyčlí, kloubní defigurace a kostní deformity, hepatomegalie a/nebo splenomegalie, opakované infekty horních cest dýchacích, obstrukční a/nebo restriktivní plicní porucha, pokles renálních funkcí, anemie a u některých typů LSD i poruchy růstu, kraniofaciální dysmorfie a porucha kognitivních funkcí. Pacienti procházeli řadou odborných ambulancí včetně revmatologie, ortopedie, ORL, neurologie, nefrologie či hematologie, což vedlo ve většině případů k pozdní diagnostice. Ačkoliv jednotlivé typy LSD se vyskytují poměrně vzácně, řada z nich je léčitelná nebo alespoň léčbou ovlivnitelná. V terapii se uplatňuje enzymová substituční terapie (enzyme replacement therapy – ERT), substrát redukující terapie (SRT) a transplantace hematopoetickými kmenovými buňkami, které mohou významně zlepšit prognózu onemocnění nebo alespoň stabilizovat jeho progresi.
Závěr: Účinnost léčby u pacientů s lyzozomálním střádavým onemocněním významně závisí na včasné diagnostice, na které by se měli podílet i odborníci v oboru revmatologie.
Klíčová slova:
lyzozomálně střádavá onemocnění – mukopolysacharidózy – Gaucherova nemoc – Fabryho nemoc – muskuloskeletální postižení
ÚVOD
Od roku 1984, kdy profesor Wagenhäuser formuloval revmatismus jako „širokou variaci klinických obrazů, jejichž společným rysem a hlavním příznakem je bolest v lokomočním systému, často spojená s poškozením a zhoršením pohybové schopnosti“ (1), se revmatologie stala širokým oborem, který se zabývá zdravotními obtížemi všech věkových skupin s velkým přesahem do řady dalších oborů. Jedním z příkladů je diagnostika lyzozomálních střádavých onemocnění (LSD), která představuje heterogenní skupinu > 60 závažných dědičných poruch metabolismu způsobených poruchou funkce jednoho nebo více lyzozomálních enzymů nebo poruchami transportu přes lyzozomální membránu (2). Z 836 pacientů s LSD, které jsme diagnostikovali v posledních 30 letech, byly nejčastěji diagnostikovány mukopolysacharidózy (n = 150), Gaucherova nemoc (n = 57) a Fabryho nemoc (74 hemizygotních mužů a 116 heterozygotních žen).
Tíže klinických projevů a progrese onemocnění u pacientů s LSD bývá variabilní, závisí na věku, typu střádavé molekuly a typu postižených tkání, na genové expresi a zbytkové aktivitě postiženého enzymu, indukci prozánětlivého stavu, oxidativním stresu, porušení endocytózy i exocytózy, sekundárním poškození mitochondrií a indukcí buněčné smrti (3). Lyzozomální střádání aktivuje či inhibuje přenos signálu, mění lokalizaci receptorů a modifikuje jejich funkce. Porušená je homeostáza vápníkových iontů, syntetická funkce buňky, maturace proteinů a buněčného transportu.
Počáteční příznaky LSD bývají zpočátku nespecifické, což významně oddaluje diagnostiku a možnost včasné terapie. Řada pacientů se při pomalu progredujícím postižení velkých a/nebo malých kloubů dostává i do revmatologických ambulancí.
MUKOPOLYSACHARIDÓZY
Mukopolysacharidózy (MPS) jsou způsobeny poruchou degradace glykosaminoglykanů (GAG, dříve označované jako mukopolysacharidy), které se podílejí na stavbě pojivové tkáně a které představují důležitou složku extracelulární matrix (4). MPS byla v naší sestavě 836 pacientů s LSD nalezena u 150 pacientů. Jde o onemocnění vzácné, výskyt MPS v České republice je cca 1 : 26 000 (5). Podle postiženého enzymu se rozlišuje několik klinických typů MPS s autozomálně recesivní dědičností, pouze u MPS II je dědičnost vázaná na chromozom X (tab. 1).
První příznaky onemocnění se sice objevují již v dětském věku, ale nejsou specifické: např. pupeční nebo tříselné kýly, hepatomegalie, splenomegalie, makrocefalie, recidivující záněty horních cest dýchacích a středouší nebo porucha sluchu. K podezření na diagnózu MPS mohou vést muskuloskeletální příznaky, se kterými se pacient dostává do revmatologické ambulance. Jedná se o kloubní kontraktury či naopak kloubní hypermobilitu, dysproporční poruchu růstu, torakolumbální kyfózu či gibbus (obr. 1a–d).
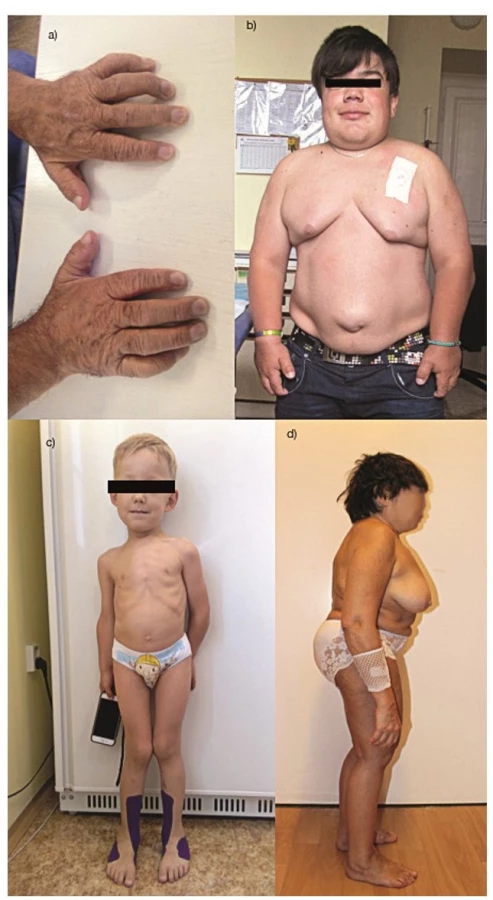
V kojeneckém věku je růst normální či dokonce urychlený, ale v průběhu batolecího věku dochází k jeho stagnaci (2), která je způsobena poruchou enchondrální osifikace a sekundárními strukturálními deformitami. Vliv na hybnost mohou mít kontraktury velkých i malých kloubů. Ztuhlost může být provázená bolestí, typicky ranní. Prsty jsou široké, falangy krátké. Spolu s kontrakturami tak vzniká obraz drápovité ruky, která významně limituje jemnou motoriku (obr. 1a) (6). U MPS IV je typická povšechná kloubní hypermobilita při zvýšené ligamentózní laxitě, s výjimkou loktů, kyčlí a kolen. Časně v dětství se objevují genua valga, kyfóza, růstová porucha s krátkým trupem a krkem a poruchy chůze s tendencí k pádům (obr. 1c)(7). Skeletální postižení zahrnuje platyspondylii, hypoplazii odontoidního výběžku, hyperlordózu, skoliózu či ulnární deviaci zápěstí. Všeobecně typický pro pacienty s MPS je rozvoj syndromu karpálního tunelu, který se jinak v dětském věku prakticky nevyskytuje.
Postupně se objevují další a více charakteristické projevy, jako jsou hrubší rysy obličeje s klenutým čelem, prominující nadočnicové oblouky, široký kořen nosu, hypertelorismus, antevertrované nostrily a plné rty (obr. 1b). Dále je přítomna makroglosie a kratší nosní průchody, které spolu s hypertrofií adenoidních vegetací a tonzilární tkáně souvisejí s rozvojem chronické rýmy, hlasitým dýcháním, ronchopatií a opakovanými záněty středouší (3). Mohou být přítomné tvarové změny zubů, široké uložení zubů, gingivální hyperplazie, hluboký a chraplavý hlas a u některých typů (MPS I, IV, VI) i zákal rohovky. Při střádání GAG na srdečních chlopních dochází k rozvoji chlopenních vad a sekundárně i ke kardiomyopatii. Plíce mohou být postiženy obstrukční nebo restrikční poruchou. Na rozvoji restrikční poruchy se podílí menší obvod hrudního koše s abnormálně tvarovanými žebry a omezená pohyblivost bránice podmíněná zvětšením sleziny a jater (8). Ke vzniku obstrukční poruchy dochází střádáním GAG v sliznici dolních cest dýchacích a přítomnosti abnormalit tracheobronchiálního stromu (8, 9). Kůže může být zhrubělá, s hypertrichózou, hustým obočím a nižší vlasovou hranicí. U řady pacientů se stanovení správné diagnózy posunuje do dospívání až dospělosti (6).
Pro těžké formy MPS I, II a III je typické opoždění a regres vývoje, ale ten se nevyskytuje u MPS IV a VI (3). Psychiatrická symptomatologie s poruchami chování (agresivita, hyperaktivita, afektivní záchvaty, střídání emocí) je typická pro MPS II a III (10). U těžkých forem MPS I, II a VI typu může dojít k rozvoji hydrocefalu a epilepsii.
Mezi nejzávažnější komplikace skeletálního postižení patří atlanto-axiální instabilita způsobená hypoplazií odontoidního výběžku, která představuje riziko vzniku život ohrožující cervikální myelopatie s následnou kvadruparézou. K včasné detekci je zásadní zobrazení magnetickou rezonancí, terapeutickým řešením je pak dekompresní chirurgický zákrok. Veliké opatrnosti je třeba při intubaci pacienta pro kontraindikaci záklonu hlavy. Jako prevence akutního poškození míchy při intubaci pacienta je přísně kontraindikován výrazný záklon hlavy a doporučováno zajištění dýchacích cest za pomoci flexibilního bronchoskopu.
Soubor radiologických nálezů charakteristický pro MPS (dysostosis multiplex) zahrnuje rozšíření diafýz dlouhých kostí, rozšíření medulární dutiny se ztenčením kortikalis, rybí obratle, hypoplazii odontoidního výběžku, veslovitá žebra, dysplastická acetabula, deformaci hlavice femurů, genua valga, krátké robustní falangy (obraz bullet shape) či sella turcica ve tvaru J (7). Diagnostika MPS je při klinickém podezření založena na screeningovém vyšetření glykosaminoglykanů v moči, ale vyžaduje i enzymatické vyšetření v izolovaných leukocytech či kultivovaných fibroblastech a molekulárně-genetické vyšetření. Prenatální diagnostika je dostupná na enzymové i molekulární úrovni.
GAUCHEROVA NEMOC
Gaucherova nemoc (GD) je způsobena poruchou kyselé β-glukocerebrosidázy (GBA), která vede v buňkách monocyto-makrofágového systému ke střádání nerozštěpeného glukocerebrosidu (4). Dědičnost je autozomálně recesivní. V České republice je výskyt Gaucherovy nemoci odhadován na 1 : 88 000 (5). V našem souboru 836 pacientů s LSD byla Gaucherova nemoc diagnostikována u 57 pacientů.
První obtíže se mohou objevit kdykoliv od novorozeneckého věku až do dospělosti. Podle závažnosti onemocnění a typu postižených tkání se Gaucherova nemoc dělí na tři základní typy. Nejčastější je non-neuropatický typ I, zatímco typy II a III s akutním nebo chronickým neurologickým postižením se v České republice vyskytuje poměrně vzácně.
Mezi první příznaky Gaucherovy nemoci (typ I) patří ataky bolestí kostí či kloubů, únava, pocit plnosti po jídle a známky zvýšené krvácivosti (2). Bolesti kostí jsou způsobeny tlakem střádaného materiálu v kostní dřeni. Kruté bolesti vznikají v důsledku lokalizované poruchy krevního zásobení s následným vznikem kostního infarktu, nejčastěji v dlouhých kostech na dolních končetinách. Bolesti mohou trvat i několik dní a jsou doprovázené teplotou a leukocytózou, takže mohou být mylně interpretovány jako počínající osteomyelitida nebo chronická multifokální osteomyelitida. Méně časté jsou aseptické nekrózy v oblasti velkých kloubů, které mohou vest k projevům předčasné artrózy. Kortikální kost dlouhých kostí dolních končetin je často tenčí v důsledku převažující aktivity osteoklastů nad osteoblasty, což se může na rtg snímku projevit rozšířením dolní diafýzy až do obrazu „Erlenmayerovy baňky“ (obr. 2). Časté jsou fraktury dlouhých kostí či kompresivní fraktury obratlů (11). Při střádání glukocerebrosidu je utlačována i kostní dřeň. Vázne tvorba krevních destiček, později i erytrocytů a leukocytů, což spolu se splenomegalií vede k projevům hypersplenismu a pancytopenie. U postižených pacientů dochází ke snadné tvorbě hematomů a protrahovanému krvácení po poranění či menších chirurgických výkonech. U pacientů s Gaucherovou nemocí typu I nedochází k poruchám kognitivních funkcí, ale zvětšení jater a sleziny se projeví zvětšením obvodu břicha a útlakem žaludku, který vede k pocitu plnosti a poklesu příjmu výživy s následným neprospíváním. U dětí dochází i k poruše růstu nebo opožděnému nástupu puberty.

Laboratorní vyšetření ukazují různě závažnou trombocytopenii, anemií či leukopenii.
Při hemokoagulačním vyšetření nacházíme mírně prodloužené APTT a zvýšené D-dimery a hladiny ferritinu a imunoglobulinů mohou být zvýšené. Střádání a aktivace v makrofágovém systému zvyšuje aktivitu tartarát-rezistentní kyselé fosfatázy (ACP), „angiotensin converting“ enzymu (ACE), chitotriosidázy a hladiny Lyso-GL-1. Diagnostika je založena na enzymovém stanovení aktivity glukocerebrosidázy v izolovaných leukocytech či v suché krevní kapce a na molekulárně genetickém vyšetření GBA genu (12). Prenatální diagnostika je dostupná na enzymové i molekulární úrovni. Vyšetření kostní dřeně s průkazem Gaucherových buněk s nahromaděním glukocerebrosidu je indikováno jen u pacientů, u kterých diferenciálně diagnostická rozvaha zahrnuje i podezření na hematoonkologické onemocnění.
FABRYHO NEMOC
Fabryho choroba (Fabry disease, Fabry-Anderso-nova choroba – FD) je pomalu progredujcí lyzozomální střádavé onemocnění ze skupiny sfingolipidóz, které je způsobeno poruchou α-galaktosidázy. Dědičnost FD je na rozdíl od většiny ostatních LSD gonozomálně recesivní, ale onemocnění se neprojevuje pouze u hemizygotních mužů, ale v poněkud mírnější formě i u heterozygotních žen. Klinická variabilita u žen je však velice široká od stejně závažných manifestací jako u mužů až po bezpříznakové nosičství, což je do velké míry dáno poměrem inaktivace mutovaného a nemutovaného chromozomu X v procesu lyonizace v časných stadiích embryonálního vývoje (13). Výskyt FD v České republice je cca 1 : 77 000 (5). V našem souboru 836 pacientů s LSD byla Fabryho nemoc diagnostikována u 74 hemizygotních mužů a 116 heterozygotních žen.
První projevy FD se mohou manifestovat kdykoliv od předškolního věku až po pozdní dospělost (14). Typickým prvním příznakem onemocnění, zejména u chlapců ve věku mezi 5. a 8. rokem, jsou kožní projevy s výsevem drobných červených angiokeratomů (15). Angiokeratomy jsou lokalizovány zejména periumbilikálně, na bocích, v oblasti genitálu, na hýždích a flexorové straně stehen. Jejich výskyt s věkem často narůstá. Střádání v oblasti kůže a potních žláz vede k poruše pocení (hypohidrosis až anhidrosis) s následným přehříváním organismu. FD tak patří do diferenciálně diagnostické rozvahy u chlapců s atakami periodických horeček nejasné etiologie. Chlapci nebo jejich rodiče často uvádějí, že zvýšená fyzická zátěž (tělocvik ve škole, sport, infekce) vede ke zvýšení teploty (až 38–39 °C). Mezi časné příznaky FD patří i parestezie rukou a nohou, které chlapci popisují jako „nepříjemné pálení“.
Do obrazu očního postižení u pacientů s FD patří zadní subkapsulární katarakta nebo přítomnost vinutých retinálních cév, dále velmi typické paprsčitě se větvící opacity v povrchových vrstvách rohovky viditelné při vyšetření na štěrbinové lampě. Tento nález, tzv. „cornea verticillata“, připomíná nežádoucí účinek amiodaronu a patří do diferenciální diagnostiky intoxikací. U menší části pacientů s FD se rozvine lymfedém (13).
Prognóza pacienta s FD je však především závislá na orgánovém postižení ledvin a srdce. Postižení ledvin s mikroalbuminurií začíná v období puberty, k poklesu glomerulární filtrace s proteinurií dochází v průběhu třetí dekády a terminální selhání ledvin se projevuje u mužů ve věku kolem 40 let a u žen kolem 50. roku věku. Většina pacientů s FD má i kardiovaskulární postižení. Typická je zejména hypertrofie levé komory, ve vyšším věku se objevují poruchy srdečního rytmu vyžadující implantaci kardiostimulátoru. Pacienti s FD jsou oproti ostatní populaci vystaveni až dvacetinásobnému riziku pro tranzitorní ischemické ataky a cévní mozkové příhody (16). Klinické projevy u heterozygotních žen s FD jsou obvykle mírnější a nastupují ve vyšším věku než u mužských pacientů.
Diagnostika FD závisí na klinickém podezření, screeningovém vyšetření α-galaktosidázy v suché kapce krve, enzymatickém vyšetření v izolovaných leukocytech periferní krve a molekulárně genetickém vyšetření genu GLA. Prenatální diagnostika je dostupná na molekulární úrovni.
TERAPIE
V současné době je jen menší část LSD léčitelná nebo alespoň léčbou ovlivnitelná. Mezi standardní metody léčby patří enzymová substituční terapie (ERT – enzyme replacement therapy), která je dostupná pro MPS I, II, IVa, VI a VII, Gaucherovu a Fabryho chorobu a některé další poruchy ze skupiny LSD (tab. 1) (2). ERT není u všech pacientů stejně účinná a její dopad u většiny nemocných nekoreluje s konkrétními mutacemi v postižených genech.

Princip ERT je založen na zjištění, že intravenózně aplikovaný enzym se pomocí endocytózy a manóza-6-fosfátových receptorů dostává v průběhu lyzozomální biogeneze do časného endozomu (early endosome), ve kterém putuje přes pozdní endosome (late endosome) až do lyzozomů (17). Nejdelší a nejlepší zkušenosti s ERT jsou u pacientů s Gaucherovou nemocí s non-neuropatickou formou, u kterých léčba vede k ústupu hepatosplenomegalie, normalizaci hematologických parametrů a úpravě skeletálních potíží. U pacientů s Fabryho nemocí na ERT dochází k ústupu subjektivních obtíží a příznivému ovlivnění renálního postižení a kardiomyopatie (2). ERT u pacientů s MPS stabilizuje nebo alespoň zpomaluje progresi onemocnění. U většiny pacientů se zmenšuje objem jater a sleziny, u části nemocných se zlepšuje fyzická výdrž objektivizovaná prodloužením délky tratě během šestiminutového testu chůze, stabilizuje se funkční vitální kapacita plic a ustupuje únava a bolesti kloubů (8). ERT je účinnější při včasné diagnostice, problematická je indikace ERT při větším postižení srdečních chlopní či chrupavky, protože velké molekuly enzymu špatně pronikají do těchto tkání. ERT nepřechází přes hematoencefalickou bariéru, a proto není indikována u pacientů s významným postižením kognitivních funkcí. Proto se výzkumně zkouší i intratekální aplikace. Nevýhodou ERT je kromě nutnosti pravidelné intravenózní aplikace léčiva také její extrémně vysoká cena.
Principem substrát redukující terapie (SRT) je využití malých molekul, které přecházejí hemato-encefalickou bariérou a které mají schopnost snižovat syntézu střádaných molekul nebo jejich prekurzorů. SRT se používá u pacientů s Gaucherovou nemocí a u pacientů s MPS III. Výhodou SRT je, že se mohou používat perorálně, limitací jsou však časté nežádoucí účinky. U některých forem LSD je ve fázi klinických studií genová terapie.
Významnou složkou léčby je symptomatická terapie. Při multisystémovém charakteru většiny LSD je zcela zásadní multioborová péče. Patří k ní nejen ortopedické a neurochirurgické a kardiochirurgické zákroky, ale také rehabilitace, psychoterapie a antiepileptika u pacientů se sekundární epilepsií. U pacientů s Fabryho nemocí se uplatňují nefroprotektivní léky (kontrola hypertenze a zmírnění proteinurie), antiagregační terapie, antiarytmika. Velmi důležitou součástí jsou dietní opatření a léčba bolesti. Pacienti s renálním selháním často dospějí k hemodialýze či transplantaci ledvin.
ZÁVĚR
Lyzozomální střádavá onemocnění představují skupinu závažných chronicky probíhajících onemocnění. Závažnou prognózu obvykle se systémovým postižením lze u řady pacientů příznivě ovlivnit, ale vyžaduje to včasnou diagnostiku, na které se musí podílet odborníci z řady oborů, včetně revmatologie.
Práce byla podpořena projektem Univerzity Karlovy PROGRES Q32/LF2 a grantem Ministerstva zdravotnictví ČR RVO-VFN 64165/2012.
Konflikt zájmů: žádný.
doc. MUDr. Martin Magner, Ph.D.
Klinika dětského a dorostového lékařství 1. LF a UK, Praha
Ke Karlovu 2, 120 00 Praha 2
e-mail: martin.magner@vfn.cz
Zdroje
1. Mozolova D, Dallos T. Detská reumatologia v obrázkoch. Bratislava: Solen 2013.
2. Malinová V, Honzík T. Lysosomální onemocnění – současné možnosti diagnostiky a terapie. Pediatr praxi 2013; 14 : 99–103.
3. Magner M, Kulhánek J, Zeman J. Enzymová substituční terapie pacientů s mukopolysacharidózami. Remedia 2016; 4 : 356–361.
4. Honzík T, Zeman J, a kol. Dědičné poruchy metabolismu v kazuistikách. Praha: Mladá fronta 2016.
5. Poupětová H, Ledvinová J, Berná L, Dvořáková L, Kožich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33 : 387–396.
6. Dostalova G, Hlubocka Z, Lindner J, Hulkova H, Poupetova H, Vlaskova H, et al. Late diagnosis of mucopolysaccharidosis type IVB and successful aortic valve replacement in a 60-years-old female patient. Cardiovasc Pathol 2018; 35 : 52–56.
7. White KK. Orthopaedic aspects of mucopolysaccharidoses. Rheumatology 2011; 50 : 26–33.
8. Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: Managment and Treatment Guidelines. Pediatrics 2009; 123 : 19–29.
9. Berger KI, Fagondes SC, Giugliani R, Hardy KA, Lee KS, McArdle C, et al. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis 2013; 36 : 201–210.
10. Simmons MA, Bruce IA, Penney S, Wraith E, Rothera MP. Otorhinolaryngological manifestations of the mucopolysaccharidos. Int J Pediatr Otorhinolaryngol 2005; 5 : 589–595.
11. Hughes D, Mikosch P, Belmatoug N, Carubbi F, Cox T, Goker-Alpan O, et al. Gaucher disease in bone: from pathophysiology to practise. J Bone Miner Res 2019; 34 : 996–1013.
12. Biegstraaten M, Cox TM, Belmatoug N, Berger MG, Collin-Histed T, Vom Dahl S, et al. Management goals for type 1 Gaucher disease: An expert consensus document from the European working group on Gaucher disease. Blood Cells Mol Dis 2018; 68 : 203–208.
13. Anderson W. A case of „angiokeratoma“. Br J Dermatol 1898;10 : 113–117.
14. Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I, et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr 2003; 162 : 767–772.
15. Larralde M, Boggio P, Amartino H, Chamoles N. Fabry disease: a study of 6 hemizygous men and 5 heterozygous women with emphasis on dermatologic manifestations. Arch Dermatol 2004; 140 : 1440–1446.
16. Schiffmann R. Fabry disease. Pharm. Ther 2009; 122 : 65–77.
17. Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet 2002; 3(12): 954–966.
Štítky
Dermatologie Dětská revmatologie RevmatologieČlánek vyšel v časopise
Česká revmatologie

2019 Číslo 4
- Ingenol-mebutát rozšiřuje možnosti léčby aktinické keratózy
- Riziko roztroušené sklerózy u pacientů s psoriázou
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
Nejčtenější v tomto čísle
- Paraneoplastické syndromy
- Doporučení České revmatologické společnosti pro léčbu dny
- Závažnost zánětlivých granulomů u revmatických chorob
- Význam dosažení remise u pacientů s revmatoidní artritidou
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy