
Doporučení České revmatologické společnosti pro diagnostiku a sledování nemocných se systémovým lupus erythematodes
Autoři:
P. Horák; D. Tegzová; J. Závada; M. Olejárová; M. Skácelová; A. Smržová; M. Žurek
Vyšlo v časopise:
Čes. Revmatol., 21, 2013, No. 2, p. 59-70.
Kategorie:
Doporučené postupy
Definice choroby
Systémový lupus erythematodes (SLE) je autoimunitní zánětlivé onemocnění postihující zejména ženy v reprodukčním věku. Je charakterizováno hyperaktivitou B buněk a nadprodukcí orgánově nespecifických autoprotilátek, z nichž mnohé se podílejí na tvorbě imunokomplexů. Jejich tkáňová či cévní depozita pak vedou k zánětlivému orgánovému postižení. Klinický obraz SLE je velmi pestrý. Jedná se o vysoce heterogenní chorobu, kterou lze dělit do řady klinicky a laboratorně definovaných podtypů, a které se může s řadou dalších jednotek překrývat (smíšené onemocnění choroba pojiva, Sjögrenův syndrom, antifosfolipidový syndrom). Průběh choroby je charakterizován střídáním remisí a exacerbací. Laboratorně je pro SLE typická tvorba orgánově nespecifických protilátek, které jsou namířeny proti nukleárním, cytoplazmatickým i povrchovým antigenům buněk vlastního těla (1). Akutního vzplanutí je zpravidla doprovázeno systémovými příznaky, jako je horečka, únava, hmotnostní úbytek. K nejčastějším projevům patří postižení kůže, kloubů, kardiovaskulárního systému, plic, glomerulů ledvin, centrálního nervového systému či krvetvorby. SLE může vyústit do selhání postiženého orgánu a závažná forma choroby je spojena s významnou mortalitou (2).
Epidemiologie
Prevalence SLE v populaci se pohybuje mezi 20–150 případy na 100 000 obyvatel (3–5). SLE je asi 10krát častější u žen než u mužů a většina jedinců je postižena onemocněním mezi 15 a 40 lety. Asi u 10–15 % se nemoc vyvine po 50 letech věku, poměr žen a mužů v této věkové skupině pak klesá na 4 : 1 (6). U mužů, zejména evropské rasy, je SLE relativně vzácné onemocnění. K chorobě jsou citlivější osoby s Klinefelterovým syndromem s karyotypem XXY. Otázka srovnání závažnosti SLE u mužů a žen nebyla dosud úplně zodpovězena, prakticky se ale jedná o prognosticky srovnatelné entity. Přibližně 20 % všech případů SLE se manifestuje před 18. rokem věku. U dětí je rovněž přítomný jiný poměr zastoupení ženského a mužského pohlaví (udává se 3 : 1) (7). Pokud bereme do úvahy údaje o prevalenci choroby v Evropě a USA (8), lze odhadovat počet nemocných se SLE v rámci České republiky na 6–10 tisíc.
Diagnostika SLE
Diagnostika choroby se opírá o správnou interpretaci řady anamnestických, klinických, laboratorních a paraklinických nálezů. Pro diagnózu SLE neexistuje žádný zlatý standard, nález, příznak či vyšetření, které by s velmi vysokou pravděpodobností diagnózu choroby potvrdilo.
Klinický obraz
Klinický obraz choroby je neobyčejně pestrý (14). SLE nejčastěji začíná následujícími příznaky:
- Systémové projevy jako jsou horečka, únava, hmotnostní úbytek, uzlinový syndrom
- Fotosenzitivní vyrážka
- Alopecie
- Artritida či artralgie
- Raynaudův fenomén
- Serozitida (pleuritida, perikarditida, aseptická peritonitida)
- Nefritický či nefrotický syndrom
- Neurologické projevy (křeče, mozková příhoda, kognitivní dysfunkce, bolest hlavy, psychóza)
- Flebotrombóza, tromboembolická choroba, arteriální trombóza
- Anémie, trombocytopenie, leukopenie
- Opakované potraty
Imunologická vyšetření
Autoprotilátky jsou protilátky namířené proti nukleárním a cytoplazmatickým antigenům a představují jeden z charakteristických nálezů u SLE a jeden z pilířů jeho diagnostiky. Jejich detekce má diagnostický význam, i když jejich význam se z hlediska specificity pro SLE liší (1).
Antinukleární protilátky (ANA). Více než 95 % nemocných se SLE má pozitivní test na antinukleární protilátky v séru v signifikantním titru (1 : 160 či více), detekované metodou nepřímé imunofluorescence na jádrech buněčných linií, jako jsou například Hep2 buňky (human epithelial cells). Různý typ imunofluorescence (homogenní, periferní, skvrnitý, nukleolární, centromerový a cytoplazmatický) odpovídá typu přítomných protilátek. Typ fluorescence závisí rovněž na množství přítomné protilátky. Nemocní s negativními ANA protilátkami mohou mít někdy pozitivní anti-Ro protilátky a méně trpí glomerulonefritidou (9).
Anti-dsDNA protilátky se dle literárních údajů vyskytují u 40–90 % nemocných se SLE (10, 11). Mnohé studie poukázaly na souvislost mezi anti--dsDNA protilátkami a přítomností ledvinného postižení (12). K určení anti-dsDNA protilátek lze využít metodu imunofluorescence. Zdrojem dvojvlákna DNA je v tomto případě prvok, Crithidia luciliae (12). V některých případech se lze také setkat s použitím radioaktivní imunoprecipitace, tzv. Farrův test. K longitudinálnímu sledování vývoje hladin anti-dsDNA protilátek je pak vhodná metoda ELISA (12).
Protilátky proti extrahovatelným nukleárním antigenům (ENA), zahrnující antigeny Ro, La, Sm, U1RNP, U2RNP, Ku se vyskytují asi u 30 až 50 % nemocných. Anti-La a anti-Ro protilátky jsou často spojeny se sekundárním Sjögrenovým syndromem, v případě anti-Ro protilátek také s fotosenzitivitou, subakutní kožní formou SLE a s lupusem neonatorum. Protilátky proti RNP se pojí se smíšeným onemocněním pojiva (MCTD). Anti-Sm protilátky jsou prakticky diagnostické pro SLE, ale vyskytují se pouze v relativně malém procentu nemocných (5 % u bílé rasy, 30 % u černochů). Ke stanovení těchto protilátek lze použít imunodifuzi a protisměrnou imunoelektroforézu, v současnosti se používají převážně enzymové eseje (13).
Protilátky proti histonům se vyskytují asi u 40 až 60 % SLE, takřka ve 100 % jsou přítomny u lupusu indukovaného léky (15).
Lupus antikoagulans a antikardiolipinové protilátky mají význam v diagnostice antifosfolipidového syndromu jako prediktory rizika trombofilie a jsou součástí klasifikačních kritérií choroby (16).
Další autoprotilátky, které se vyskytují v průběhu SLE, mají z hlediska diagnostiky menší význam.
Komplement. Hypokomplementemie sama o sobě není specifická pro SLE, odráží všeobecně přítomnost imunitně mediované choroby doprovázené spotřebou komplementu, dědičnou deficienci komplementu či jeho poškozenou syntézu. V kontextu SLE jsou nízké hladiny komplementu vyjádřením aktivity choroby a možného postižení ledvin. Podobně jako anti-dsDNA protilátky i hladiny C3 a C4 složky komplementu či aktivita CH50 mohou sloužit u řady nemocných jako ukazatelé aktivity choroby a nově jsou i součástí klasifikačních kritérií (17, 20).
Biochemická vyšetření a reaktanty akutní fáze
Sedimentace erytrocytů je často zvýšená u nemocných s aktivní chorobou, hladiny CRP bývají zvýšeny jen mírně. Lze využít stanovení CRP k odlišení infekční komplikace od vzplanutí aktivity vlastní choroby. Vyšší hladiny CRP se však vyskytují v případě pleuritidy či perikarditidy.
Rutinní biochemický screening (testy jaterního souboru, urea, kreatinin, proteinurie, výpočet glomerulární filtrace dle MDRD apod.) odráží přítomnost a stupeň orgánového postižení. V poslední době narůstá všeobecně důraz na sledování parametrů aterogeneze, která zejména v pozdních fázích SLE představuje klinicky významný problém.
Hematologie
Anémie se vyskytuje u 50 % nemocných zejména během období vyšší aktivity procesu. Ve většině případů se jedná o anémii chronických chorob, normocytární anémii s nízkou hladinou sérového železa, nízkou vazebnou kapacitou a zvýšenými nebo normálními hladinami feritinu a nízkými hodnotami solubilních transferinových receptorů v séru. U více než třetiny nemocných se SLE se vyskytuje pozitivita přímého Coombsova testu, skutečná hemolytická anémie se projeví asi u 10 % nemocných. Pozitivita Coombsova testu je zahrnuta do SLICC kritérií choroby (20).
Leukopenie. Zejména období vystupňované aktivity choroby jsou spojena s leukopenií, přičemž nejčastěji se vyskytuje lymfopenie způsobená přítomností antilymfocytárních protilátek, může se však objevit i protilátkami mediovaná neutropenie. Výskyt protilátek proti kmenovým buňkám je poměrně vzácný.
Trombocytopenie se vyskytuje často v rámci antifosfolipidového syndromu či trombotické trombocytopenické purpury či hemolyticko-uremického syndromu (TTP/ HUS). Protilátky proti trombocytům jsou velmi častým nálezem.
Prodloužení parciálního tromboplastinového času je způsobeno přítomností protilátek proti jednotlivým komponentám koagulační kaskády (VII, IX, XII). Jedná se o častý nález rovněž u antifosfolidového syndromu.
Bioptické metody
Z popisu možných orgánových manifestací choroby vyplývá význam histologického vyšetření postižené tkáně či orgánu, v případě SLE zejména význam biopsie kožních lézí (biopsie neosvětlené kůže, lupus band test) či biopsie ledvin. Bioptický vzorek ledvinné tkáně se vyšetřuje jak metodou světelné mikroskopie, tak imunofluorescenčně a elektronovým mikroskopem.
Paraklinická vyšetření
V diagnostickém procesu je indikována řada běžných zobrazovacích a vyšetřovacích metod (například EKG, radiogram hrudníku a postižených kloubů, sonografie břicha). V rámci možných mnohočetných orgánových manifestací choroby je třeba využít i řady elektivních vyšetření (například echokardiografie, magnetická rezonance mozku, vyšetření mozkomíšního moku, kostní denzitometrie pro riziko osteoporózy). Jedná se o onemocnění, které snad jako žádné jiné překračuje hranice řady medicínských oborů, a proto je diagnostický proces výrazně multidisciplinární záležitostí (revmatologie, nefrologie, neurologie, dermatologie, kardiologie, pneumonie, oftalmologie, hematologie, imunologie, psychiatrie, porodnictví, atd.), který by však měl být koordinovaný lékařem se zkušeností v diagnostice a léčbě systémových chorob pojiva.
Klasifikační kritéria SLE
The American Rheumatism Association (nyní American College of Rheumatology, ACR) publikovala klasifikační kritéria v roce 1971, která byla revidovaná v roce 1982 (18). Další revize byla přijata v roce 1997 (19). Všeobecně přijímaná kritéria měla původně sloužit spíše pro klasifikaci choroby pro účely výzkumu nežli pro rutinní diagnostiku, jsou však takto často používaná. Klasifikační kritéria jsou založena na hodnocení jedenácti položek. Pro klasifikační účely je diagnóza SLE podmíněna přítomností čtyř a více z těchto kritérií současně nebo v průběhu sledování nemocného (tab. 1).

V roce 2012 byla validizována a publikována nová klasifikační kritéria lupusu SLICC (Systemic Lupus International Collaborating Clinic), která se skládají ze dvou částí – z klinických a laboratorních kritérií (20). Jsou oproti kritériím ACR rozšířena, především co se týká neurologických, hematologických i imunologických nálezů (tab. 2). Dle pravidel SLICC pro klasifikaci SLE se přítomnost definitivní choroby opírá o pozitivitu nejméně 4 kritérií, z nichž musí být alespoň jedno klinické a jedno laboratorní, nebo pacient musí mít biopticky prokázanou lupusovou nefritidu se současnou přítomností ANA či anti-dsDNA protilátek. Tato nová kritéria byla validizována ve dvou skupinách pacientů. V derivační skupině 702 nemocných prokázala větší senzitivitu (94 % vs. 86 %, p < 0,0001) a stejnou specificitu (92 % vs. 93 %, p = 0,39) v porovnání s ACR kritérii. Počet chybných klasifikací byl nižší (49 vs. 70). Ve validační skupině (690 pacientů se SLE) prokázala větší senzitivitu (97 % vs. 83 %, p < 0,0001), menší množství chybných klasifikací (62 vs. 74), ale nižší specificitu (84 % vs. 96 %, p < 0,0001) (20). V současnosti lze pro klasifikaci SLE použít oba dva systémy.
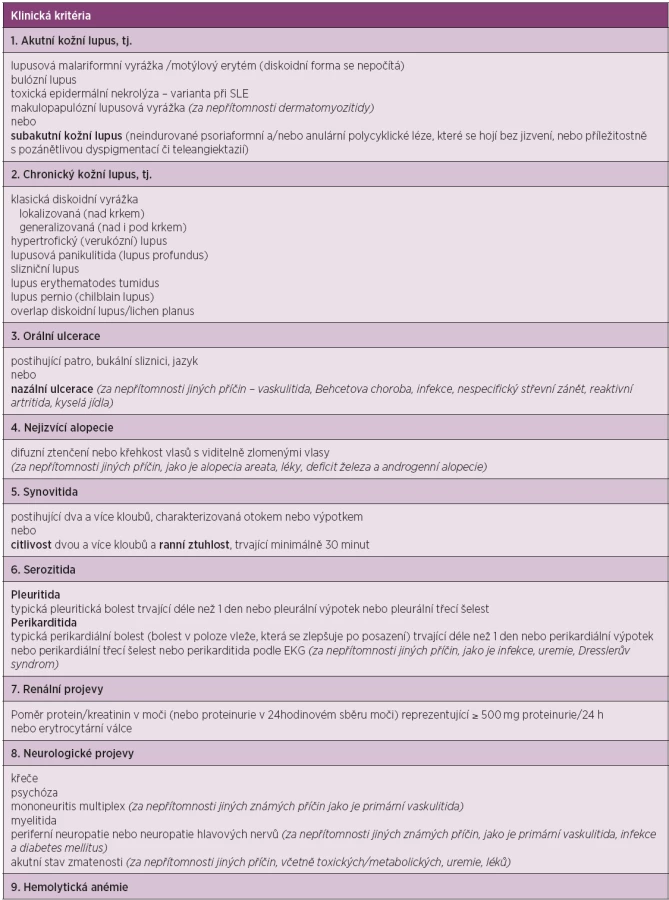

Klasifikace kožně-slizničních projevů choroby
Škála kožního postižení je nesmírně široká a sahá od klasického motýlovitého exantému, přes projevy diskoidních lézí, fotosenzitivitu, alopecii až k jizevnatým či bulózním lézím. Lupusové kožní projevy lze dle Gilliamovy klasifikace dělit na akutní, subakutní a chronické, vaskulární, nejizvící se a ostatní (21, 22).
Klasifikace neuropsychiatrických manifestací
American College of Rheumatology (ACR) formulovala definice a standardy pro diagnostiku 19 neuropsychiatrických syndromů v rámci SLE (23). Jsou děleny na a) difuzní psychiatrické a neuropsychiatrické syndromy, b) centrální nervové syndromy, c) periferní nervové syndromy.
Jejich plné definice a rozsáhlý popis jsou čtenáři k dispozici na webové stránce:
www.rheumatology.org/publications/ar/1999/aprilappendix.asp?aud=mem
V roce 2010 byla publikována i doporučení EULAR pro diagnostiku a léčbu neuropsychiatrického lupusu (24).
Histologická klasifikace lupusových nefritid
Při provedení biopsie lze nalézt jistý stupeň postižení ledvin téměř u všech nemocných se SLE. Histologická klasifikace rozlišuje 6 základních tříd glomerulonefritid a je v současnosti doplněna o indexy aktivity a chronicity. Revize histologického členění lupusových glomerulonefritid z roku 2004 (ISN/RPS) nahradila starší klasifikaci (tab. 3) (25).

Hodnocení aktivity nemoci
Posouzení stupně aktivity nemoci je důležité nejen pro účelnou, individuálně přizpůsobenou léčbu, ale také se zásadním způsobem uplatňuje v definici terapeutické intervence v klinických studiích s novými léčivy. Stupeň aktivity lze posuzovat podle klinických projevů nemoci a výsledků laboratorních testů (26, 27). V posledních desetiletích byla vytvořena řada skórovacích systémů, které byly v průběhu doby validizovány na různých populacích pacientů a byly vyzkoušeny v praxi v řadě klinických studií (28). Většina z nich hodnotí aktivitu lupusu globálně (výsledkem je porovnatelná číselná hodnota aktivity), pouze systém BILAG (29) hodnotí individuální orgánové postižení. Dokument EULAR z roku 2008 obsahuje doporučení k volbě některého z validizovaných skórovacích systémů pro sledování aktivity SLE (30). V našich doporučeních panuje nejvýraznější shoda ohledně použití systému SLEDAI (31), který je na jednu stranu poměrně jednoduchý, na stranu druhou je nejčastěji používán i v rámci klinických studií.
Systém SLEDAI-2000 (také SLEDAI-2K) (SLE Diasease Activity Index) se skládá z 24 kritérií, která postihují 9 orgánových systémů včetně imunitního (hladiny komplementu, anti-dsDNA protilátky). Hodnotí přítomnost projevů v posledních 10 dnech. Nejvyšší bodové ohodnocení mají postižení CNS, očí a nález vaskulitidy. Za aktivní chorobu lze považovat dosažení skóre 6 a více (tab. 4) (31).

Hodnocení poškození
SLICC/ACR DI (Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index) je systém, který hodnotí přítomnost poškození orgánu či orgánového systému, pokud je přítomno 6 a více měsíců. Poškození u SLE narůstá s délkou trvání choroby a je důležitým prognostickým faktorem mortality (tab. 5) (32).

Doporučení pro sledování pacientů s lupusem
Pacienti se SLE vyžadují dlouhodobé pečlivé sledování aktivity choroby a dalších zdravotních rizik souvisejících se základní nemoci i s rizikem vyplývajícím z léčby. Na základě doporučení EULAR (33) a publikovaných indikátorů kvality péče u SLE (34) doporučuje ČRS věnovat pozornost zejména následujícím oblastem:
- Pečlivé sledování pacientů se SLE
- a. Hodnocení stupně aktivity choroby při návštěvách pacientů (doporučujeme využít systém SLEDAI-2000)
- b. Hodnocení orgánového postižení (možno využít systém SLICC)
- c. Při kontrolním vyšetření hodnotit komorbidity a případnou toxicitu léčby
- d. Hodnocení kvality života
- Hodnocení kardiovaskulárního rizika při diagnóze, následně nejméně 1x ročně
- a. Kouření, kardiovaskulární příhody, fyzická aktivita, rodinná anamnéza
- b. Přítomnost hypertenzní nemoci, její případná monitorace
- c. Hodnocení lipidového spektra a glykemie
- Hodnocení rizika osteoporózy při diagnóze, následně 1x ročně
- a. Hodnocení adekvátnosti příjmu vápníku, vitaminu D, pohybu a kouření
- b. Screening a sledování osteoporózy dle národních doporučení pro postmenopauzální ženy a pro pacienty léčené dlouhodobě glukokortikoidy (35–37)
- Hodnocení infekčního rizika na počátku choroby
- a. Na počátku choroby se doporučuje provést screening HIV, HCV, HBV
- b. Zvážit screening TBC dle osobní a rodinné anamnézy a při potřebě vysokých dávek glukokortikoidů
- c. Vakcinace: doporučuje se očkování neživými vakcínami (chřipka, pneumokok)
- d. Hodnocení infekčního rizika při kontrolních návštěvách (těžká neutropenie < 500/mm3, lymfopenie < 500/mm3), snížení IgG (< 5 g/l)
- Hodnocení rizika očního poškození (glaukom, katarakta, retinální postižení)
- a. U pacientů léčených antimalariky či glukokortikoidy vyšetření při zahájení léčby
- b. Antimalarika, glukokortikoidy
- nízké riziko – druhé vyšetření po pěti letech léčby, při dalším pokračováním léčby ročně
- vysoké riziko – ročně
- Frekvence návštěv
- a. U pacientů bez klinické aktivity, orgánového poškození a komorbidit interval 6–12 měsíců
- b. U pacientů aktivních, s orgánovým poškozením, komorbiditami přizpůsobit frekvenci aktuálnímu stavu (ve většině případů 2–3 měsíce)
- Doporučená laboratorní a paraklinická hodnocení
- a. Při diagnóze
- krevní obraz s diferenciálním rozpočtem leukocytů
- sedimentace erytrocytů a CRP
- moč chemicky a sediment
- AST, ALT, GMT, ALP, bilirubin, LDH, albumin, celková bílkovina
- s-kreatinin, s-urea, GF dle MDRD
- proteinurie nebo poměr proteinurie/kreatininu
- ANA, anti-dsDNA, anti-Sm, anti-Ro, anti-La, anti-RNP
- antifosfolipidové protilátky
- hladiny komplementu C3, C4 (CH50)
- celkový cholesterol, LDL, HDL, triglyceridy, glykemie
- koagulace (INR, aPTT)
- zvážení provedení Coombsova testu
- zobrazovací vyšetření: rtg hrudníku, EKG, echokardiografie, abdominální ultrazvuk
- oční pozadí
- b. Při kontrole (intervaly přizpůsobit aktuálnímu stavu, u zcela neaktivních 1x za 6–12 měsíců)
- krevní obraz s diferenciálním rozpočtem leukocytů
- sedimentace erytrocytů a CRP
- moč chemicky a sediment
- AST, ALT, albumin
- s-kreatinin, s-urea, GF dle MDRD
- proteinurie nebo poměr proteinurie/kreatininu
- komplement, anti-dsDNA protilátky
- a. Při diagnóze
- Vyšetření při různých klinických formách choroby
- a. Při postižení muskuloskeletálního systému
- revmatoidní faktor, anti-citrulinové protilátky
- radiogramy postižených kloubů
- b. Kožní postižení
- dermatologické vyšetření
- zvážení biopsie kůže a histologické vyšetření (lupus band test)
- c. Kardiovaskulární manifestace
- EKG, echokardiografie
- d. Ledvinné manifestace
- s-kreatinin, s-urea, GF dle MDRD, kultivace moči
- proteinurie nebo poměr proteinurie/kreatininu
- ultrazvuk ledvin (velikost)
- zvážení biopsie ledvin k hodnocení aktivity a chronicity postižení
- e. Plicní postižení
- radiogram hrudníku
- zvážení spirometrie, difuze, CT plic, scintigrafie
- zvážení cytologického vyšetření aspirátu získaného při bronchoalveolární laváži
- vyšetření pleurálního výpotku
- echokardiografie
- f. Neuropsychiatrické postižení
- neurologické a psychiatrické vyšetření
- psychologické vyšetření
- zvážení EEG, CT či MR mozku či páteře, EMG, vyšetření likvoru
- g. Hematologické manifestace
- krevní obraz, diferenciální rozpočet, retikulocyty, schistocyty, LDH, volný hemoglobin, haptoglobin
- abdominální ultrazvuk
- Coombsův test či kompletní imunohematologické vyšetření
- zvážit aspiraci kostní dřeně, protilátky proti trombocytům, přežívání trombocytů
- antifosfolipidové protilátky, hladiny koagulačních faktorů
- a. Při postižení muskuloskeletálního systému
Diskuse – závěr
Autoři navrhli tato doporučení jako podklad pro diagnostiku a sledování pacientů se systémovým lupusem v klinické praxi. Jsou si vědomi jejich limitací. Jak už bylo opakovaně zdůrazněno, SLE je choroba s enormní variabilitou klinických manifestací a průběhu. Sledování pacienta je třeba přizpůsobit klinické situaci. Na druhou stranu tato doporučení zdůrazňují několik oblastí, které jsou určitě hodny většího zájmu než dosud (kardiovaskulární riziko, riziko postižení očí, osteoporóza, infekce), zavádějí však také nová doporučení ohledně sledování aktivity pomocí skórovacího systému SLEDAI a hodnocení orgánového poškození systémem SLICC. Umožňují rovněž využití nových SLICC klasifikačních kritérií choroby. Výhodou SLICC klasifikace SLE je zařazení více klinických i laboratorních nálezů, což zvyšuje jejich senzitivitu. Potřeba nových doporučení pro diagnostiku a následně i pro terapii SLE odráží vývoj na straně léčby systémového lupus, kdy zavádění nových biologických léčiv se sebou nese potřebu nově a srovnatelně definovat aktivitu choroby, průkazu efektu léčby, jejího nedostatečného účinku či selhání. Doporučení standardů v diagnostice SLE přispěje k možnosti vzájemného srovnání pacientů v rámci vznikajícího registru a umožní provádění observačních studií. Výčet doporučených položek je třeba brát jako optimální, je však třeba jej přizpůsobit běžné klinické praxi. Navržené skórovací systémy a laboratorní či zobrazovací metody by měly být zváženy podle aktuální klinické situace, možností daného zdravotnického zařízení i potřeb a stavu konkrétního pacienta.
Standardizace diagnostického a léčebného procesu povede k odstranění určité variability péče o nemocné se SLE a jak jsme přesvědčeni, k jejímu zkvalitnění. Byli jsme toho svědky i u revmatoidní artritidy či ankylozující spondylitidy.
Podpořeno grantem IGA Ministerstva zdravotnictví ČR č. NT/13707-4 a projektem MZ ČR koncepčního rozvoje výzkumné organizace 023728
Adresa pro korespondenci:
Prof. MUDr. Pavel Horák, CSc.
III. interní klinika FN a LF UP
I. P. Pavlova 6
772 00 Olomouc
e-mail: horakp@fnol.cz
Zdroje
1. Sherer Y, Gorstein A, Fritzler M, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: More than 100 different antibodies found in SLE patients. Sem Arthritis Rheum 2004; 34 : 501-7.
2. Urowitz MB, Gladman DD, Tom BD, et al. Changing patterns in mortality and disease outcomes for patients with systemic lupus erythematosus. J Rheumatol 2008; 35 : 2152-8.
3. Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus 2006; 15 : 308-18.
4. Rus V, Maury EE, Hochberg MC. The epidemiology of systemic lupus erythematosus. In: Dubois' Lupus Erythematosus. Wallace DJ, Hahn BH (Eds), Lippincott Williams and Wilkins, Philadelphia 2002.
5. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum 1998; 41 : 778-99.
6. Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum 2007; 56 : 2092-4.
7. Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum 2010; 39 : 257-68.
8. Jonsson H, Nived O, Sturfelt G, Silman A. Estimating the incidence of systemic lupus erythematosus in a defined population using multiple sources of retrieval. Br J Rheumatol 1990; 29 : 185-8.
9. von Mühlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin Arthritis Rheum 1995; 24 : 323-58.
10. Smeenk R, Brinkman K, van den Brink H, et al. Antibodies to DNA in patients with systemic lupus erythematosus. Their role in the diagnosis, the follow-up and the pathogenesis of the disease. Clin Rheumatol 1990; 9 : 100-10.
11. Eaton RB, Schnneider G, Schur PH. Enzyme immunoassay for antibodies to native DNA. Specificity and quality of antibodies. Arthritis Rheum 1983; 26 : 52-62.
12. Aviña-Zubieta JA, Galindo-Rodriguez G, Kwan-Yeung L, et al. Clinical evaluation of various selected ELISA kits for the detection of anti-DNA antibodies. Lupus 1995; 4 : 370-4.
13. Munves EF, Schur PH. Antibodies to Sm and RNP. Prognosticators of disease involvement. Arthritis Rheum 1983; 26 : 848-53.
14. Dostál C. Klinické projevy SLE. In Dostál C, Vencovský J, et al. Systémový lupus erythematosus. Medprint 1997 : 79-166.
15. Fritzler MJ, Tan EM. Antibodies to histones in hrug-Induced and idiopathic lupus erythematosus J Clin Invest 1978; 62 : 560–67.
16. Hughes GR. Thrombosis, abortion, cerebral disease, and the lupus anticoagulant. Br Med J 1983; 287 : 1088–99.
17. Walport MJ. Complement and systemic lupus erythematosus. Arthritis Res 2002; 4:(Suppl. 3) 279-93.
18. Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classificatin of systemic lupus erythematosus. Arthritis Rheum 1982; 25 : 1271-77.
19. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997; 40 : 1725-6.
20. Petri M, Orbai AM, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012; 64 : 2677–86.
21. Gillian JN, Sontheimer RD. Skin manifestations of SLE. Clin Rheum Dis 1981;8 : 207-18.
22. Sontheimer, RD. The lesions of cutaneous lupus erythematosus-a review and personal perspective on nomenclature and classification of cutaneous manifestations of lupus erythematosus. Lupus 1997; 6 : 84-95.
23. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum 1999;42 : 599-608.
24. Bertsias G, Ioannidis JPA, Aringer M, et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: report of a task force of the EULAR standing committee for clinical affairs. Ann Rheum Dis 2010; 69 : 2074–82.
25. Weening JJ, D'Agati VD, Melvin M, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney International 2004; 65 : 521-30.
26. Merrill JT. Measuring disease activity in systemic lupus: progress and problems. J Rheumatol 2002; 29 : 2256-7.
27. Illei GG, Tackey E, Lapteva L, Lipsky PE. Biomarkers in systemic lupus erythematosus. I. General overview of biomarkers and their applicability. Arthritis Rheum 2004; 50 : 1709-20.
28. Liang MH, Socher SA, Larson MG, Schur PH. Reliability and validity of six systems for the clinical assessment of disease activity in systemic lupus erythematosus. Arthritis Rheum 1989; 32 : 1107-8.
29. Yee CS, Farewell V, Isenberg DA, et al. British Isles Lupus Assessment Group 2004 index is valid for assessment of disease activity in systemic lupus erythematosus. Arthritis Rheum 2007; 56 : 4113-9.
30. Bertsias G, Ioannidis JPA, Boletis J, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis 2008; 67 : 195–205.
31. Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol 2000; 29 : 288-91.
32. Gladman DD, Goldsmith CH, Urowitz MB, et al. The Systemic Lupus International Collaborating Clinics/American College of Rheumatology (SLICC/ACR) Damage Index for Systemic Lupus Erythematosus International Comparison. J Rheumatol 2000; 27 : 373-6.
33. Mosca M, Tani A, Aringer M, et al. European League Against Rheumatism recommendations for monitoring patients with systemic lupus erythematosus in clinical practice and in observational studies. Ann Rheum Dis 2010; 69 : 1269-74.
34. Mosca M, Tani C, Aringer M, et al. Development of quality indicators to evaluate the monitoring of SLE patients in routine clinical practice. Autoimmunity Reviews 2011;10 : 383–8.
35. Rosa J, Bayer M, Jeníček J. Doporučené postupy pro diagnostiku a terapii postmenopauzální osteoporózy II. Osteol Bull 2007; 12 : 74-81.
36. Růžičková O, Bayer M, Pavelka K, Palička V. Doporučení pro prevenci a léčbu glukokortikoidy indukované osteoporózy u pacientů s revmatickým onemocněním. Osteol Bull 2004; 9 : 78-85.
37. Štěpán J. Algoritmus léčby glukokortikoidy indukované osteoporózy – hledání východisek – editorial. Vnitř Lék 2009; 55 : 448-54.
Štítky
Dermatologie Dětská revmatologie RevmatologieČlánek vyšel v časopise
Česká revmatologie

2013 Číslo 2
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Doporučení k perioperační úpravě terapie zánětlivých revmatických onemocnění
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
- Přidání perorálního acetazolamidu (Diluran) k topické léčbě dorzolamidem může vést k dalšímu snížení nitroočního tlaku u některých dětí s glaukomem
Nejčtenější v tomto čísle
- Průběh těhotenství u žen se systémovým lupus erythematodes
- Eozinofilní fasciitida
- Doporučení České revmatologické společnosti pro diagnostiku a sledování nemocných se systémovým lupus erythematodes
- Co bychom měli vědět o diagnostických testech
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy