
Prognostické faktory idiopatické plicní fibrózy (IPF) – analýza Českého registru IPF
Idiopathic pulmonary fibrosis prognostic factors – analysis of the Czech registry
Idiopathic pulmonary fibrosis (IPF) is a rare, progressive and usually fatal form of idiopathic interstitial pneumonia. IPF is characterized by failure of alveolar re-epithelization, persistence of fibroblasts, deposition of extracellular matrix, and distortion of lung architecture, which ultimately results in respiratory failure.
We analysed 202 consecutive patients with IPF diagnosed at the Departments of Pulmonary Diseases and Tuberculosis in the Czech Republic, who they were included in the nationwide Czech IPF registry. Our aim was to determine prognostic factors of IPF and outcome of the disease.
There were 129 males and 73 females who were the median age 67 years. IPF was biopsy-proven in 66 (33 %) of patients. Median time from the first symptom to diagnosis was 12 months. Diagnosis was made in 57 patients (28.3 %) within 6 months from the onset of respiratory symptoms. 8 (4 %) patients had an acute exacerbation during the course of the disease.
In uniparametric (univariate) analysis as prognostic factors associated with poorer survival were found: higher age, higher degree dyspnea scores, clubbing fingers, comorbidities (arterial hypertension, osteoporosis), patients without histology biopsy, and bronchoalveolar increased neutrophil count. We found these positive prognostic factors: higher levels of VC (vital capacity), TLC (total lung capacity) and DLCO (diffusing capacity for carbon monooxide).
In multiparametric (multivariate) analysis as prognostic factors associated with mortality were found: higher age, higher degree of dyspnoe score. Increased lymphocytes in bronchoalveolar fluid, higher level of VC a DLCO were associated with better survival. There was no difference in survival of patients by sex, by smoking status. No significant difference in survival rates was found between IPF with and without emphysema, between the extent of fibrosis on HRCT (high resolution computed tomography) of thorax and mortality. Median survival was 51.6 months. 58 (28.7 %) patients died. The most frequent reason of dead was IPF progression with respiratory failure.
Key words:
Idiopathic pulmonary fibrosis; prognosis; treatment
Autoři:
Martina Doubková 1; Michal Uher 2; Vladimír Bartoš 3; Martina Šterclová 4; Ladislav Lacina 5; Vladimíra Lošťáková 6; Ilona Binková 1; Martina Plačková 7; Monika Žurková 6; Radka Bittenglová 8; Jana Pšikalová 9; Lenka Šišková 10; Pavlína Lisá 11; František Petřík 11; Jaroslav Polák 12; Vladimír Řihák 10; Jana Skřičková 1; Martina Vašáková 4
Působiště autorů:
Klinika nemocí plicních a TBC LF MU a FN Brno
1; Institut biostatistiky a analýz LF MU Brno
2; Plicní klinika LF UK a FN Hradec Králové
3; Pneumologická klinika 1. LF UK a Thomayerovy nemocnice v Praze
4; Klinika pneumologie a hrudní chirurgie Nemocnice Na Bulovce v Praze
5; Klinika plicních nemocí a tuberkulózy LF UP a FN Olomouc
6; Klinika plicních nemocí a tuberkulózy LF OU a FN Ostrava
7; Klinika pneumologie a ftizeologie LF UK a FN Plzeň
8; Pneumologicko-alergologické oddělení Kroměřížské nemocnice
9; Plicní oddělení Krajské nemocnice T. Bati Zlín
10; Pneumologická klinika 2. LF UK a FN Motol v Praze
11; Dopravní zdravotnictví a. s., Nemocnice s poliklinikou Praha Italská
12
Vyšlo v časopise:
Čas. Lék. čes. 2016; 155: 188-194
Kategorie:
Původní práce
Souhrn
Idiopatická plicní fibróza (IPF) je progresivní a obvykle fatální forma idiopatické intersticiální pneumonie (IIP). IPF je charakterizována selháním alveolární reepitelizace, perzistencí fibroblastů, depozicí extracelulární matrix a poruchou alveolární architektoniky, která vede k respiračnímu selhání. Cílem naší práce bylo zjistit klinické charakteristiky, průběh nemoci a prognostické faktory u pacientů s IPF v běžné klinické praxi.
Analyzovali jsme 202 pacientů, kteří byli pro IPF sledování v síti center pro diagnostiku a léčbu intersticiálních plicních procesů v České republice. Diagnostika IPF vycházela z doporučení American Thoracic Society (ATS)/European Respiratory Society (ERS). Naším cílem bylo ověřit prognostické faktory nemoci a osud našich pacientů.
Do analýzy bylo zahrnuto 73 mužů a 129 žen, s mediánem věku 67 let. IPF byla histologicky ověřena u 66 (33 %) pacientů. Medián času od prvních klinických příznaků do stanovení diagnózy byl 12 měsíců. U 57 nemocných (28,3 %) byla diagnóza stanovena do 6 měsíců od začátku symptomů. Osm (4 %) pacientů mělo akutní exacerbaci.
V jednorozměrné analýze byly zjištěny následující faktory, negativně ovlivňující přežití v době diagnózy: vyšší věk, paličkovité prsty, vyšší stupeň dušnosti dle NYHA (New York Heart Association), průkaz neutrofilní alveolitidy v bronchoalveolární tekutině (BALT), vyšší věk bez histologické verifikace, kardiovaskulární komorbidity, diabetes a osteoporóza. Jako příznivé prognostické faktory byly zjištěny: lepší hodnoty vitální kapacity (VC), celkové plicní kapacity (TLC) a plicní difuze (DLCO, KCO).
Vícerozměrná analýza ukázala, že nepříznivá prognóza nemoci je asociována s vyšším věkem a vyšším stupněm dušnosti. Průkaz lymfocytární alveolitidy v BALT a lepší hodnoty vstupních parametrů VC a DLCO byly spojeny s lepším přežitím. Nebyl zjištěn žádný rozdíl v přežití mezi pohlavími a mezi kuřáky a nekuřáky. Přítomnost emfyzému neměla vliv na mortalitu a ani na rozsah plicní fibrózy na HRCT hrudníku.Medián přežití dosahoval 51,6 měsíce od diagnózy a nejčastější příčinou smrti bylo respirační selhání.
Klíčová slova:
Idiopatická plicní fibróza, prognóza, léčba
Úvod
Idiopatická plicní fibróza (IPF) je nejběžnějším typem idiopatické intersticiální pneumonie a má velmi špatnou prognózu, která je způsobená progresí rozsáhlé fibrotické přestavby plicního parenchymu. Řadí se mezi méně běžná plicní onemocnění a její etiologie není přesně známa. Základním etiopatogenetickým mechanismem IPF je opakované poškození alveolárního epitelu a abnormální proliferace fibroblastů s tvorbou extracelulární matrix. Zánět může být přítomen sekundárně (1).
Medián věku při diagnóze IPF se pohybuje mezi 60 a 70 roky (1, 2). Rizikovými faktory jsou kouření cigaret, expozice organickým i anorganickým prachům, gastroezofageální reflux a infekce (1–3). Pacienti obvykle trpí postupně se zhoršující dušností, neproduktivním kašlem a umírají na respirační selhání po 2–4 letech od stanovení diagnózy (1–3). Průběh nemoci a přežití jsou ovlivněny přítomností akutních exacerbací, které jsou spojené se skokovým zhoršením plicních funkčních parametrů a progresí plicního nálezu (1).
Stanovení diagnózy IPF vyžaduje nález obvyklé intersticiální pneumonie (UIP – usual interstitial pneumonia) na HRCT hrudníku (high resolution computed tomography – výpočetní tomografie s vysokým rozlišením) v korelaci s klinickým obrazem nebo histologickým ověřením UIP plicní biopsií. Plicní biopsie již není zlatým standardem pro diagnostiku. IPF je spojena s UIP, ale ne každá UIP značí IPF. UIP může být pozorována jako plicní postižení i u jiných onemocnění, například revmatických (1).
IPF trpí kolem 5 milionů lidí na celém světě (4). Epidemiologická data nejsou přesně známa, ale nemoc má stoupající výskyt. Prevalence se ve světě pohybuje mezi 13 a 20/100 000 a incidence činí 6,8–16,3/100 000 obyvatel (4).
V ČR není aktuální epidemiologická situace známa. Studie odhadovaly incidenci na 1/100 000 a prevalenci na 6,5–12/100 000 obyvatel, nicméně údaj je zřejmě podhodnocen vzhledem k poddiagnostikovanosti této nemoci (5). IPF se u nás věnoval již v roce 1953 docent Ladislav Levinský. První sdělení o IPF publikoval na stránkách Časopisu lékařů českých (6).
Materiál a metody
Celkem bylo analyzováno 202 pacientů zadaných do Českého registru IPF. Zadávajícími centry podle počtu pacientů byly: Hradec Králové, Thomayerova nemocnice v Praze, Brno, Nemocnice Na Bulovce v Praze, Olomouc, Ostrava, Plzeň, Kroměříž, Zlín a Fakultní nemocnice Motol v Praze.
Diagnostika a pacienti
IPF byla diagnostikována podle konsenzu American Thoracic Society/European Respiratory Society (ATS/ERS) z roku 2011 (1). Kritéria IPF jsou následující:
- vyloučení jiné známé příčiny intersticiálního plicního procesu;
- plicní funkční vyšetření s restriktivní ventilační poruchou a/nebo snížením plicní difuze;
- jinak nevysvětlitelná dušnost;
- potíže trvající více než 3 měsíce;
- bilaterálně bazálně při plicní auskultaci slyšitelné krepitace (velcro crackles, připodobňované rozepínání suchého zipu);
- charakteristický HRCT nález v plicní tkáni (změny maximálně subpleurálně bazálně typu retikulárních a lineárních opacit, voštinovitá plíce – honeycomb lung, trakční bronchiektazie, mimimum opacit mléčného skla – ground glass);
- negativní laboratorní testy na průkaz autoprotilátek s negativním revmatologickým nálezem (3).
Všechny sporné nálezy HRCT byly ověřovány druhým čtením v Thomayerově nemocnici v Praze.
Zdrojem statistických dat byl Český registr IPF spuštěný v roce 2012. Jedná se o pilotní data.
Metody
U 202 pacientů zařazených do celorepublikového registru jsme analyzovali klinické, fyziologické a radiologické charakteristiky v době diagnózy. Jako prognostické faktory byly v době diagnózy (bazální ukazatele) hodnoceny:
- věk;
- pohlaví;
- klinické příznaky (dušnost, kašel, fenotypový projev paličkovitých prstů s nehty tvaru hodinového skla);
- fyziologické parametry v klidu:
- spirometrie: FVC – forsírovaná vitální kapacita, forced vital capacity; FEV1 – jednosekundová forsírovaná vitální kapacita, forced expiratory volume in 1 second;
- pletysmografie: VC – vitální kapacita, vital capacity; TLC – totální plicní kapacita, total lung capacity; plicní difuze (DLCO – difuzní kapacita plic, diffusing lung capacity for carbon monooxid; KCO – transfer factor);
- hodnoty krevních plynů (parciální tlak kyslíku, saturace kyslíku);
- status kuřáctví;
- body mass index (BMI);
- přítomnost akutní exacerbace;
- HRCT nález – alveolární a intersticiální skóre, přítomnost emfyzému na HRCT hrudníku,
- cytologické vyšetření bronchoalveolární tekutiny (BALT) získané bronchoalveolární laváží (BAL);
- léčebné modality.
Plicní funkční testy
Spirometrie, pletysmografie a plicní difuze byly prováděny ve shodě s doporučením ATS/ERS (7–9).
HRCT nálezy
U všech nemocných bylo provedeno HRCT hrudníku v době diagnózy a v průběhu sledování. HRCT skóre alveolární (0–5) a intersticiální (0–5) dle Gay et al. a Šterclové et al. (10, 11) bylo hodnoceno na úrovních aortálního oblouku, bifurkace trachey, maximální šíře pravé komory, kupole pravé poloviny bránice. Čím vyšší skóre, tím větší rozsah postižení. Hodnoceno bylo zesílení interlobárních sept, rozsah voštiny a opacity mléčného skla.
Bronchoalveolární laváž
Cytologické vyšetření bronchoalveolární tekutiny (BALT) získané bronchoalveolární laváží bylo provedeno v době diagnózy u všech nemocných (12). Normální nálezy byly definovány takto: lymfocyty < 15 %, neutrofilní granulocyty < 3 %, eozinofilní granulocyty < 0,5 %, makrofágy > 80 % (12).
Statistické metody
Byla provedena jednorozměrná analýza přežití, kde je každý faktor hodnocen samostatně a jeho vliv na přežití je kvantifikován jako HR (hazard ratio – jakým způsobem daný parametr ovlivňuje riziko úmrtí: HR > 1 zvyšuje riziko úmrtí a naopak HR < 1 snižuje riziko úmrtí).
Dále byla provedena vícerozměrná analýza, která hodnotí všechny faktory najednou, přičemž méně významné jsou z modelu postupně vyřazeny (nicméně základní demografické proměnné jako je věk a pohlaví a primární léčbu v modelu ponecháváme vždy). Hladina významnosti p < 0,05.
Výsledky
Základní charakteristiky
Analyzována byla data 73 (36 %) žen a 129 (64 %) mužů. 66 (33 %) mělo diagnózu potvrzenu histologicky. Průměrný věk činil 67 let. 8 pacientů (3,5 %) mělo familiární formu IPF (rodinný výskyt). Anamnéza kuřáctví byla přítomná u 105 nemocných – 28 (14 %) současných a 77 (38 %) bývalých kuřáků. Hlavními symptomy byly dušnost u 193 (96 %) a kašel u 160 (79,2 %) pacientů. Fenotypový projev paličkovitých prstů s nehty tvaru hodinového skla byl zaznamenán u 113 (56 %). Medián času mezi začátkem symptomů a stanovením diagnózy dosahoval 12 měsíců. Základní charakteristiky pacientů s IPF jsou uvedeny v tab. 1. Mezi nejčastější komorbidity patřily arteriální hypertenze, diabetes mellitus a hyperlipidemie (viz tab. 2).


Restrikční ventilační porucha a snížení plicní difuze bylo nejčastější plicní funkční abnormalitou.
HRCT hrudníku bylo provedeno u všech pacientů; průměrné intersticiální skóre činilo 2,72 a alveolární 1,20. Výskyt emfyzému byl pozorován u 16 pacientů (7,9 %).
BAL byl proveden u všech nemocných v čase diagnózy; neutrofilní alveolitida byla zjištěna u 169 (83,7 %), lymfocytární u 46 (22,8 %), eozinofilní alveolitidou u 155 (76,7 %) pacientů.
Klinický průběh a léčba
33 (16,3 %) pacientů bylo léčeno kombinací kortikosteroidů s azathioprinem a N-acetylcysteinem, 59 (29 %) pirfenidonem, 79 (39 %) N-acetylcysteinem s inhibitory protonové pumpy, 31 (15,3 %) neužívalo žádnou léčbu (viz tab. 1). 10 pacientů je zařazeno na transplantační listinu. U 8 (4 %) pacientů byla zaznamenána akutní exacerbace.
Přežití
Medián přežití od stanovení diagnózy dosahoval 51,6 měsíce (4,3 roku). Během sledování 58 (28,7 %) subjektů zemřelo. Nejčastější příčinou smrti bylo respirační selhání (v 66 % případů).
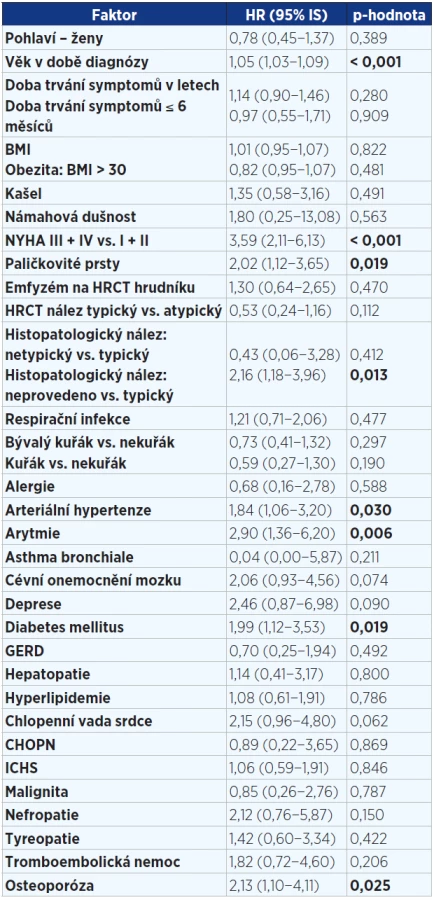
V jednorozměrné analýze (viz tab. 3 a 4) jsme prokázali prognostické faktory mající negativně vliv na přežití v době diagnózy: vyšší věk, paličkovité prsty, závažnější stupeň dušnosti dle NYHA, alveolitida s převahou neutrofilních granulocytů v BALT, pacienti bez histologického ověření, komorbidity – kardiovaskulární (arytmie, hypertenze) a osteoporóza. Jako parametry snižující riziko úmrtí byly prokázány: vyšší hodnoty plicních funkcí (VC, TLC, DLCO, KCO) v době diagnózy.
Ve vícerozměrné analýze byly zjištěny tyto faktory ovlivňující negativně přežití: vyšší věk, vyšší stupeň dušnosti. Naopak lymfocytární alveolitida v BALT, lepší vstupní hodnoty VC a DLCO byly spojeny s nižším rizikem mortality (viz tab. 5). Nebyl zjištěn žádný signifikantní rozdíl mezi pohlavími, mezi kuřáky a nekuřáky, mezi rozsahem plicní fibrózy na HRCT hrudníku a přežitím. Žádná z léčebných modalit statisticky významně neovlivnila přežití.



Diskuse
Nové poznatky o etiopatogenezi IPF zcela změnily pohled na léčebné možnosti. IPF se vyskytuje s vyšší četností u mužů (13). Průměrný věk u našich pacientů při stanovení diagnózy 67 let je v souladu s celosvětovými daty (1). Etiologie nemoci není známa, ale kouření cigaret je asociováno s IPF (3, 14). Současné studie ukazují lepší přežití u nekuřáků (15), my jsme však tuto souvislost nepozorovali. Familiární (rodinný) výskyt je pozorován do 5 % případů IPF, což je ve shodě s naším výsledkem (3). Průměrná doba stanovení diagnózy od prvních příznaků se pohybuje od 6 měsíců do 2 let (16). Nejčastějšími symptomy v době diagnózy jsou suchý kašel a střední až těžký stupeň dušnosti při zátěži. Paličkovité prsty jsou přítomné u 50 % pacientů (1). Etiopatogeneze paliček (clubbing finger) není přesně známa, ale předpokládá se vliv chronické aktivace makrofágů a produkce profibrotických reparačních faktorů (17).
Nejčastějšími komorbiditami v zahraničních publikacích jsou diabetes mellitus, ischemická choroba srdeční a gastroezofageální reflux (18). GER je zvažován jako možný rizikový faktor vzniku a progrese IPF (18). Tyto závěry potvrdila i naše studie.
Klinický průběh IPF neumíme jasně předpovídat a kritéria progrese IPF nejsou zcela dobře definována (19). Nicméně geny a epigenetické faktory (zevní prostředí, kouření) mají vliv na vznik, vývoj a klinický fenotyp IPF (20, 21). Známe tři rozdílné průběhy nemoci (22–24). Větší část pacientů vykazuje pozvolný pokles plicních funkcí, někteří jsou dlouhodobě stabilní (zejména osoby starší 75 let) a asi 10–20 % má rychlý pokles plicních funkčních parametrů (16, 20). Selman et al. ve své práci definují pacienta s rychlou progresí jako toho, u něhož doba mezi začátkem symptomů a diagnózou byla kratší než 6 měsíců a byla asociována se zhoršením klinickým, funkčním a radiologickým (20). V této studii byl zjištěn významný rozdíl v přežití mezi těmito dvěma skupinami: pacienti s rychlou progresí přežívali 27 měsíců, zatímco pacienti s pomalou progresí 93 měsíců (16, 20) – tyto dvě skupiny pravděpodobně reprezentují rozdílné fenotypy IPF. V naší práci nebyl dostatek dat pro potvrzení těchto závěrů.
Pro prognostické účely IPF se užívají různé klinické a fyziologické determinanty. Prediktory mortality rozdělujeme na základní (bazální v době diagnózy) a dynamické (longitudinální po dobu sledování) (22, 25).
Ve shodě se zahraničními daty nebyl prokázán vliv pohlaví (26, 27). Věk v novějších studiích není prognostickým faktorem (22, 28), ale v naší práci byl vyšší věk spojen s horším přežitím. Histologicky neověření pacienti měli horší prognózu než ti s plicní biopsií, pravděpodobným důvodem je opět věk a špatný zdravotní stav způsobený pokročilým plicním procesem. Stupeň dušnosti a paličkovité prsty jsou označovány ve shodě s našimi závěry jako prognostické ukazatele a jsou obvykle dávány do vztahu k déletrvajícímu fibrotizujícímu onemocnění, delší době do stanovení diagnózy a pokročilosti onemocnění (24).
Několik studií popsalo jako další negativní prognostický faktor přežití horší vstupní plicní funkční ukazatele FVC, VC, TLC a DLCO (29, 30), což je v souladu s našimi závěry. Nižší hodnoty těchto parametrů, zejména DLCO, jsou způsobeny větším rozsahem plicní fibrózy a závažností onemocnění. Další možný prediktor mortality je hodnota PaO2 (parciální tlak kyslíku) v klidu (31), ačkoli existují i práce, které toto nepotvrdily (32). Údaje pro analýzu krevních plynů v našem souboru byly nedostatečné, proto jsme je nemohli hodnotit.
Prognostickým je i 6MWT (six-minute walk test – šestiminutový test chůze), absolvovaná vzdálenost a nejnižší desaturace kyslíku během chůze (33). Bohužel v naší kohortě chyběl dostatek dat pro analýzu 6MWT.
Pro IPF je charakteristické zmnožení neutrofilních granulocytů obvykle s malou příměsí eozinofilů v BALT, lymfocyty bývají zvýšeny minimálně. V naší kohortě byla lymfocytární alveolitida spojena s lepším přežitím a medián lymfocytů v BALT činil 22 %. Neutrofilie v BALT je popisována jako špatný prognostický faktor ve vztahu k přežití v uniparametrických analýzách (34), multiparametrické analýzy to však nepotvrzují (35, 36). Tyto výsledky jsou ve shodě s našimi.
Plicní hypertenze (PH) je uváděna jako další prediktor mortality v longitudinálních studiích a její přítomnost vede ke zhoršení plicních funkčních parametrů (28, 37). My jsme pro hodnocení vlivu PH na mortalitu neměli dostatečné množství dat. Vztah mortality a BMI (body mass index) popisují ve své studii Alakhas et al. – nejlepší přežití vykazovali pacienti s BMI > 30, i když vysoké BMI je samo o sobě relativní kontraindikací k provedení plicní transplantace (38). My jsme vliv BMI na přežití nepotvrdili.
HRCT nález rozsahu plicní fibrózy je zvažován jako jeden z možných prognostických faktorů (39). Kombinace plicní fibrózy a emfyzému (CPFE – syndrome of combined pulmonary fibrosis and emphysema) byla poprvé popsána v roce 2005 (40). Emfyzém je zaznamenán v zahraničních publikacích u 28–35 % nemocných (41, 42). Sugino et al. ve své studii popisují špatnou prognózu pacientů s CPFE oproti IPF. Pacienti s CPFE s paraseptálním emfyzémem a vyšším systolickým plicním arteriálním tlakem měli kratší dobu přežití (43). V naší práci toto zjištěno nebylo, pravděpodobně to souvisí s malým množstvím dat.
AE-IPF (akutní exacerbace) je popsána u 5–10 % všech IPF pacientů (23, 44), mortalita je vysoká, pohybující se mezi 50 a 100 % (16, 45). Výskyt AE-IPF nemá vztah k závažnosti vstupního plicního funkčního postižení (16). V naší kohortě bylo AE pozorováno u 8 (4 %) pacientů, což je v korelaci s výše uvedenými zahraničními daty. AE je asociována se zhoršením klinickým, funkčním i radiologickým (46).
Dalším hodnoceným faktorem byla terapie. Od roku 2012 již není pro IPF doporučována léčba trojkombinací kortikoidy + azathioprin + N-acetylcystein (47). Dle výsledků studie z roku 2014 nebyl zaznamenán ani pozitivní vliv léku s antioxidačním účinkem N-acetylcysteinu na plicní funkční parametry ve srovnání s placebem (48). V naší kohortě bylo touto trojkombinací léčeno jen 16 % nemocných. V léčbě se pokračovalo přechodně jen u pacientů, kteří z ní zaznamenali profit. Od roku 2011 máme možnost v ČR podávat pacientům s IPF a mírnou až středně těžkou formou onemocnění (FVC 50–80 %, DLCO > 35 %) antifibrotický lék pirfenidon ovlivňující primární proces fibrogeneze. U pacientů léčených pirfenidonem bylo zaznamenáno zpomalení deklinace plicních funkcí v čase a lepší přežití oproti skupině neléčených (49). Žádná léčebná možnost v naší studii neměla vztah k přežití, důvodem byla krátká doba sledování.
Medián přežití, který je v různých studiích uváděn od 2 do 4 let od stanovení diagnózy, je ve shodě s našimi výsledky (19, 50). Příčinou smrti u nemocných s IPF je obvykle respirační selhání (51). Tento závěr potvrdila i naše analýza. Dalšími příčinami jsou kardiovaskulární choroby (srdeční selhání, ischemická choroba srdce), bronchogenní karcinom, infekce, plicní embolie (26, 52).
Závěr
IPF je méně běžné onemocnění se špatnou prognózou. IPF registr nám umožňuje získat první data o českých pacientech a srovnávat je se zahraničními.
V jednorozměrné analýze byly námi zjištěné negativní faktory ve vztahu k přežití v době diagnózy následující: vyšší věk, vyšší stupeň dušnosti, paličkovité prsty, nález zvýšených hodnot neutrofilních granulocytů v BALT, kardiovaskulární komorbidity, diabetes a osteoporóza, pacienti bez histologického ověření. V jednorozměrné analýze jsme zjistili jako pozitivní prognostický ukazatel lepší hodnoty plicních funkčních parametrů (VC, TLC, DLCO, KCO) v době diagnózy.
Ve vícerozměrné analýze byly v době diagnózy potvrzeny tyto negativní faktory ovlivňující přežití: vyšší věk, vyšší stupeň dušnosti. Lymfocytární alveolitida v BALT a lepší vstupní funkční ukazatele (VC, KCO) byly spojeny s delším přežitím.
Adresa pro korespondenci
MUDr. Martina Doubková
Klinika nemocí plicních a TBC Lékařské fakulty MU a Fakultní nemocnice Brno
Jihlavská 20
625 00 Brno
Tel.: 532 232 565
e-mail: doubkovamartina@seznam.cz
Zdroje
1. Raghu G, Collard HR, Egan JJ et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183 : 788–824.
2. American Thoracic Society;European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2011. Am J Respir Crit Care Med 2002; 165 : 277–304.
3. Vašáková M, Šterclová M. Idiopatická plicní fibróza – doporučený postup pro diagnózu, léčbu a sledovní (2. aktualizace). http://www.pneumologie.cz/guidelines
4. Fernández Pérez ER, Daniels CE, Schroeder DR et al. Incidence, prevalence and clinical course of idiopathic pulmonary fibrosis. Chest 2010; 137 : 129–137.
5. Kolek V. Epidemiology of cryptogenic fibrosing alveolitis in Moravia and Silesia. Acta Palacki Olomuc Fac Med 1994; 137 : 49–50.
6. Levinský L. Chronická difuzní intersticiální plicní fibróza (idiopatická). 1. sdělení. Čas Lék Čes 1953; 92 : 976–981.
7. Miller MR, Hankinson J, Brusasco V et al. Standardisation of spirometry. Eur Respir J 2005; 26 : 319–338.
8. Wanger J, Clausen JL, Coates A et al. Standardisation of the measurement of lung volumes. Eur Respir J 2005; 26 : 511–522.
9. Macintyre N, Crapo JE, Viegi G et al. Standardisation of the single-breath determination of carbon monoxide uptáme in the lung. Eur Respir J 2005; 26 : 720–735.
10. Gay SE, Kazerooni EA, Toews GB et al. Idiopathic pulmonary fibrosis – predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157 : 1063–1072.
11. Šterclová M, Vašáková M. Kvantifikace rozsahu postižení u nemocných s fibrotizujícími intersticiálními procesy. Čes Radiol 2013; 67 : 204–208.
12. Skřičková J, Kolaříková R. Standardní postup při provádění bronchoalveolární laváže (BAL) a vyšetřování bronchoalveolární tekutiny (BAT). http://www.pneumologie.cz/soubory/BAL_Standard_Studia_po_recenzi_8_2004.pdf
13. Raghu G, Weycker D, Edelsberg J et al. Incidence a prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006; 174 : 810–816.
14. Baumgartner KB, Samet JM, Stidley CA et al. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155 : 242–248.
15. Antoniou KM, Hansell DM, Rubens MB et al. Idiopathic pulmonary fibrosis outcome in relation to smoking status. Am J Respir Crit Care Med 2008; 177 : 190–194.
16. Kim DS, Collard HR, King TE jr. et al. Classification and natural history of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006; 3 : 285–292.
17. Toovey OT, Eisenhauer HJ et al. A new hypothesis on the mechanism of digital clubbing secondary to pulmonary pathologies. Med Hypothese 2010; 75 : 511–513.
18. Gribbin J, Hubbard R, Smith C et al. Role of diabetes mellitus and gastro-oesophageal reflux in the aetiology of idiopathic pulmonary fibrosis. Respir Med 2009; 103 : 927–931.
19. Mura M, Poretta M, Bargagli E et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J 2012; 40 : 101–109.
20. Selman M, Carrillo G, Estrada A et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One 2007; 2: e482.
21. Noth I, Zhang Y, Ma SF et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med2013; 1 : 309–317.
22. Barlo NP, van Moorsel Ch, van den Bosch et al. Predicting prognosis in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27 : 85–95.
23. Martinez FJ, Safrin S, Weycker D et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 2005; 142 : 963–967.
24. King TE jr., Tooze JA, Schwarz MI et al. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med 2001; 164 : 1171–1181.
25. Carbone R, Balleari J, Grosso M et al. Predictors of mortality of idiopathic pulmonary fibrosis. Eur Rev Med Pharmacol Sci 2008; 12 : 97–104.
26. Lee SH, Shim HS, Cho SH et al. Prognostic factors for idiopathic pulmonary fibrosis: clinical, physiologic, pathologic and molecular aspects. Sarcoidosis Vasc Diffuse Lung Dis 2011; 28 : 102–112.
27. Doubková M, Binková I, Jančíková J et al. Jak včasná je diagnostika idiopatické plicní fibrózy a jak úspěšná je její terapie? Stud Pneumol Phtiseol 2007 : 67 : 113–119.
28. Nadrous HF, Myers JL, Decker PA et al. Idiopathic pulmonary fibrosis in patients younger than 50 years. Mayo Clin Proc 2005; 80 : 37–40.
29. Collared HR, King TE JR, Bartelson BB et al. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168 : 538–542.
30. Jegal Y, Kim DS, Shim TS et al. Physiology is a stronger predictor of survival than patology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171 : 639–644.
31. Stephan S, de Castro Pereira, Coletta EM et al. Oxygen desaturation during a 4-minute step test: predicting survival in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2007; 24 : 70–76.
32. Miki K, Maekura R, Hiraga T et al. Prognosis of idiopathic pulmonary fibrosis. Respir Med 2003; 97 : 482–490.
33. Flaherty KR, Andrei AC, Murray S et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174 : 803–809.
34. Kinder BW, Brown KK, Schwarz MI et al. Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest 2008; 133 : 226–232.
35. Tabuena RP, Nagai S, Tsutsumi T et al. Cell profiles of bronchoalveolar lavage fluid as prognosticators of idiopathic pulmonary fibrosis/usual interstitial pneumonia among Japanese patients. Respiration 2005; 72 : 490–498.
36. Veeraraghavan S, Latsi PI, Wells AU et al. BAL findings in idiopathic nonspecific interstitial pneumonia and usual interstitial pneumonia. Eur Respir J 2003; 22 : 239–244.
37. Lettieri CJ, Nathan SD, Barnett SD et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 129; 746–752.
38. Alakhras M, Decker PA, Nadrou HF et al. Body mass index and mortality in patients with idiopathic pulmonary fibrosis. Chest 2007; 131 : 1448–1453.
39. Wells AU, Desai SR, Rubens MB et al. Idiopathic pulmonary fibrosis. A Composite Physiologic Index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 2003; 167 : 962–969.
40. Cottin V, Nunes H, Brillet PY et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 2005; 26 : 586–593.
41. Kurashima K, Takayanagi N, Tsuchiya N et al. The effect of emphysema on lung function and survival in patients with idiopathic pulmonary fibrosis. Respirology 2010; 15 : 843–848.
42. Mejía M, Carillo G, Rojas-Serrano J et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest 2009; 136 : 10–15.
43. Sugino K, Ischida F, Kikuchi N et al. Comparison of clinical characteristics and prognostic factors of combined pulmonary fibrosis and emphysema versus idiopathic pulmonary fibrosis alone. Respirology 2014; 19 : 239–245.
44. Lazor R, Bonetti A, Nicod LP et al. Acute exacerbations of idiopathic pulmonary fibrosis. Rev Med Suisse 2010; 6 : 2228–2230, 2232.
45. Song JW, Hong SB, Lim CM et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir 2011; 37 : 356–363.
46. Hyzy R, Huang S, Myers J et al. Acute exacerbation of idiopathic pulmonary fibrosis. Chest 2007; 132 : 1652–1658.
47. Raghu G, Anstrom KJ, King TE et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366 : 1968–1977.
48. Martinez FJ, de Andrade JA, Anstrom KJ et al. Randomized trial of acetylcystein in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370 : 2093–2101.
49. King TE jr., Bradford JZ, Catro-Bernardini S et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370 : 2083–2092.
50. Ley B, Collard HR, King TE JR et al. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183 : 431–440.
51. Saydain G, Islam A, Afessa B et al. Outcome of patients with idiopathic pulmonary fibrosis admitted to the intensive care unit. Am J Respir Crit Care Med 2002; 166 : 839–842.
52. Panos R, Mortenson RL, Nicolli SA. Clinical deterioration in patients with idiopathic pulmonary fibrosis: cause and assessment. Am J Med 1990; 88 : 396–404.
Štítky
Adiktologie Alergologie a imunologie Angiologie Audiologie a foniatrie Biochemie Dermatologie Dětská gastroenterologie Dětská chirurgie Dětská kardiologie Dětská neurologie Dětská otorinolaryngologie Dětská psychiatrie Dětská revmatologie Diabetologie Farmacie Chirurgie cévní Algeziologie Dentální hygienistkaČlánek vyšel v časopise
Časopis lékařů českých

- Jak a kdy u celiakie začíná reakce na lepek? Možnou odpověď poodkryla čerstvá kanadská studie
- Jaké zdravotní benefity může mít popíjení kávy nebo čaje?
- Doc. Jitka Fricová: V USA nasazovali fentanyl poměrně nekriticky, v Česku je situace jiná
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Když léky přinášejí jinou chuť Vánoc...
Nejčtenější v tomto čísle
- Význam HPV vakcinace mužů
- Dermatomyozitída
- Hodnocení hemodynamické významnosti koronárních stenóz metodou frakční průtokové rezervy
- Matrix Gla protein jako přirozený inhibitor vaskulárních kalcifikací a potenciální léčebný cíl
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy