
Spojenie akromegálie a Klinefelterovho syndrómu u jedného pacienta
The combination of acromegaly and Klinefelter syndrome in one patient
Acromegaly is a rare disorder usually caused by a benign tumour of the pituitary gland. Long-term presence of elevated growth hormone (GH) and insulin like growth factor 1 (IGF1) levels accompanying this disease is associated with complications such as cardiomyopathy, diabetes mellitus, sleep apnoea and arthropathy. Incidence of acromegaly is 3–4 patients per million per year. Klinefelter syndrome (KS) is the most common sex chromosome disorder occuring in about 1/500 live male births. Common physical features include particularly small testes, among other symptoms are tall stature, reduced muscle tone, delayed pubertal development, lack of secondary male sex characteristics and gynecomastia. We present a 32-year-old man suffering from both acromegaly and 47, XXY Klinefelter syndrome. The patient with typical acromegalic features. Laboratory tests revealed high level of GH which was not suppressed after glucose administration, high level of IGF1, low testosterone concentration with high concentation of luteinizing hormone and follicle stimulating hormone. A magnetic resonance imaging scan revealed a 25 × 18 × 18 mm macroadenoma involving the pituitary gland. A diagnosis of acromegaly was established. After this examination trans-sphenoidal resection was performed. Histopathologic and immunohistochemical findings revealed growth hormoneproducing pituitary adenoma. The presence of infertility with clinical features such as small testes, lack of secondary male sex characteristics and laboratory findings revealed hypergonadotropic hypogonadism that could not be explained by the diagnosis of acromegaly. A chromosomal karyotyping revealed a 47, XXY, confirming the diagnosis of KS. Testosterone replacement therapy wasn´t begun because of patient disagreement Postoperatively elevated plasma concentration of GH and IGF1 levels persist. Treatment by somatostatin analogues (lanreotid) was initiated at dose 120 mg every 28 days. Control magnetic resonance imaging of the sella demonstrated a residue of pituary adenoma size 14 × 14 × 7 mm. The patient is currently undergoing endoscopic revision of the residue.
acromegaly – growth hormone – IGF1 – Klinefelter syndrome – testosterone
Autoři:
Ivana Ságová 1; Dušan Pávai 2; Daniela Kantárová 1; Anton Vaňuga 2,3; Jurina Sadloňová 1; Peter Vaňuga 2; Milan Dragula 1
Působiště autorů:
I. interná klinika JLF UK a UNM, Martin, Slovenská republika
1; Endokrinologické oddelenie Národného endokrinologického a diabetologického ústavu Ľubochňa, Slovenská republika
2; Alphamedical, s. r. o., Bratislava, Slovenská republika
3
Vyšlo v časopise:
Vnitř Lék 2019; 65(1): 51-54
Kategorie:
Kazuistiky
Souhrn
Akromegália je zriedkavé ochorenie, ktorého najčastejšou príčinou je adenóm hypofýzy. V dôsledku dlhodobej elevácie rastového hormónu (RH) a inzulínu podobného rastového faktora 1 (IGF1) pri tomto ochorení dochádza k rozvoju komplikácií ako kardiomyopatia, diabetes mellitus, syndróm spánkového apnoe a artropatia. Incidencia akromegálie je 3–4 pacienti na 1 000 000 za rok. Klinefelterov syndróm (KS) je najčastejšou príčinou mužského hypogonadizmu, vyskytujúcou sa u približne 1 z 500 živo narodených chlapcov. Medzi klinické charakteristiky KS patria najmä malé semenníky, spomedzi ďalších príznakov je to vysoká postava, znížený svalový tonus, oneskorená puberta s nedostatočným vývojom sekundárnych pohlavných znakov a gynekomastia. V našej kazuistike prezentujeme 32-ročného muža s kombináciou akromegálie a 47,XXY Klinefelterovho syndrómu. Pacient klinicky s typickými akromegalickými znakmi. Laboratórne bola potvrdená vysoká hladina rastového hormónu bez supresie v orálnom glukózovom tolerančnom teste, vysoká hladina IGF1, nízka hladina testosterónu s vysokými hladinami luteinizačného hormónu (LH) a folikuly stimulačného hormónu (FSH). Doplnená magnetická rezonancia s nálezom makroadenómu hypofýzy veľkosti 25 × 18 × 18 mm. Na základe uvedeného bola potvrdená diagnóza akromegálie. Následne realizovaná transsfenoidálna resekcia makroadenómu hypofýzy. Histopatologické a imunohistochemické nálezy odhalili rastový hormón produkujúci adenóm hypofýzy. Prítomnosť klinických znakov – malých, tuhých semenníkov, nedostatku sekundárnych pohlavných mužských znakov, ako aj laboratórne vyšetrenia suponovali hypergonadotropný hypogonadizmus, ktorý nemohol byť vysvetlený diagnózou akromegálie. Chromozomálny karyotyp 47,XXY potvrdil diagnózu KS. Substitučná liečba testosterónom nebola zahájená z dôvodu nesúhlasu pacienta. Pooperačne pretrvávala zvýšená plazmatická koncentrácia RH a IGF1. Bola zahájená liečba somatostatínovými analógmi (lanreotid) v dávke 120 mg každých 28 dní. Kontrolná magnetická rezonancia hypofýzy preukázala reziduum veľkosti 14 × 14 × 7 mm. Aktuálne je pacient po endoskopickej revízii rezidua.
akromegália – IGF1 – Klinefelterov syndróm – rastový hormón – testosterón
Úvod
Klinefelterov syndróm (KS) je najčastejšou chromozomálnou aberáciou. Až 80 % postihnutých má karyotyp 47,XXY. V dôsledku nedostatočne vyjadrených príznakov prepubertálne zostáva často KS v tomto období nediagnostikovaný [1]. Najčastejšími dôvodmi vedúcimi k diagnóze sú najmä infertilita a malé testes. Ďalšie príznaky, ktoré môžu byť prítomné sú vysoká postava, gynekomastia, prejavy hypoandrogenizácie, oneskorená puberta, problémy s učením [2]. Laboratórnym nálezom pri KS je hypergonadotropný hypogonadizmus s vyššou hladinou FSH ako LH. Diagnóza je založená na overení karyotypu. Androgénna nedostatočnosť sa lieči substitúciou testosterónom.
Akromegália je ochorenie charakterizované hypersekréciou rastového hormónu najčastejšie na podklade adenómu hypofýzy. Klinický obraz ochorenia sa rozvíja postupne v priebehu niekoľkých rokov až desaťročí. Medzi najčastejšie príznaky a symptómy akromegálie patrí zvýšené potenie, parestézie, dysmorfia, artralgie, cefalea, slabosť, syndróm karpálneho tunela a poruchy zraku. Diagnostika sa zakladá na potvrdení vysokej hladiny rastového hormónu, najmä jeho nesupresibilite v orálnom glukózovom tolerančnom teste ako aj vysokej hladine IGF1. Zo zobrazovacích vyšetrení má dominantné postavenie MRI hypofýzy a celej selárnej oblasti. V súčasnosti sú 3 možnosti liečby akromegálie, a to chirurgická, rádioterapia a farmakologická liečba.
Kazuistika
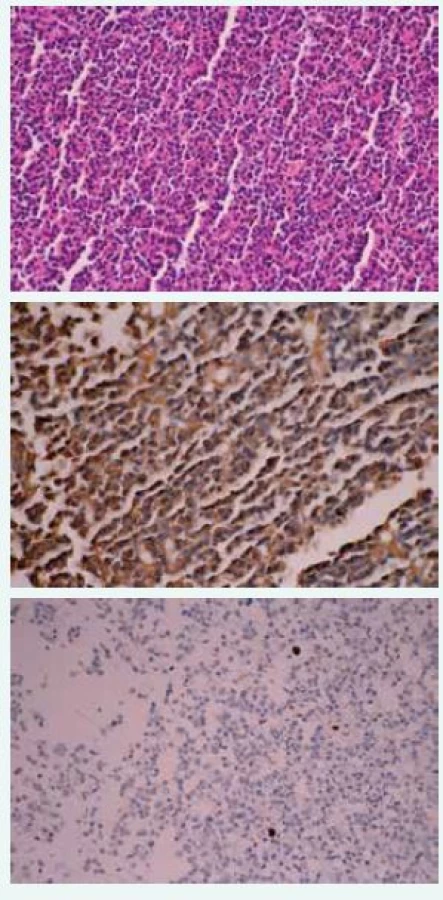
Uvádzame prípad 32-ročného muža (narodený v roku 1985) s kombináciou akromegálie a 47, XXY Klinefelterovho syndrómu. Pacient bol prijatý na oddelenie endokrinológie Národného endokrinologického a diabetologického ústavu v Ľubochni s klinickými prejavmi, a to vysokou postavou (telesná výška 195 cm, hmotnosť 90 kg), zväčšenými akrálnymi časťami tela (ruky, nohy, nos, nadočnicové oblúky, jazyk), opuchmi prstov na rukách. Z celkových klinických symptómov dominovala bolesť hlavy, zvýšené potenie, tŕpnutie prstov rúk. Laboratórne testy odhalili vysokú hladinu rastového hormónu bez supresie v orálnom glukózovom tolerančnom teste, vysokú hladinu IGF1, IGFBP3 (IGF viažuci proteín 3), nízku koncentráciu testosterónu s vysokými koncentráciami luteinizačného hormónu (LH) a folikulostimulačného hormónu (FSH), tab. Na základe uvedeného bola potvrdená diagnóza akromegálie. Perimeter bol bez výpadku v zornom poli, magnetická rezonancia s nálezom intraselárneho tumoru hypofýzy heterogénnej štruktúry s dominantnou cystoidnou komponentou s dediferenciáciou tkaniva adenohypofýzy a neurohypofýzy s deviáciou infundibula doľava s odtlačením paraselárnych cievnych štruktúr vs makroadenóm hypofýzy veľkosti 25 × 18 × 18 mm (obr. 1). Doplnené USG abdomenu bez nálezu hepatomegálie, echokardiografia bez prítomnosti kardiomyopatie, denzitometria v norme pre daný vek a pohlavie. Následne bola vykonaná transsfenoidálna resekcia makroadenómu hypofýzy. Histopatologické a imunohistochemické nálezy odhalili tumor so solídnym rastom, bez výraznejších nukleárnych atypií so stratou retikulínu, produkujúci rastový hormón, prolaktín slabo/sporne pozitívny, FSH, tyreoidálny stimulačný hormón (TSH), LH a adrenokortikotropný hormón (ACTH) negatívny. Proliferačná aktivita (index Ki-67) menej ako 1 %, E-kadherín pozitívny, P-53 negatívny, CHRA pozitívny (chromogranín A), SY-38 pozitívny (obr. 2). Prítomnosť klinických znakov, a to malých, tuhých semenníkov, nedostatku sekundárnych pohlavných mužských znakov (riedke telesné ochlpenie) ako aj laboratórne vyšetrenia, ktoré potvrdili hypergonadotropný hypogonadizmus, suponovali možný Klinefelterov syndróm. Pacient neuvádzal problémy so sexuálnymi funkciami, klinicky penis primeranej veľkosti, bez prítomnosti gynekomastie. Bolo realizované USG vyšetrenie testes s dlhou osou 28 mm, bez ložiskových zmien, bez prítomnosti patologického obsahu v skróte. Vyšetrenie spermiogramu potvrdilo azoospermiu. Na základe cytogenetického vyšetrenia z kultivovaných buniek periférnej krvi bol stanovený karyotyp 47,XXY, ktorý potvrdil diagnózu KS. Substitučná liečba testosterónom nebola zahájená z dôvodu nesúhlasu pacienta. Pooperačne pretrvávala zvýšená plazmatická koncentrácia hladín RH bez supresability v orálnom glukózovom tolerančnom teste ako aj elevovaná plazmatická koncentrácia IGF1 a IFGBP3. Bola zahájená liečba somatostatínovými analógmi (lanreotid) v dávke 120 mg každých 28 dní. Kontrolná magnetická rezonancia hypofýzy po 6 mesiacoch od operačného výkonu preukázala solídno-cystickú léziu s významnou regresiou veľkosti viac než 50 % v porovnaní s MRI vyšetrením pred zákrokom vs reziduum veľkosti 14 × 14 × 7 mm (obr. 3). Následne bola po chirurgickej konzultácií realizovaná endoskopická revízia rezidua. Pozákrokovo došlo k znormalizovaniu plazmatických hladín rastového hormónu, IGF1, IGF BP3, čo poukázalo na priaznivý efekt chirurgickej liečby. Liečba somatostatínovými analógmi bola vysadená. V súčasnosti je po súhlase pacienta zahájená substitučná liečba testosterónom (testosterón-undekanoát) i.m. v dávke 1 000 mg à 3 mesiace.



Diskusia
V našej kazuistike popisujeme prítomnosť makroadenómu hypofýzy produkujúceho rastový hormón u pacienta s Klinefelterovým syndrómom, ktorý nebol dovtedy diagnostikovaný. Oneskorená diagnostika KS býva spôsobená rozmanitosťou v klinických prejavoch ochorenia ako aj absenciou vyšetrenia genitálu pri rutinnom klinickom vyšetrení. Včasná diagnostika ochorenia môže viesť k zlepšeniu kvality života pacientov s KS a predchádzať závažným následkom [3]. Náš pacient mal diagnostikovaný KS na základe prítomnosti malých semenníkov, laboratórne verifikovaného periférneho hypogonadizmu (nízka plazmatická koncentrácia testosterónu s elevovanou hladinou gonadotropínov) a následného genetického vyšetrenia s potvrdením chromozomálnej aberácie 47,XXY. Mužský hypogonadizmus sa delí na hypogonadotropný – centrálny a hypergonadotropný – periférny. Centrálny hypogonadizmus môže byť získaný, spôsobený léziami v oblasti hypotalamu a hypofýzy, alebo vrodený, vznikajúci v dôsledku genetických mutácií [4]. Najčastejšou príčinou periférneho hypogonadizmu je Klinefelterov syndróm. Incidencia KS je približne 1 z 500 živo narodených chlapcov. Až 80 % postihnutých má karyotyp 47,XXY, zvyšných 20 % má karyotyp iný (mozaika 46,XY/47,XXY, 48,XXXY, 48,XXYY, 49,XXXXY) [5]. Väčší počet pacientov s Klinefelterovým syndrómom zostáva nediagnostikovaných. Abramsky et al prezentovali výsledky, podľa ktorých 10 % prípadov býva identifikovaných prenatálne, 26 % je diagnostikovaných v detstve alebo v dospelosti z dôvodu hypogonadizmu, gynekomastie alebo neplodnosti, pričom 64 % zostáva nediagnostikovaných [6]. Hlavnou klinickou charakteristikou KS je nález malých, tuhých testes v dôsledku hyalinizácie semenotvorných kanálikov pri zvyčajne normálne vyvinutom vonkajšom mužskom genitále. 99 % pacientov s KS má azoospermiu vedúcu k infertilite. Môžu byť prítomné poruchy neurokognitívnych funkcií (oneskorený vývoj reči, znížené jazykové schopnosti, poruchy učenia), ako aj znížený intelekt. Fenotyp KS ovplyvňuje SHOX gén na chromozóme X. Gén je umiestnený v pseudoautozomálnej oblasti 1 na Xp. SHOX haplo insuficiencia je spájaná s retardáciou rastu kostí pri Turnerovom syndróme a dyschondrosteóze Leriho-Weilla [5]. Pacienti s KS mávajú vyššiu postavu, ktorá nie je dôsledkom deficitu androgénov, ale nadpočetného pohlavného chromozómu, a tým pádom aj tretej kópie SHOX génu, ktorý zohráva úlohu v raste dlhých kostí [7]. Habitus pri KS môže byť gynoidný s redukovaným ochlpením a slabšie vyvinutým svalstvom. Zvýšené koncentrácie LH stimulujú Leydigove bunky k zvýšenej sekrécii estradiolu a jeho prekurzorov. Vysoký pomer estradiol/testosterón má za následok feminizáciu a gynekomastiu rôzneho stupňa. Riziko rakoviny prsníka je u pacientov s KS 20-krát vyššie [7,8]. S KS sa spája zvýšené riziko autoimunitných ochorení ako Sjögrenov syndróm, systémový lupus erythematodes, Addisonova choroba, diabetes mellitus 1. typu, sclerosis multiplex, reumatoidná artritída a autoimunitná hypotyreóza [9]. Laboratórnym nálezom pri KS je hypergonadotropný hypogonadizmus s vyššou hladinou FSH ako LH. Diagnóza je založená na overení karyotypu. Androgénna nedostatočnosť sa lieči substitúciou testosterónom.
Primárnym dôvodom vyšetrenia nášho pacienta boli charakteristické známky akromegálie. Laboratórne bola potvrdená elevovaná hladina rastového hormónu s nesupresibilitou v oGTT teste s vysokou plazmatickou koncentráciou IGF1. Na MRI vyšetrení bol prítomný makroadenóm hypofýzy. Akromegália je zriedkavé ochorenie vznikajúce v 99 % na podklade adenómu hypofýzy, ktoré je charakterizované nadmernou sekréciou IGF1 vyvolanou nadprodukciou rastového hormónu. Kontogeorgos et al definovali adenómy hypofýzy vylučujúce gonadotropín ako tie, ktoré vykazujú hladinu expresie FSH, alebo LH o viac ako 10 % [10]. U nášho pacienta bolo imunohistochemické farbenie negatívne pre FSH ako aj LH. Gonadotropinóm alebo adenóm so zmiešanou tvorbou gonadotropínov a rastového hormónu sa nepotvrdil. Incidencia akromegálie sú 3–4 pacienti/milión za rok [11]. Od nástupu symptómov až po diagnostiku ochorenia je zvyčajne oneskorenie 5–10 rokov [12]. Diagnostika je založená na prítomnosti klinických prejavov ochorenia, laboratórnej diagnostike a zobrazovacích vyšetreniach. RH stimuluje fibroblastickú proliferáciu, ktorá spôsobuje zhrubnutie mäkkého tkaniva. Antiinzulínový a nátrium-retenčný efekt spôsobuje zvýšenie hladiny sérového nátria a vody. Táto retencia prispieva k opuchu mäkkých tkanív s rozvojom neuropatie (syndróm karpálneho tunela). V počiatočných štádiách ochorenia býva prítomný opuch prstov, hyperhidróza, zdrsnenie čŕt tváre, zvýšená unaviteľnosť a polyartralgie. Neskôr sa objavuje typická akromegalická dysmorfia, a to zhrubnuté pery, makroglosia, zväčšený nos a ušnice, prominencia mandibuly s rozostupom zubov, zvýraznené nadočnicové oblúky, zväčšenie rúk a chodidiel, artropatia. Pacienti mávajú často sprievodné ochorenia ako artériovú hypertenziu, porušenú glukózovú toleranciu, prípadne diabetes mellitus, syndróm spánkového apnoe, polypózu hrubého čreva [13]. Zo samotného rastu tumoru môže byť prítomná bolesť hlavy pri supraselárnom raste, poruchy zrakových nervov s následnými výpadkami zorného poľa. Laboratórna diagnostika je založená na potvrdení vysokej hladiny rastového hormónu, najmä jeho nesupresibilite v orálnom glukózovom tolerančnom teste a vysokej hladine IGF1. Zo zobrazovacích vyšetrení má dominantné postavenie magnetická rezonancia hypofýzy a celej selárnej oblasti. Možnosti liečby zahŕňajú chirurgický zákrok, farmakologickú liečbu a rádioterapiu. Transsfenoidálna chirurgia je prioritná liečba u pacientov s akromegáliou. Avšak u 40–60 % pacientov s akromegáliou sa po operácii vyskytne pretrvávajúce alebo rekurentné ochorenie, čo si vyžaduje ďalšiu liečbu [14]. Ak sa operačným výkonom nedosiahne biochemická kontrola ochorenia, pristupuje sa k stereotaktickej rádioneurochirurgii. Využíva sa najmä lineárny urýchľovač a Leksellov gama nôž. Nástup účinku tejto liečby môže trvať aj niekoľko rokov. Analógy somatostatínu sú liečbou prvej línie pre pacientov s akromegáliou. Ich predoperačné podávanie môže zvýšiť úspešnosť samotného chirurgického zákroku. Antagonisti receptora pre rastový hormón predstavujú alternatívu pri zlyhaní somatostatínovej liečby. Agonisti dopamínu majú doplnkový účinok najmä pri súčasnej sekrécií prolaktínu.
Záver
Koincidencia mnohých endokrinopatií je zriedkavá, ale stále možná. Jedna diagnóza, a to akromegália, nevylučuje iné endokrinologické ochorenie – Klinefelterov syndróm, ktorý pre nedostatočne vyjadrené príznaky, ako aj chýbanie vyšetrenia genitálu pri bežnom vyšetrení, zostáva v mnohých prípadoch nediagnostikovaný. Včasné rozpoznanie, diagnostika a manažment týchto ochorení sú dôležitými faktormi pre zlepšenie prognózy a kvality života pacientov.
MUDr. Ivana Ságová, PhD.
I. interná klinika JLF UK a UNM, Martin, Slovenská republika
Doručeno do redakce 4. 6. 2018
Přijato po recenzi 2. 7. 2018
Zdroje
- Groth KA, Skakkebaek A, Host C et al. Clinical review: Klinefelter syndrome – a clinical update. J Clin Endocrinol Metab 2013; 98(1): 20–30. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2012–2382>.
- Benaiges D, Pedro-Botet J, Hernandez E et al. Different clinical presentation of Klinefelter‘s syndrome in monozygotic twins. Andrologia 2015; 47(1): 116–120. Dostupné z DOI: <http://dx.doi.org/10.1111/and.12219>.
- Sabir AA, Zagga AD, Sahabi SM et al. Klinefelter‘s syndrome: report of a case from Sokoto, Northern Nigeria and review of literature. Sahel Med J 2013; 16(1): 32–34. Dostupné z DOI: <http://dx.doi.org/10.4103/1118–8561.112070>.
- Marek J, Hana V et al. Endokrinologie. Galén: Praha 2017. ISBN 978–80–7262–484–3.
- Ottesen AM, Aksglaede L, Garn I et al. Increased number of sex chromosomes affects height in a nonlinear fashion: a study of 305 patients with sex chromosome aneuploidy. Am J Med Genet A 2010; 152A(5): 1206–1212. Dostupné z DOI: <http://dx.doi.org/10.1002/ajmg.a.33334>.
- Abramsky L, Chapple J. 47,XXY (Klinefelter syndrome) and 47, XYY: estimated rates of and indication for postnatal diagnosis with implications for prenatal counselling. Prenat Diagn 1997; 17(4): 363–368.
- Veselý O. Opoždená puberta a gynekomastie jako projev Klinefelterova syndromu. Pediatr Praxi 2017; 18(2): 131–134.
- Germine LD. Klinefelter syndrome clinical presentation. History, Physical, Causes. MedScape 2018. Dostupné z WWW: https://emedicine.medscape.com/article/945649-clinical.
- Liberato D, Granato S, Grimaldi D et al. Intelligence, traits of personality and personality disorders in a cohort of adult KS patients with the classic 47, XXY karyotype. J Endocrinol Invest 2017; 40(11): 1191–1199. Dostupné z DOI: <http://dx.doi.org/10.1007/s40618–017–0674–2>.
- Kontogeorgos G, Kovacs K, Horvath E et al. Null cell adenomas, oncocytomas, and gonadotroph adenomas of the human pituitary: An immunocytochemical and ultrastructural anafysis of 300 cases. Endocr Pathol 1993; 4: 20. Dostupné z DOI: <https://doi.org/10.1007/BF02914485>.
- Capatina C, Wass JH. 60 Years Of Neuroendocrinology: Acromegaly. J Endocrinol 2015; 226(2): 141–160. Dostupné z DOI: <http://dx.doi.org/10.1530/JOE-15–0109>.
- Roelfsema F. Treatment of acromegaly: Are we satisfied with the outcome? EBioMedicine 2014; 2(1): 5–6. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ebiom.2014.12.010>.
- Biju Baby J, Veena SN, Jaishankar HP et al. Acromegaly – a case report. J Clin Biomed Sci 2012; 4(1): 247–250.
- Melmed S, Colao A, Barkan A et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab 2009; 94(5): 1509–1517. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2008–2421>.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2019 Číslo 1
- Není statin jako statin aneb praktický přehled rozdílů jednotlivých molekul
- S profesorem Vladimírem Tesařem o léčbě CKD dle aktuálních doporučení: praktický lékař jako klíč k renoprotekci
- Porovnání empagliflozinu a dapagliflozinu v léčbě srdečního selhání
- Jak souvisí index systémového zánětu s kardiovaskulární mortalitou pacientů s hypertenzí?
Nejčtenější v tomto čísle
- Kreatin ve vnitřním lékařství a jeho vliv na ledvinné funkce – editorial
- Systémová zánětlivá reakce s vysokými hodnotami CRP jako dominantní příznak mnohočetného myelomu
- Diagnostická úskalí celiakie
- Neceliakální glutenová/pšeničná senzitivita: stále více otázek než odpovědí