
Vybrané závažné „hematologické“ syndromy u dospělých pacientů v intenzivní péči
Selected severe „haematological“ syndromes in adult intensive care patients
Haemophagocytic syndrome, diffuse alveolar haemorrhage, catastrophic antiphospholipid syndrome and various types of thrombotic microangiopathies are rare conditions with significant morbidity and mortality. A common feature is late diagnosis, which can affect the success of treatment. The aim of this review article is to summarize the basic diagnostic and therapeutic steps of the present subpopulation of critically ill patients.
Keywords:
vasculitis – thrombotic thrombocytopenic purpura – thrombotic microangiopathy – plasma exchange – thrombosis – immunosuppressive therapy – diffuse alveolar haemorrhage – rituximab – eculizumab – hemophagocytic lymphohistiocytosis – antiphospholipid syndrome – hemophagocytic syndrome – HLH – macrophage activation syndrome – cytokine storm syndrome – catastrophic antiphospholipid syndrome – haemolytic‑uremic syndrome – etoposide – recombinant factor VIIa – caplacizumab – ravulizumab
Autoři:
Jaromír Gumulec 1,4; Ivo Demel 1; Klára Lančová 1; Eva Drbohlavová 2; Alicia Piegzová 3; Zdeněk Kořístek 1,4; Milan Navrátil 1,4; Vladimír Černý 5
Působiště autorů:
Klinika hematoonkologie Fakultní nemocnice Ostrava
1; Klinická hematologie Krajské nemocnice Liberec, a. s.
2; Gynekologicko‑porodnická, klinika Fakultní nemocnice Ostrava
3; Lékařská fakulta Ostravské univerzity
4; Klinika anesteziologie, perioperační a intenzivní medicíny Fakulty zdravotnických studií Univerzity J. E. Purkyně, v Ústí nad Labem a Krajské zdravotní, a. s. – Masarykovy nemocnice v Ústí nad Labem, o. z.
5
Vyšlo v časopise:
Vnitř Lék 2022; 68(8): 498-507
Kategorie:
Hlavní téma
doi:
https://doi.org/10.36290/vnl.2022.107
Souhrn
Hemofagocytární syndrom, difuzní alveolární hemoragie, katastrofický antifosfolipidový syndrom a různé typy trombotických mikroangiopatií patří mezi vzácné stavy s významnou morbiditou a mortalitou. Společným rysem bývá pozdní stanovení diagnózy, což může ovlivnit úspěšnost léčby. Cílem přehledného článku je shrnutí základních diagnostických a léčebných kroků předmětné subpopulace kriticky nemocných.
Klíčová slova:
trombóza – vaskulitida – rituximab – trombotická trombocytopenická purpura – eculizumab – imunosupresivní terapie – trombotická mikroangiopatie – difuzní alveolární hemoragie – antifosfolipidový syndrom – hemofagocytární syndrom – hemofagocytární lymfohistiocytóza – HLH – syndrom z aktivace makrofágů – syndrom z uvolnění cytokinů – katastrofický antifosfolipidový syndrom – hemolyticko‑uremický syndrom – etopozid – výměnná plazmaferéza – rekombinantní aktivovaný FVII – kaplacizumab – ravulizumab
Úvod
Hemofagocytární syndrom, difuzní alveolární hemoragie, katastrofický antifosfolipidový syndrom a různé typy trombotických mikroangiopatií patří mezi vzácné stavy s významnou morbiditou a mortalitou. Protože se vyskytují vzácně, často unikají pozornosti lékařů a bývají diagnostikovaná pozdě. Navíc zpravidla chybí data o léčbě z randomizovaných klinických studií. Publikované zkušenosti obvykle pocházejí z jednotlivých kazuistických sdělení nebo z malých souborů pacientů, jen výjimečně z klinických registrů. Cílem přehledu je shrnout základní pravidla včasné diagnostiky a iniciální léčby pacientů s těmito chorobami.
Hemofagocytární syndrom
Stručný popis případu: 63letý polymorbidní muž s prohlubující s trombocytopenií (71 × 109/L) a leukopenií (1,7 × 109/L) zjištěnou přibližně dva měsíce před plánovanou implantací mechanické náhrady aortální chlopně. Po operaci došlo k rozvoji sepse komplikované těžkým krvácením díky trombocytopenii a antikoagulaci warfarinem. Laboratorně dominovala leukopenie (0,16 × 109/L), trombocytopenie (14 × 109/L), anémie (koncentrace hemoglobinu po masivní substituci 98 g/L), hyperferitinemie (4116,8 ug/L), elevace aktivity transamináz a laktátdehydrogenázy (LDH 5,07 ukat/L), hyperbilirubinemie (29,4 umol/L) a zvýšení koncetrace CRP (77 mg/L). Na CT byla popsána splenomegalie. Pacient byl zajištěn empiricky antibiotiky (mikrobiologicky jen PCR pozitivita viru Epstein‑Barrové (EBV) a pro riziko krvácení kontinuální aplikací profylaktické dávky LMWH.
Vzhledem k anamnéze užívání mesalazinu pro Crohnovu nemoc byl zvažován polékový útlum krvetvorby. Postupně byly vyloučeny heparinem indukovaná trombocytopenie, trombotická mikroangiopatie, paroxysmální noční hemoglobinurie, systémová autoimunitní onemocnění. V aspirátu kostní dřeně byla těžká hypocelularita bez zmnožení blastů, mírné dysplastické změny, četnější histiocyty, ojediněle fagocytóza normoblastů a cytogeneticky delece genu ETV6. Diferenciálně diagnosticky byla proto nově zvažována hemofagocytóza, hypoplastická forma myelodysplastického syndromu a aplastická anémie.
V dalším průběhu se rozvinula hypofibrinogenemie (1,1 g/L), zvýraznila hyperferitinemie (14357,1 ug/L), prokázali jsme hypertriacylglycerolemii (3,97 mmol/L) a trval febrilní stav. Teprve výsledek druhého čtení histologie z trepanobiopsie potvrdil přítomnost makrofágů s fagocytovanými krevními elementy a zásadně přispěl k závěru hemofagocytární lymfohistiocytóza (dohromady naplněno 6 z 8 kritérií HLH-24, resp. HScore 249 bodů). Léčba dexametazonem a etopozidem podle protokolu HLH - 94 měla jen dílčí a přechodný účinek bez úpravy cytopenie. Přidaly se nové infekční komplikace a difuzní hemoragická kolitida refrakterní na i.v. imunoglobuliny a podpůrnou péči. K úpravě nedošlo ani po léčbě alemtuzumabem a pacient zemřel pod obrazem multiorgánového selhání.
Hemofagocytární syndrom (hemofagocytární lymfohistiocytóza, HLH) je vzácné onemocnění s významným uvolněním cytokinů, aktivací makrofágů, cytotoxických T lymfocytů a NK buněk vedoucí k multiorgánovému selhání (MODS) a smrti (1, 2). Mortalita dospělých pacientů se pohybuje v rozmezí od 26,4 do 74,8 % (1).
HLH se dělí na primární a sekundární. Primární HLH se nejčastěji objevuje u dětí s genetickou predispozicí, sekundární u dospělých v souvislosti s infekcí, malignitami a autoimunními nemocemi. V patogenezi HLH dospělých pacientů hraje významnější roli neadekvátní odpověď na spouštěcí faktory spíše než vrozená vloha. Spouštěče HLH je nutné aktivně vyhledávat, protože nepoznané a neléčené mohou udržovat aktivitu nebo rekurenci HLH (viz Tab. 1) (2).

U řady dospělých pacientů se HLH manifestuje triádou horečka + bicytopenie s krvácivými projevy + splenomegalie. Kromě toho mohou HLH pacienti mít kožní exantém, bolesti kloubů, hepatomegalii a lymfadenopatii, v rozvinutém stavu otoky, dušnost, průjem, obraz napodobující sepsi (2). V diagnostice se využívají kritéria HLH-2004 (viz Tab. 2) (2) nebo tzv. HScore (HLH‑probability calculator dostupný na https://www. mdcalc.com/hscore‑reactive‑hemophagocytic‑syndrome – viz Tab. 2). Ve srovnání s HLH-2004 má HScore u dospělých HLH pacientů vyšší sensitivitu (100 %, resp. 90 %) a specificitu (80 %, resp. 79 %) (3).

Morfologický průkaz hemofagocytózy je nespecifický, lze jej najít u dalších kriticky nemocných pacientů bez HLH a pomůže jen v případech se silným klinickým podezřením.
Nicméně závěr HLH bez morfologického nálezu hemofagocytózy je třeba dělat opatrně a event. odběr materiálu opakovat. Prevalence morfologického nálezu
hemofagocytózy v aspirátu kostní dřeně osciluje mezi 25 a 100 %
Na rozdíl od dětských pacientů není u dospělých všeobecně vyžadováno funkční vyšetření cytotoxicity lymfocytů nebo molekulárně genetické vyšetření. Tyto testy se doporučuje provádět u pacientů s pozitivní rodinnou anamnézou HLH, s albinismem, s relabujícím onemocněním, u mladých mužů s lymfoproliferací asociovanou s infekcí EBV a v případech bez jasného spouštěče (2).
V terapii dospělých pacientů s HLH je doporučován tzv. protokol HLH-94 s event. intratekální aplikací etopozidu (trvá‑li neurologická symptomatologie nebo patologický nález v mozkomíšním moku) nebo alogenní transplantací krvetvorných buněk (AlloTx – primární nebo relabující HLH, HLH u vybraných hematologických malignit). Vzhledem k heterogenitě HLH u dospělých pacientů je doporučováno vést léčbu individuálně a zohlednit charakter základního onemocnění (infekce, nádory, autoimunitní nemoci…) – viz tabulka 3 (2).

Zásadní informace pro klinickou praxi: Nevysvětlená cytopenie v jedné nebo více řadách u pacienta s projevy systémové zánětové odpovědi, splenomegalií, hyperferitinemií, hypofibrinogenémií nereagující na antibiotickou léčbu a progredující do MODS má vést k podezření na HLH.
Difúzní alveolární hemoragie
Difúzní alveolární hemoragie (DAH) je vzácný život ohrožující klinický syndrom manifestovaný difuzním krvácením z acinární části plic, nově vzniklými plicními infiltráty na rentgenovém snímku plic a anemií (4).
Mortalita hospitalizovaných pacientů kolísá mezi 20 a 50 %, je zvýšena u pacientů s hemoragickým šokem, vysokou aktivitou LDH, akutní poruchou funkce ledvin, ve skupině imunitně podmíněných DAH a je‑li diagnostikována pozdě (5). I po zvládnutí akutní fáze zůstává úmrtnost vysoká (kolem 16 %).
DAH syndromy se dělí do čtyř skupin: 1) imunitně podmíněná DAH, 2) DAH při městnavém srdečním selhání, 3) heterogenní skupina DAH různého původu a 4) idiopatická DAH – viz tabulka 4 (6).
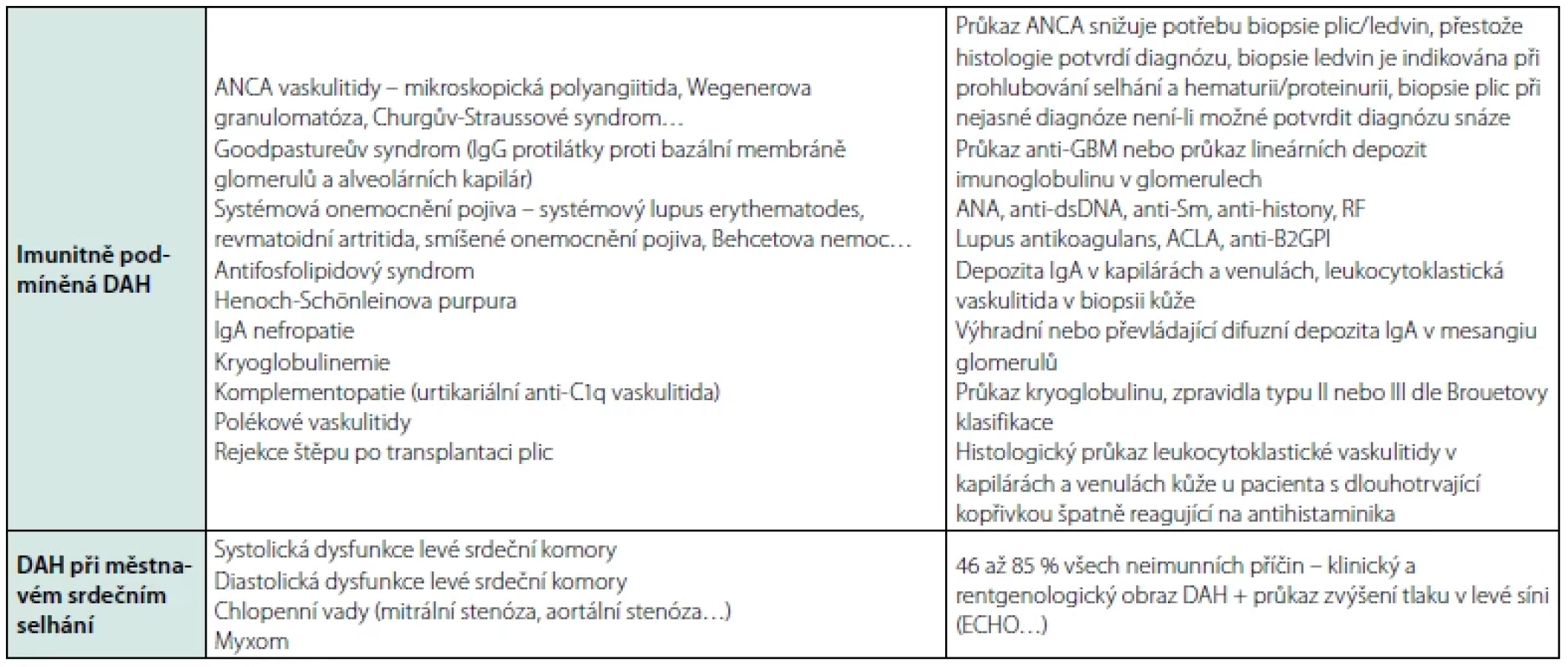

Hlavní patologické nálezy u DAH jsou protilátkami zprostředkovaná plicní kapilaritida, difuzní alveolární poškození a alveolární krvácení – viz tabulka 5 (7, 8).

Vedle klasické triády provází DAH nespecifické projevy jako kašel, dušnost a horečka. Asi u 30–40 % nemocných se hemoptýza objevovat nemusí (9). Pacienti s imunitně podmíněnou DAH mívají častěji postižení kůže, kostí, kloubů, oblasti nosu a hrtanu a/nebo ledvin (9). DAH různého původu a DAH u systémového lupus erythematodes (SLE) se rozvíjí prudce, u ostatních imunitně podmíněných DAH je rozvoj obvykle subakutní (9). S prudkým rozvojem souvisí méně častý nález siderofágů při bronchoalveolární laváži (BAL) (8). Naproti tomu pacienti s idiopatickou DAH mívají BAL na siderofágy bohatou, což může být důsledek chronického onemocnění.
Diferenciální diagnostika DAH je široká – viz tabulka 4. Pro potvrzení diagnózy DAH a vyloučení jiné příčiny krvácení je rozhodující vyšetření BAL provedené během prvních 48 hodin (7). Podíl siderofágů v BAL ≥ 20 % je pro DAH diagnostický. Role transbronchiální biopsie je vzhledem k nerovnoměrnému rozložení postižených úseků nejistá, proto je u všech pacientů s DAH nejasného původu doporučováno provedení plicní biopsie (10). Na rentgenovém snímku srdce a plic bývají fokální nebo difuzní bilaterální alveolární opacity nebo konsolidace především bazálně. Rekurentní epizody mohou vést k fibróze s retikulární intersticiální kresbou. U 20–50 % případů akutní DAH může být RTG obraz negativní. Počítačová tomografie (CT) hrudníku ozřejmí detaily a zpřesní diferenciální diagnostiku. Echokardiografie srdce (ECHO) přispěje k vyloučení onemocnění srdce (mitrální stenóza).
Kromě obecných postupů orgánové podpory (podpora oběhu, umělá plicní ventilace apod.) je součástí léčby podání imunosupresiv ke kontrole aktivity základního onemocnění, výměnná plazmaferéza (PEX) k odstranění autoprotilátek a lokální hemostáza rekombinantním aktivovaným FVII (rFVIIa). Je ‑ li DAH projevem systémového onemocnění, může včasné zahájení cílené terapie kortikosteroidy zabránit rozvoji postižení ledvin (5). U pacientů s neimunními typy DAH (kardiální příčiny, infekce) není imunosupresivní terapie indikována (9).
Ke kontrole zánětlivé aktivity jsou indikované vysoké dávky kortikosteroidů co nejdříve od stanovení diagnózy (10). Methylprednisolon i.v. 500 mg až 2 g/den nebo 30 mg/kg/den po dobu 3–5 dní s postupným vysazováním během čtyřech týdnů. Přes tuto terapii umírá v akutní fázi více než 50 % nemocných. Nízce dávkovaný methylprednisolon (< 250 mg/den) má signifikantně menší mortalitu v akutní fázi bez vlivu na celkovou úmrtnost.
Výměnná plazmaferéza je užitečná k odstranění autoprolilátek et cetera u vybraných onemocnění – anti‑GBM u Goodpastureova syndromu, ANCA u ANCA vaskulitid a nejrůznějších protilátek u SLE (11). Podle americké společnosti pro aferézu je u ANCA‑DAH PEX indikovaná při hypoxemické respirační insuficienci vyžadující high‑flow oxygenaci nebo mechanickou ventilaci (12).
Rituximab je efektivní u DAH provázející autoimunitní onemocnění. U těžké ANCA vaskulitidy je rituximab srovnatelně účinný jako cyklofosfamid, nicméně efektivněji snižuje rekurenci, rozvoj terminální renální insuficience nebo alveolární hemoragie (13). Rituximab má lepší dlouhodobý efekt než udržovací azathioprin.
Podmínkou rychlé a funkční hemostázy je úprava trombocytopenie a koagulopatie. K dispozici jsou destičkové transfuzní přípravky, plazma nebo Octaplas LG®, tranexamová kyselina i.v. a/nebo intrapulmonálně i jako aerosol a rFVIIa. Pro dosažení účinné dávky rFVIIa v alveolárním prostoru je nutné udržovat vysokou koncentraci v systémové cirkulaci, což může zvýšit riziko trombotických komplikací (14). Intrapulmonálně aplikovaný rFVIIa je účinný i v menší dávce (50 ug/kg) a nižší frekvenci aplikací (14). Podle publikovaných zkušeností lze použít i.v. aplikace 35 - 200 ug/kg jednorázově nebo opakovaně v intervalu 2–4 hodin, resp. intrapulmonální aplikace v rámci bronchoskopie s celkovou dávkou 50–90 ug/kg rFVIIa ředěného ve fyziologickém roztoku jednorázově nebo při opakovaném krvácení opakovaně po 24 hodinách (14).
Zásadní informace pro klinickou praxi: Nevysvětlená hemoptýza s nově vzniklými plicními infiltráty na snímku plic při současném zhoršování klinického stavu má vést k podezření na DAH.
Katastrofický antifosfolipidový syndrom
Stručný popis případu: 36letý muž po ischemické mozkové příhodě užíval protidestičkovou léčbu acetylsalicylovou kyselinou (ASA) a pro hypertenzi telmisartan. Vyšetřením byl prokázán lupus antikoagulans, vysoký titr antifosfolipidových protilátek (antikardiolipinové protilátky IgG 467 GPL/mL, anti‑B2GPI IgG nad 532 U/mL) a trombocytopenie kolem 100 × 109/L. Pro vysoké riziko byl k ASA přidán warfarin s cílovým rozmezím 2,0–3,0 INR (International Normalized Ratio). Po měsíci léčby se po úderu do hlavy při INR 3,9 rozvinul subdurální hematom s nutnou kraniotomií a evakuací hematomu. Týden po operaci došlo ke zhoršení stavu s progresí trombocytopenie 90 × 109/L, rozvojem poruchy funkce ledvin (koncentrace kreatininu 170 μmol/L) a systémové zánětlivé odpovědi (C reaktivní protein 183 mg/L). Laboratorním vyšetřením byla mj. vyloučena heparinem indukovaná trombocytopenie. V následujících dnech se rozvinula mikroangiopatická hemolytická anémie a hepatorenální insuficience (kreatinin 262 μmol/L, ALT 7,54 μkat/L, AST 5,56 μkat/L, GMT 15,5 μkat/L). Pro uvažovaný katastrofický antifosfolipidový syndrom (CAPS) byla zahájena série PEX a léčba kombinací prednisonu (1 mg/kg/den) s cyklofosfamidem (50–100 mg/ den). Série čtyř aferéz s podáním celkem 100 balení Octaplas LG® byla ukončena po vzestupu počtu destiček na 148×109/l, odeznění hemolýzy, poklesu koncentrace kreatininu na 149 μmol/l a normalizaci aktivity transamináz. V antitrombotické profylaxi bylo pokračováno kombinací ASA a vyšší profylaktické dávky nízkomolekulárního heparinu (LMWH). Cyklofosfamid byl vysazen pro jaterní toxicitu a postupně byl vysazován prednison. Po ukončení imunosuprese je léčen kombinací ASA 100 mg/ den s enoxaparinem 0,4 mL/den. Klinicky i laboratorně je bez známek trombotické mikroangiopatie, hepatopatie a nefropatie, trvá přítomnost antifosfolipidových protilátek (lupus antikoagulans, ACLA IgG 317 GPL/ mL, anti‑B2GPI IgG nad 499 U/mL). V průběhu léčby CAPS se stal otcem a těší se ze svého syna.
Antifosfolipidový syndrom (APS) je heterogenní autoimunitní onemocnění charakterizované současnou přítomností antifosfolipidových protilátek a žilních nebo tepenných trombotických příhod nebo porodnických komplikací (15). Katastrofický antifosfolipidový syndrom je život ohrožující manifestace APS s prudkým rozvojem trombotického postižení predominantně drobných cév u současně třech a více orgánů nebo tkání během jednoho týdne s možným rozvojem MODS, typicky bez trombóz velkých cév a mortalitou až 50 % (16). Objevuje se u 1 % pacientů s APS a až ve 40 % případů může být jeho první manifestací.
Antifosfolipidové protilátky, obzvláště anti‑B2GPI mohou vést k protrombotickému nastavení různými cestami – aktivací endotelu, destiček, monocytů, neutrofilů, systému adhezivních molekul a/nebo prozánětlivých cytokinů, spotřebou oxidu dusného, zásahem do dráhy tkáňového faktoru, fibrinolýzy, aktivací komplementu nebo NETózy. CAPS může být spouštěn infekcí, graviditou, operacemi, malignitami nebo jiným autoimunitním onemocněním, které mohou vést k poškození endotelu a/nebo CRS a/nebo k syndromu systémové zánětlivé odpovědi.
Kombinace antikoagulační léčby (nízkomolekulární heparin s cílovým rozmezím 0,6–1,0 IU/mL nebo nefrakcionovaný heparin s cílovým anti‑Xa 0,3–0,7 IU/mL) s intravenózně podávanými kortikosteroidy (methylprednisolon 500–100 mg/den po dobu 3–5 dní), výměnnou plazmaferézou (start při podezření na CAPS, podle léčebné odpovědi nejméně 5 dní po sobě) a/nebo intravenózně podávanými imunoglobuliny (IVIG – 1 g/kg/den po dobu až třech dnů) je spojována s přežitím 69–78 % pacientů.
Pro pacienty s CAPS při SLE je doporučován cyklofosfamid 750 mg/ m2 v měsíčních intervalech po dobu aktivity nemoci nebo do neakceptovatelné toxicity. Hydroxychloroquin, sirolimus, rituximab nebo eculizumab jsou indikované u refrakterní choroby. Návrh terapeutického algoritmu je uveden v tabulce číslo 6 (17).
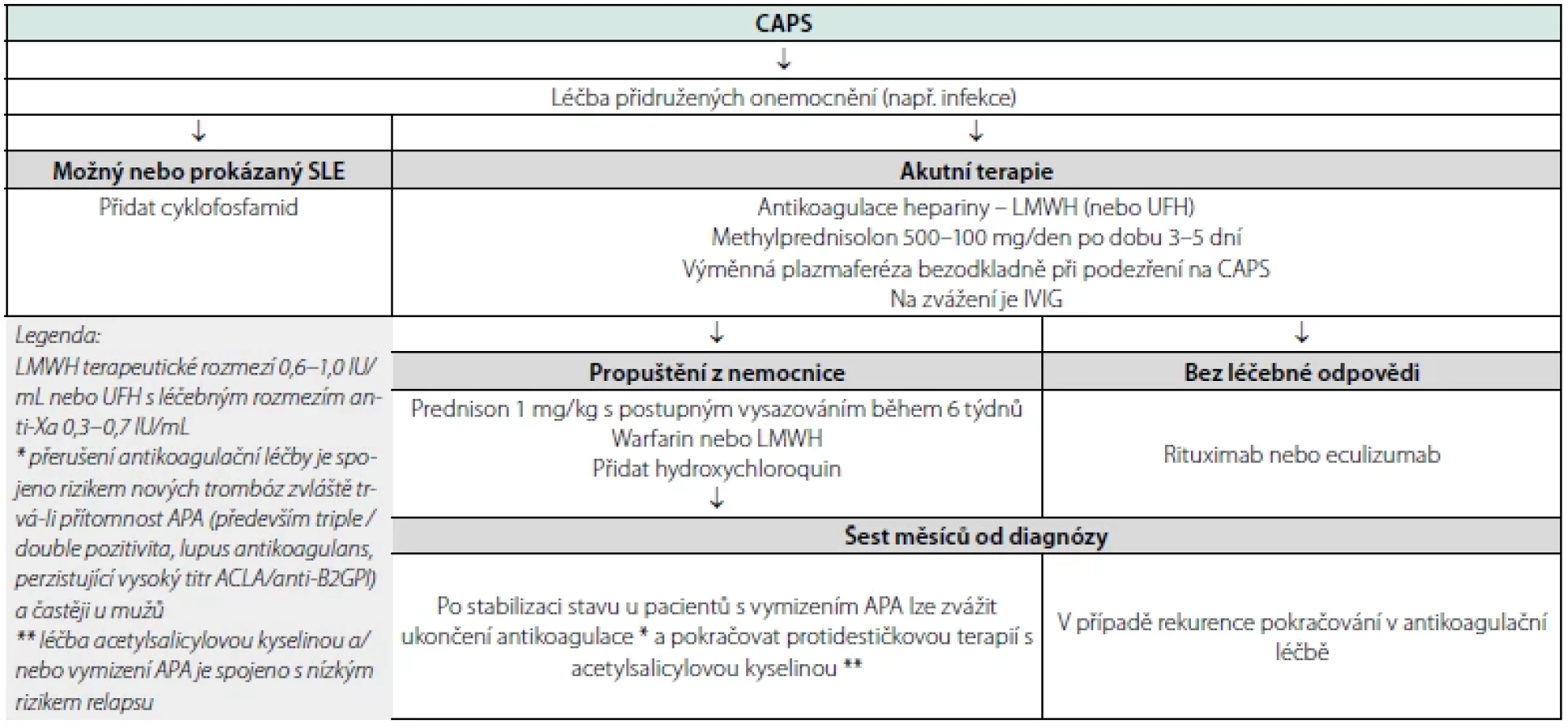
Zásadní informace pro klinickou praxi: Nevysvětlený rychlý rozvoj (multi)orgánového selhání, zvláště u pacientů s anamnézou žilních nebo tepenných trombóz a/nebo se známou přítomností antifosfolipidových protilátek má vést k podezření na CAPS.
Trombotické mikroangiopatie
Stručný popis případu: 39letá těhotná byla přijata na porodnici ve 37. týdnu těhotenství s gestační hypertenzí a známkami intrauterinní restrikce růstu plodu s vysokým poměrem sFlT-1/PlGF 137. Uzavřeno jako gestační hypertenze a těhotenství bylo ukončeno akutním císařským řezem pro hrozící hypoxii plodu a abrupci placenty. Přibližně čtyři hodiny po nekomplikovaném císařském řezu se u pacientky prudce rozvinul syndrom akutní respirační tísně, těžká porucha vědomí, anurické akutní poškození ledvin, trombocytopenie (trombocyty 33 × 109/L) a mikroangiopatická hemolytická anémie (hemoglobin 79 g/L, LDH 55,27 μkat/L, počet schistocytů 0,048 a přímý antiglobulinový test negativní). Po vyloučení plicní embolie byl stav uzavřen jako možná ataka postpartálního hemolyticko‑uremického syndromu (CM‑HUS). V té době byla aktivita ADAMTS13 34 %, exprese CD46 (membránový kofaktorový protein) na granulocytech byla 13,3 (referenční rozmezí 16,5–22,6 MFI) a C3 složka komplementu 0,62 g/L (referenční rozmezí 0,90–1,80 g/L). Pro těžkou hemodynamickou nestabilitu byla místo série výměnných plazmaferéz bezprostředně zahájena léčba eculizumabem a komplexní podpůrná péče včetně kontinuální hemodialýzy. I přesto se u pacientky rozvinulo terminální renální selhání. Celková délka pobytu na jednotce intenzivní léčbě trvala 26 dní, celková hospitalizace 55 dní. Dvacet dva měsíců po porodu pacientka podstoupila úspěšnou transplantaci ledviny. Genetické testování odhalilo „pouze“ takzvané modifikátory, které sice zvyšují penetraci a/nebo závažnost CM‑HUS, ale samostatně bez jiné patogenní mutace nebo silného provokačního momentu nemusí vyústit v CM‑HUS. Žádná patogenní varianta genů C3, CD46, CFB, CFH, CFHR5, CFI, THBD, VTN and / or DGKE prokázána nebyla. Opakovaným testováním byl vyloučen antifosfolipidový syndrom.
Trombotické mikroangiopatie (TMA) jsou vzácné patologické stavy s vysokým rizikem závažného průběhu a smrti. Hlavním patogenetickým mechanismem orgánového selhání je porucha mikrocirkulace, mikroangiopatická hemolytická anémie a trombocytopenie.
Za normálních okolností destičky, leukocyty i erytrocyty nerušeně cirkulují v neporušené cévě spolu s plazmatickým von Willebrandovým faktorem (vWF) uvolňovaným z endotelových buněk. ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) štěpí ULM‑vWF na menší a méně trombogenní části. Pokud ADAMTS13 chybí nebo je‑li díky excesivnímu uvolňování multimerů vWF (ULM‑vWF) překročena jeho kapacita štěpit ULM‑vWF, mohou se v prostředí zvýšeného střihového napětí multimery vWF navazovat na endotel arteriol a kapilár v podobě dlouhých vláken, která účinně vážou destičky. Výsledkem je vaskulární mikrotrombotický syndrom manifestovaný neimunní mikroangiopatickou hemolytickou anemií (MAHA), konsumpční trombocytopenií a ischemickým poškozením tkání a orgánů, tzn. TMA (18).
Nejcitovanější formy TMA jsou trombotická trombocytopenická purpura (TTP) a hemolyticko‑uremický syndrom (HUS). Všechny typy TMA spojuje vysoké riziko smrti zejména při pozdní nebo neadekvátní léčbě (19). Mortalita neléčené TTP je 90% a k polovině úmrtí dochází během prvních 24 hodin. Včasná PEX sníží mortalitu na 10 až 20 % (20). U CM‑HUS vede PEX ke kompletní odpovědi jen v 50 % případů, nicméně pomůže zvládnout fulminantní průběh, minimalizovat poškození orgánů a snížit rekurenci (21).
Vedle TTP a HUS existuje řada sekundárních TMA provázejících infekční, nádorová nebo autoimunitní onemocnění, transplantace krvetvorných buněk nebo orgánů, těhotenství a postpartální období nebo jsou nežádoucím účinkem léků – přehled dominantních patofyziologických dějů je v tabulce 7.


Diagnostický závěr TMA představuje průkaz MAHA s rozvinutou nebo rozvíjející se trombocytopenií u pacienta s dynamicky se měnícím klinickým a laboratorním obrazem postižení nejméně jednoho orgánového systému. Diagnostický algoritmus je jednoduchý a realizovatelný kdekoli a kdykoli během 3–6 hodin – viz tabulka 8 (22–25). V krevním obraze nacházíme trombocytopenii, anémii a známky hemolýzy. Biochemické testy upozorní na postižení ledvin nebo jater. Základní koagulační testy mohou být v úvodu v referenčním rozmezí, později se může rozvíjet obraz diseminované intravaskulární koagulopatie. V uvedených případech je třeba vždy indikovat stanovení aktivity LDH, vyšetření počtu schistocytů a přímý antiglobulinový test (PAT). Velmi výrazně zvýšená aktivita LDH je projevem hemolýzy a ischemického poškození tkání a orgánů. Zvýšení počtu schistocytů nad 1 % je typická pro MAHA u TTP. Počet schistocytů může ale být u různých typů TMA v různých fázích nemoci různý. Negativní PAT je důkazem neimunní podstaty hemolýzy (22, 26, 27).


Vysoká mortalita a morbidita neléčené TMA znamená, že je nezbytné zahájit adekvátní léčbu bezprostředně po průkazu TMA, ideálně během prvních 4–8 hodin od prvního kontaktu s pacientem kombinací kortikoterapie a PEX. Není‑li to v daném zařízení možné, je nezbytný transport na pracoviště, kde PEX provádět lze. Před zahájením PEX (event. před podáním plazmy) je třeba nabrat krev k vyšetření aktivity ADAMTS13, specifických anti‑ADAMTS13 a k archivaci materiálu pro detailnější vyšetření, která by mohla být zavedenou léčbou zkreslená. Přehled pravidel léčby vybraných typů TMA viz tabulka 7.
Zásadní informace pro klinickou praxi: Prudký rozvoj trombocytopenie a hemolytické anemie u pacienta s rozvíjející se poruchou orgánových funkcí (především mozek a ledviny) má vést k podezření na TMA.
Závěry
Hemofagocytární syndrom, difuzní alveolární hemoragie, katastrofický antifosfolipidový syndrom a skupina trombotických mikroangiopatií patří mezi vzácné stavy s významnou morbiditou a mortalitou. Na možnou přítomnost některého z uvedených onemocnění je nutno myslet vždy při výskytu jednoho nebo více nejčastějších klinických či laboratorních příznaků (syndrom dechové tísně, akutní selhání ledvin, splenomegalie, hemoptýza, cytopenie), které nelze vysvětlit jinou, v aktuálním klinickém kontextu více pravděpodobnou příčinou. Klíčovou podmínkou klinického úspěchu je včasná diagnostika a včasné zahájení léčby, přestože názory na optimální léčebný postup nejsou vždy jednotné ani v odborné literatuře. Konziliární role hematologa je pro stanovení přesného diagnosticko ‑ léčebného postupu na nehematologických pracovištích intenzivní péče zásadní.
Kompletní seznam odkazů na literaturu je dostupný u prvního autora
KORESPONDENČNÍ ADRESA AUTORA:
MUDr. Jaromír Gumulec
jaromir.gumulec@fno.cz
Klinika hematoonkologie FN Ostrava a LF Ostravské univerzity
17. listopadu 1790, 708 52 Ostrava-Poruba
Cit. zkr: Vnitř Lék. 2022;68(8):498-507
Článek přijat redakcí: 22. 9. 2022
Článek přijat po recenzích: 16. 11. 2022
Zdroje
1. Yildiz H, Van Den Neste E, Defour JP, Danse E, Yombi JC. Adult haemophagocytic lymphohistiocytosis: a Review. QJM: monthly journal of the Association of Physicians. 2020 Jan 14. eng. Epub 2020/01/17. doi:10.1093/qjmed/hcaa011. Cited in: Pubmed; PMID 31943120.
2. La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, Gil‑Herrera J, Girschikofsky M, Jordan MB, Kumar A, van Laar JAM, Lachmann G, Nichols KE, Ramanan AV, Wang Y, Wang Z, Janka G, Henter JI. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019 Jun 6;133(23):2465-2477. eng. Epub 2019/04/18. doi:10.1182/blood.2018894618. Cited in: Pubmed; PMID 30992265.
3. Debaugnies F, Mahadeb B, Ferster A, Meuleman N, Rozen L, Demulder A, Corazza F. Performances of the H‑Score for Diagnosis of Hemophagocytic Lymphohistiocytosis in Adult and Pediatric Patients. Am J Clin Pathol. 2016 Jun;145(6):862-70. eng. Epub 2016/06/15. doi:10.1093/ajcp/aqw076. Cited in: Pubmed; PMID 27298397.
4. Collard HR, Schwarz MI. Diffuse alveolar hemorrhage. Clinics in chest medicine. 2004 Sep;25(3):583-92, vii. eng. Epub 2004/08/28. doi:10.1016/j.ccm.2004. 04. 007. Cited in: Pubmed; PMID 15331194.
5. Levy JB, Turner AN, Rees AJ, Pusey CD. Long‑term outcome of anti‑glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Annals of internal medicine. 2001 Jun 5;134(11):1033-42. eng. doi:10.7326/0003-4819 - 134-11-200106050-00009. Cited in: Pubmed; PMID 11388816.
6. Alexandre AT, Vale A, Gomes T. Diffuse alveolar hemorrhage: how relevant is etiology? Sarcoidosis Vasc Diffuse Lung, DiS. 2019;36(1):47-52. eng. Epub 2019/01/01. doi:10.36141/ svdld.v36i1.7160. Cited in: Pubmed; PMID 32476936.
7. Lara AR, Schwarz MI. Diffuse alveolar hemorrhage. Chest. 2010 May;137(5):1164-71. eng. Epub 2010/05/06. doi:10.1378/chest.08-2084. Cited in: Pubmed; PMID 20442117.
8. Maldonado F, Parambil JG, Yi ES, Decker PA, Ryu JH. Haemosiderin ‑ laden macrophages in the bronchoalveolar lavage fluid of patients with diffuse alveolar damage. Eur Respir J. 2009 Jun;33(6):1361-6. eng. Epub 2009/01/09. doi:10.1183/09031936.00119108. Cited in: Pubmed; PMID 19129275.
9. de Prost N, Parrot A, Cuquemelle E, Picard C, Antoine M, Fleury‑Feith J, Mayaud C, Boffa JJ, Fartoukh M, Cadranel J. Diffuse alveolar hemorrhage in immunocompetent patients: etiologies and prognosis revisited. Respir Med. 2012 Jul;106(7):1021-32. eng. Epub 2012/05/01. doi:10.1016/j.rmed.2012. 03. 015. Cited in: Pubmed; PMID 22541718.
10. Newsome BR, Morales JE. Diffuse alveolar hemorrhage. South Med J. 2011 Apr;104(4):269 - 74. eng. Epub 2011/05/25. doi:10.1097/SMJ.0b013e3182126d3b. Cited in: Pubmed; PMID 21606695.
11. Park JA. Treatment of Diffuse Alveolar Hemorrhage: Controlling Inflammation and Obtaining Rapid and Effective Hemostasis. International journal of molecular sciences. 2021 Jan 14;22(2). eng. Epub 2021/01/21. doi:10.3390/ijms22020793. Cited in: Pubmed; PMID 33466873.
12. Szczepiorkowski ZM, Winters JL, Bandarenko N, Kim HC, Linenberger ML, Marques MB, Sarode R, Schwartz J, Weinstein R, Shaz BH. Guidelines on the use of therapeutic apheresis in clinical practice--evidence‑based approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apher. 2010;25(3):83-177. eng. Epub 2010/06/23. doi:10.1002/jca.20240. Cited in: Pubmed; PMID 20568098.
13. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK, Webber L, Ding L, Sejismundo LP, Mieras K, Weitzenkamp D, Ikle D, Seyfert‑Margolis V, Mueller M, Brunetta P, Allen NB, Fervenza FC, Geetha D, Keogh KA, Kissin EY, Monach PA, Peikert T, Stegeman C, Ytterberg SR, Specks U. Rituximab versus cyclophosphamide for ANCA‑associated vasculitis. N Engl J Med. 2010 Jul 15;363(3):221 - 32. eng. Epub 2010/07/22. doi:10.1056/NEJMoa0909905. Cited in: Pubmed; PMID 20647199.
14. Park JA, Kim BJ. Intrapulmonary recombinant factor VIIa for diffuse alveolar hemorrhage in children. Pediatrics. 2015 Jan;135(1):e216-20. eng. Epub 2014/12/31. doi:10.1542/ peds.2014-1782. Cited in: Pubmed; PMID 25548333.
15. Garcia D, Erkan D. Diagnosis and Management of the Antiphospholipid Syndrome. N Engl J Med. 2018 May 24;378(21):2010-2021. eng. doi:10.1056/NEJMra1705454. Cited in: Pubmed; PMID 29791828.
16. Asherson RA. The catastrophic antiphospholipid syndrome. J Rheumatol. 1992 Apr;19(4):508-12. eng. Epub 1992/04/01. Cited in: Pubmed; PMID 1593568.
17. Mesa CJ, Rife EC, Espinoza LR. Catastrophic antiphospholipid syndrome: is life ‑ long anticoagulation therapy required? Clin Rheumatol. 2020 Jul;39(7):2115-2119. eng. Epub 2020/02/29. doi:10.1007/s10067-020-04997-6. Cited in: Pubmed; PMID 32107665.
18. Chang JC. TTP ‑ like syndrome: novel concept and molecular pathogenesis of endotheliopathy‑associated vascular microthrombotic disease. Thromb J. 2018;16 : 20. doi:10.1186/s12959-018-0174-4. Cited in: Pubmed; PMID 30127669.
19. Tsai HM, Lian EC. Antibodies to von Willebrand factor‑cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998 Nov 26;339(22):1585-94. doi:10.1056/ NEJM199811263392203. Cited in: Pubmed; PMID 9828246.
20. Sarode R, Bandarenko N, Brecher ME, Kiss JE, Marques MB, Szczepiorkowski ZM, Winters JL. Thrombotic thrombocytopenic purpura: 2012 American Society for Apheresis (ASFA) consensus conference on classification, diagnosis, management, and future research. J Clin Apher. 2014 Jun;29(3):148-67. doi:10.1002/jca.21302. Cited in: Pubmed; PMID 24136342.
21. Loirat C, Garnier A, Sellier‑Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Seminars in thrombosis and hemostasis. 2010 Sep;36(6):673-81. doi:10.1055/s-0030-1262890. Cited in: Pubmed; PMID 20865645.
Další literatura u autorů a na www.casopisvnitrnilekarstvi.cz
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2022 Číslo 8
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Omeprazol a mechanismus hojení krvácení z peptického vředu
- Porovnání farmakologických vlastností mikronizovaného diosminu a hesperidinu v terapii chronické žilní insuficience a hemoroidů
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
Nejčtenější v tomto čísle
- Vybrané závažné „hematologické“ syndromy u dospělých pacientů v intenzivní péči
- Dnešní pohled na dědičné trombofilie
- Alergenová imunoterapie v léčbě alergického eozinofilního astmatu
- Antitrombotická léčba a digestivní endoskopie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy