
Sledování vrozených a získaných, strukturních a funkčních změn fibrinogenu
Studies of structural and functional changes of fibrinogen
At the Institute of Hematology and Blood Transfusion, we have been studying hereditary dysfibrinogenemia for more than ten years. During this period we have described more than 30 families in the Czech Republic with inherited mutations in fibrinogen. This paper provides an overview of 8 interesting cases of dysfibrinogenemia which we have characterized. Individual cases differ mainly in their clinical manifestations. The study of congenital fibrinogen disorders provides scientists and clinicians important information on structural and functional aspects of the fibrinogen molecule during various physiological processes, especially hemostasis. For roughly one third of the patients with thrombosis the causes of their thrombotic complications have not been found yet. It is therefore possible that at least some of them might be the result of mutations in fibrinogen molecule, especially if these changes do not have to affect basic coagulation tests. Mutations Bβ Arg237Ser, γ Tyr363Asn, and Aα Asn106Asp have thrombotic manifestation. The carriers of these mutations reported both deep vein thrombosis and pulmonary embolism. Mutations Aα Gly13Glu, Aα Arg16Cys, γ Tyr262Cys, and γ Arg275His have bleeding manifestation with varying intensity. A seven year old carrier of the Aα Arg16His mutation has been asymptomatic; however clinical manifestation of the mutation in the future cannot be excluded because the mutation is situated in the site of fibrinopeptide A release. Posttranslational modified fibrinogen is linked with various diseases. These diseases are associated with oxidative stress which leads to uncontrolled production of oxidants. The oxidants modify structure as well as affect function of fibrinogen. We used several oxidative reagents mimicking various (patho)physiological states. We characterized the structural changes with quantification of carbonyls groups and SDS-PAGE followed by immunodetection. Tyrosyl radicals were also detected by SDS-PAGE with immunodetection and by fluorescent determinations. To determine the extent of oxidative nature of the fibrinogen we used OFR (Oxidative Fibrinogen Reactivity). We also studied the influence of these structural changes on the fibrin network architecture (scanning electron microscopy), the interaction of fibrinogen with thrombin (fibrinopeptides release, turbidimetric monitoring of fibrin network formation) and platelets (static and dynamic adhesion of platelets). New carbonyl groups and tyrosyl radicals were formed in fibrinogen. Most modifications occured both to influence fibrin network as well as its interaction with platelets. The overall effect of the functional changes depended on the nature and intensity of the oxidizing agent and what is very important, ranged from protitrombogennic to significantly thrombogenic.
Key words:
fibrinogen – dysfibrinogenemia – oxidative stress
Autoři:
J. Štikarová; R. Kotlín; J. Suttnar; T. Riedel; J. E. Dyr
Působiště autorů:
Oddělení biochemie Ústavu hematologie a krevní transfuze Praha, vedoucí oddělení biochemie prof. Ing. Jan E. Dyr, DrSc.
Vyšlo v časopise:
Vnitř Lék 2012; 58(Suppl 2): 70-83
Kategorie:
60 let Ústavu hematologie a krevní transfuze Praha
Souhrn
V Ústavu hematologie a krevní transfuze se problematikou vrozených poruch ve fibrinogenu zabýváme již více než 10 let a během této doby se nám podařilo nalézt mutaci v molekule fibrinogenu u více než 30 rodin. Tato práce přináší přehled 8 vybraných strukturálně nebo klinicky zajímavých případů dysfibrinogenemie, které se nám podařilo charakterizovat. Jednotlivé případy se liší především svými klinickými projevy. Studium vrozených poruch ve fibrinogenu přináší důležité informace o strukturálních a funkčních částech molekuly fibrinogenu, které se aktivně podílejí na nejrůznějších fyziologických procesech, především na hemostáze. U zhruba 1/3 pacientů s trombózami nebyla doposud nalezena příčina rozvoje trombotických komplikací, je možné, že některé jsou skryty za doposud neodhalenými mutacemi fibrinogenu. Mutace Bβ Arg237Ser, γ Tyr363Asn a Aα Asn106Asp se manifestovaly tromboticky. U nositelů těchto mutací byly zaznamenány jak hluboké žilní trombózy, tak i plicní embolie. V případě mutací Aα Gly13Glu, Aα Arg16Cys, γ Tyr262Cys a γ Arg275His byla zjištěna manifestace krvácivá, s různou intenzitou. Mutace Aα Arg16His se u 7letého pacienta zatím nemanifestovala klinickými příznaky, není však vyloučeno, protože se jedná o mutaci v místě odštěpování fibrinopeptidu A, že se mutace klinicky projeví v pozdějším věku. Výskyt posttranslačně modifikovaného fibrinogenu byl popsán ve spojitosti s řadou různých onemocnění. Ta bývají především spojena s oxidačním stresem, kdy dochází k nekontrolovatelnému nárůstu oxidantů. Oxidanty mění strukturu i funkci fibrinogenu. Námi zvolené modelové systémy reprezentovaly podmínky oxidačního stresu při různých onemocněních. K popisu strukturních změn jsme využili metodu stanovení karbonylů, SDS-PAGE s následnou imunodetekcí. K detekci vzniku tyrosylových radikálů jsme mimo SDA-PAGE s imunodetekcí použili i stanovení pomocí fluorescence. K určení míry oxidačního charakteru fibrinogenu jsme použili OFR (oxidative fibrinogen reactivity). Dále jsme sledovali vliv oxidativních změn na architekturu fibrinové sítě (skenovací elektronová mikroskopie), dále na interakci fibrinogenu s trombinem (odštěpování fibrinopeptidů, turbidimetrické sledování vzniku fibrinové sítě) a krevními destičkami (statická a dynamická adheze krevních destiček). Působením modifikačních látek za podmínek oxidačního stresu vznikají v molekule fibrinogenu nové karbonylové skupiny a tvoří se tyrosylové radikály. U většiny modifikací dochází jak k ovlivnění tvorby fibrinové sítě, tak i interakce s krevními destičkami. Celkové funkční vyznění vzniklých změn závisí na charakteru a intenzitě oxidačního činidla a, což je velmi důležité, pohybuje se od protitrombogenního až po výrazně trombogenní.
Klíčová slova:
fibrinogen – dysfibrinogenemie – oxidační stres
Úvod
Plazmatický glykoprotein fibrinogen hraje důležitou roli v procesu krevní koagulace, agregace krevních destiček, angiogeneze, adheze a migrace buněk, zánětu a dalších. Molekula fibrinogenu je tvořena 3 páry odlišných polypeptidových řetězců označovaných Aα, Bβ a γ. Katalytickým působením serinové proteázy trombinu dochází k hydrolytickému odštěpení N-koncových fibrinopeptidů A a B, čímž dochází k odkrytí polymeračního místa, a vzniklý fibrinový monomer následně spontánně polymeruje a vytváří fibrinovou síť [1].
Vrozené defekty v molekule fibrinogenu dávají vzniknout onemocněním hypofibrinogenemie a dysfibrinogenemie. Zatímco hypofibrinogenemie je charakterizovaná sníženou hladinou fibrinogenu pod 1,5 g/l, dysfibrinogenemie je charakterizovaná funkčními poruchami fibrinogenu. Obecně je přijímán fakt, že v případě hypofibrinogenemií způsobuje záměna aminokyselinových zbytků poruchy v syntéze, skládání nebo sekreci fibrinogenu, což má za následek degradaci či ukládání nesprávně sestavených molekul fibrinogenu v játrech [2].
V případě dysfibrinogenemií jsou molekuly se zaměněnými aminokyselinovými zbytky sekretovány do krevního řečiště, kde způsobují větší či menší poruchy tvorby fibrinu.
V současné době je celosvětově evidováno více než 400 rodin s vrozenými mutacemi v jednotlivých genech fibrinogenu [3]. Nejvíce mutací bylo popsáno v Aα řetězci – 248 případů.
K pozměnění molekuly fibrinogenu nedochází jen při genetické mutaci, ale i po jeho uvolnění do krevního oběhu. K těmto modifikacím proteinů může dojít i během tzv. oxidačního stresu. Hlavní roli v tomto ději hrají volné radikály. Tyto sloučeniny se běžně podílejí na buněčné signalizaci, na regulaci buněčných funkcí, na reakci imunitního systému a na aktivaci krevních destiček. Pokud však nastane jejich nekontrolovaná produkce nebo je omezena antioxidační obrana, dochází k jejich nadbytku. Pak reagují s dalšími molekulami za vzniku reaktivních produktů, které se účastní dalších reakcí. Výsledkem tohoto řetězce jsou modifikované proteiny, peroxidy lipidů nebo poškozená DNA. Modifikace proteinů může zahrnovat různé změny ve struktuře molekuly. Ať už se jedná o fragmentaci nebo prokřížení molekul, či vnášení nových skupin do molekuly (např. vznik karbonylových skupin, nitrace, chlorace). Modifikovaný fibrinogen se odlišuje svou schopností interagovat s krevními destičkami a tvořit fibrinovou síť [4–6].
V poslední době bývá modifikovaný fibrinogen spojován s mnoha patologickými stavy organizmu. Nejčastěji jsou zmiňována kardiovaskulární onemocnění, dále diabetes nebo zánětlivé stavy. Byla prokázána řada důsledků vzniklých modifikací pro správné působení fibrinogenu. U pacientů s kardiologickými problémy byly zjištěny rozdíly v tvorbě, architektuře a rozpouštění fibrinové sítě. Výsledkem byl celkový protrombotický charakter modifikovaného fibrinogenu [7–12]. K podobnému zjištění dospěly i pokusy s fibrinogenem izolovaným z plazmy pacientů s diabetem [10]. Stupeň modifikace může odrážet i stav a další prognózu pacienta [10,13]. Také lze modifikovaný fibrinogen využít k diagnostice jako marker rakoviny pankreatu [14].
Pro pochopení mechanizmu modifikace a následného vlivu tohoto pozměnění jsme využili in vitro reakci molekuly fibrinogenu s oxidačním činidlem. Jako modelové systémy jsme vybrali takové sloučeniny, které napodobují (pato)fyziologické reakce probíhající v organizmu. Námi zvolené systémy byly systémy obsahující:
- a) železo s kyselinou askorbovou (Fe3+/kyselina L-askorbová, Fe3+/kyselina L-askorbová/H2O2) [15], představující situaci v organizmu trpícím přetížením železem;
- b) malondialdehyd (MDA) [16], který je jedním z produktů lipoper-oxidace;
- c) chlornan sodný (NaOCl) [17,18], simulující působení chlornanových aniontů uvolňovaných při aktivaci buněk imunitního systému;
- d) sydnonimin (SIN-1) [19–21], jehož rozkladem vzniká peroxynitrit schopný nitrovat okolní molekuly proteinů;
- e) metmyoglobin (MetMb/H2O2) [19], reprezentující působení peroxyproteinradikálů na ostatní molekuly, a konečně;
- f) S-nitrozoglutation (GSNO) [22], který je oxidovanou formou glutationu (GSH), vznikajícího působením oxidu dusnatého, který se účastní S-nitrozylace a glutaionylace bíl-kovin.
Metody
Odběr vzorků krve
Vzorky krve byly získány od informovaných zdravých dárců nebo od vybraných pacientů a s jejich souhlasem. Krev byla odebírána do 3,8% roztoku citrátu sodného v objemovém poměru 1 : 9.
Příprava plazmy
Plazma chudá na destičky byla získána odstředěním krve pacientů a zdravých dárců odebrané do citrátu (2 313 × g, 10 min, 25 °C). Horní vrstva plazmy byla stažena do nové zkumavky, šetrně promíchána, rozpipetována po malých alikvotech, šokově zmražena ve směsi suchého ledu s denaturovaným etanolem a uchovávána při –80 °C. Krevní buňky byly využity k izolaci DNA.
Rutinní koagulační vyšetření
Rutinní koagulační vyšetření bylo provedeno v citrátové plazmě pomocí automatického koagulačního analyzátoru STA-R (Diagnostica Stago, Asnieres sur Seine, France).
Genetická analýza
Genomická DNA byla izolována a přečištěna vysolovací metodou dle Millera et al [23]. Přečištěná genomická DNA byla amplifikována pomocí polymerázové řetězové reakce (PCR) za pomoci specifických primerů. Následná sekvenace probíhala pomocí Dye Terminator Cycle Sequencing with Quick Start kitu a genetického analyzátoru CEQ 8000 (Beckman Coulter Inc., Fullerton, CA, USA).
Polymerace fibrinu
Polymerace fibrinu byla sledována turbidimetrickou metodou při vlnové délce 350 nm. Polymerace byla iniciována buď přídavkem trombinu (0,1 NIH U/ml), nebo reptilázy (625krát ředěná TEClot BAT Reptilase, TECO GmbH, Neufahrn, Germany) k citrátové plazmě pacientů ředěné 1 : 3 50 mM TRIS 0,1 M NaCl pufrem pH 7,4 [24–26]. Pro modifikovaný fibrinogen byl tento postup upraven. Fibrinogen byl naředěn na koncentraci 1 mg/ml PBS pufrem o pH 7,4. Reakce byla spuštěna přídavkem 0,5 NIH U/ml trombinu.
Fibrinolýza
Fibrinolýza byla sledována po přídavku trombinu (0,5 NIH U/ml), plasminogenu (0,15 U/ml, Sigma Aldrich Co., St. Louis, MO, USA), CaCl2 (8 mM) a tPA (0,3 μg/ml, Sigma Aldrich Co., St. Louis, MO, USA) k plazmě pacientů ředěné 1 : 3 50 mM TRIS 0,1 M NaCl pufrem pH 7,4. Nárůst a následný pokles turbidity byl sledován při 350 nm pomocí spektrofotometru Synergy HT (BioTek Instruments, Winooski, VT, USA) [27].
Kinetika odštěpování fibrinopeptidů
Vzorky plazmy pacientů byly naředěny 50 mM TRIS 0,1 M NaCl pufrem pH 7,4 tak, aby výsledná koncentrace fibrinogenu byla 1 g/l. Ke vzorkům byl přidán inhibitor dekarboxyláz 0,01 M o-fenantrolin hydrát. Odštěpování fibrinopeptidů bylo měřeno jako funkce času – alikvotní podíly naředěné plazmy byly inkubovány při laboratorní teplotě s trombinem (0,9 NIH U/ml) po dobu 0; 0,5; 1; 3; 5; 10; 20; 30 a 60 min. Probíhající reakce byla zastavena přídavkem 10% trichloroctové kyseliny. Vzorky byly následně odstředěny (17 000 otáček, 30 min, 4 °C) a hladina fibrinopeptidů v supernatantu byla stanovena vysokoúčinnou kapalinovou chromatografií na reverzní fázi (RP-HPLC) pomocí kolony CGC SGX C18 (150 × 3 mm) dle Suttnara et al [28].
Skenovací elektronová mikroskopie
Fibrin byl připraven ze vzorků plazmy pacientů přídavkem trombinu (2 U/ml) [27]. Po 2hodinové inkubaci při laboratorní teplotě byly vzorky promyty a vysušeny pomocí etanolu. Po vysušení metodou kritického bodu byly vzorky potaženy 5nm vrstvou platiny (Safina, Vestec). Mikroskopické pozorování probíhalo ve skenovacím elektronovém mikroskopu VEGA Plus TS 5135 (Tescan, s. r. o., Brno). Šířky jednotlivých fibrinových vláken byly měřeny pomocí programu ImageJ 1,33u (National Institutes of Health, Bethesda, USA). Naměřené hodnoty byly následně statisticky vyhodnoceny pomocí t-testu na hladině významnosti 0,05.
Modifikace fibrinogenu
Všechny modifikace zvolenými systémy probíhaly při 37 °C. Výsledná koncentrace fibrinogenu v roztoku byla 4 mg/ml. Oxidace fibrinogenu systémem Fe3+/ASCprobíhala dle Schacterové et al [15]. Výsledné koncentrace v reakční směsi byly 25 mM L-askorbát (Sigma Aldrich, Česká republika) a 100 μM FeCl3 v 25 mM Hepes (Sigma Aldrich, Česká republika). Doba modifikace byla 0–240 min. Tento systém byl dále rozšířen o působení peroxidu vodíku o výsledné koncentraci 100 μM H2O2.
Reakce s MDA o finální koncentraci 10 mM probíhala v temnu po dobu 0–120 min. Zásobní roztok MDA byl připraven kyselou hydrolýzou 1,1,3,3--tetrametoxypropanu (TEP; Sigma Aldrich, Česká republika), koncentrace MDA byla stanovena spektrofotometricky (ε245 = 13 700 l.mol–1.cm–1) [29].
Oxidace pomocí NaOCl (Sigma Aldrich, Česká republika) o výsledné koncentraci 1,25 mM probíhala 0–20 min. Pro modifikaci byl používán roztok chlornanu sodného, který obsahoval 6–14 % aktivního chloru. Přesná koncentrace byla stanovena spektrofotometricky (ε290 = 350 l.mol–1.cm–1) [30].
Modifikace fibrinogenu pomocí systému s metmyoglobinem o výsledné koncentraci 10 mM metMb a 100 μM H2O2 probíhala po dobu 0–240 min. MetMb byl připraven reakcí myoglobinu (Sigma Aldrich, Česká republika) s hexakyanoželezitanem draselným. Výsledný produkt byl přečištěn gelovou filtrací na koloně naplněné Sephadexem G-10 (23 × 2,5 cm; Pharmacia, Švédsko). Koncentrace a čistota byly stanoveny spektrofotometricky [19].
Reakce fibrinogenu s SIN-1 o finální koncentraci 100 μM probíhala 0–60 min a každých 10 min byly vzorky vortexovány. SIN-1 byl připraven podle Vadseth et al [19].
U modifikace GSNO jsme se nezaměřili na časový efekt, ale na efekt koncentrační. GSNO byl připraven reakcí GSH v prostředí HCl s NaNO2. Ve všech pokusech byl používán fibrinogen modifikovaný 50, 100 nebo 500 μM S-nitrozoglutationem (GSNO), 1 hod, 37 °C, ve tmě [22].
Stanovení obsahu karbonylových skupin
Jedna z metod stanovení obsahu vzniklých karbonylových skupin byla popsána Levinem et al [31]. Je založena na reakci modifikovaných proteinů s 2,4-dinitrofenylhydrazinem (DNPH; Sigma Aldrich, Česká republika). DNPH se kovalentně váže na vznikající karbonylové skupiny. Jejich koncentrace je určena spektrofotometricky a vztažena na obsah proteinu ve vzorku (mol karbonylových skupin/mol proteinu).
Dalším způsobem, jak jsme stanovili modifikované skupiny, byla SDS-PAGE s následnou imunochemickou detekcí. Tímto způsobem jsme detekovali nejen karbonylové skupiny [32], ale i tyrosylové radikály [33].
Stanovení tyrosylových radikálů
Tyrosylové radikály u modifikovaného a kontrolního fibrinogenu byly stanoveny pomocí fluorescence. Bylo proměřeno emisní spektrum od 340 do 500 nm při excitační vlnové délce 325 nm [34]. Fluorescence byla vyjádřena v relativních jednotkách za použití rovnice I = I415 – (I350 + I500)/2.
Fluorescence byla změřena ve spolupráci s RNDr. K. Kuželovou, Ph.D., na Oddělení buněčné biochemie ÚHKT.
Reaktivita oxidovaného fibrinogenu
Reaktivita oxidovaného fibrinogenu byla stanovena podle modifikovaného postupu publikovaného Selmeci et al. K modifikovanému fibrinogenu (1 mg/ml) byl přidán PBS, roztok KI a kyselina octová. Po následné inkubaci při 37 °C byl sledován nárůst absorbance při 358 nm po dobu 300 s [13].
Adheze krevních destiček na povrchově sorbovaný modifikovaný fibrinogen
Adheze krevních destiček na modifikovaný fibrinogen byla provedena podle Vanickova et al [35,36]. Suspenze promytých krevních destiček byla získána z krve zdravých dárců, kteří alespoň 2 týdny neužívali léky ovlivňující krevní destičky (aspirin, ibuprofen). Krev byla odebrána do ACD (v poměru 8,1 : 1,9). Poté byla odstředěna (250 × g, 15 min, 37 °C), k horní vrstvě (bohatá plazma, PRP) byl po stažení do nové zkumavky přidán prostaglandin PGE1 (1 μl/ml PRP; Sigma Aldrich, Česká republika), který je inhibitorem aktivace destiček. Po 10 min inkubace byly destičky odstředěny (1 000 × g, 10 min, 37 °C) a peleta dále resuspendována ve stejném objemu Tyrodového pufru I vytemperovaného na 37 °C a obsahujícího PGE1 (1 μl/ml PRP). Po dalším odstředění (600 × g, 10 min, 37 °C) byla peleta resuspendována v Tyrodovém pufru II, jehož objem byl vypočítán tak, aby výsledný počet destiček byl 200 × 103 destiček/μl. Před adhezí byly destičky inkubovány 30 min ve vodní lázni (37 °C). Pro sledování statické adheze krevních destiček byl fibrinogen o koncentraci 20 μg/ml sorbován na mikrotitrační destičku. Míra adheze krevních destiček na povrchově vázaný modifikovaný fibrinogen byla porovnávána s adhezí krevních destiček na kontrolní fibrinogen. Adheze krevních destiček byla vyhodnocována spektrofotometricky pomocí stanovení aktivity kyselé fosfatázy (ACP, EC 3.1.3.2; Sigma Aldrich, Česká republika). Za těchto podmínek bylo určeno množství adherovaných krevních destiček na 1 cm2.
Dynamická adheze krevních destiček
Dynamická adheze krevních destiček probíhala v přítomnosti modifikovaného a kontrolního fibrinogenu. Izolace krevních destiček byla provedena stejně jako v předchozí metodě. Pro izolaci erytrocytů byla plná krev odebraná do citrátu stočena při 250 × g, po dobu 15 min a při 37 °C. PRP byla použita pro izolaci krevních destiček. Dále byly erytrocyty 2krát promyty fyziologickým roztokem (220 × g, 10 min, 25 °C). Poslední promytí fyziologickým roztokem bylo při 2 000 × g, 10 min, 25 °C. Výsledný počet krevních destiček a erytrocytů ve vzorku byl 200 000/μl, resp. 4 000 000/μl. K izolovaným krevním destičkám a erytrocytům byl přidán modifikovaný nebo kontrolní fibrinogen o výsledné koncentraci 1 mg/ml. Takto vzniklý vzorek byl umístěn na polystyrénovou misku s dokonale padnoucím teflonovým víčkem. Po inkubaci (10 s) bylo aplikováno smykové tření (1 800 s–1, 2 min) pomocí analyzátoru „Cone and plate(let), Impact-R“ (DiaMed; Eurex Medica, Česká republika). Po opláchnutí deionizovanou vodou byly misky obarveny May-Grünwald (Merck, Česká republika). Vzorky byly vyhodnoceny systémem analýzy obrazu. Adheze a agregace krevních destiček byly vyhodnoceny jako procento pokrytí povrchu, velikost objektů a jejich počet [37].
Výsledky
Pacienti s dysfibrinogenemií
Všichni pacienti, popřípadě jejich zákonní zástupci, vyjádřili svůj souhlas s touto studií podpisem informovaného souhlasu. Veškeré vzorky pacientů byly získány postupy schválenými etickou komisí Ústavu hematologie a krevní transfuze v Praze.
Pacient I: 36letá žena s výrazným krvácením během 1. těhotenství v roce 2000. Další těhotenství v letech 2007 a 2008 byla ukončena spontánním abortem během 1. trimestru. Oba potraty byly komplikovány masivním krvácením a hematomy ve stěně dělohy. Pacientka měla výrazně prodloužený trombinový čas a sníženou hladinu funkčního fibrinogenu (tab. 1).
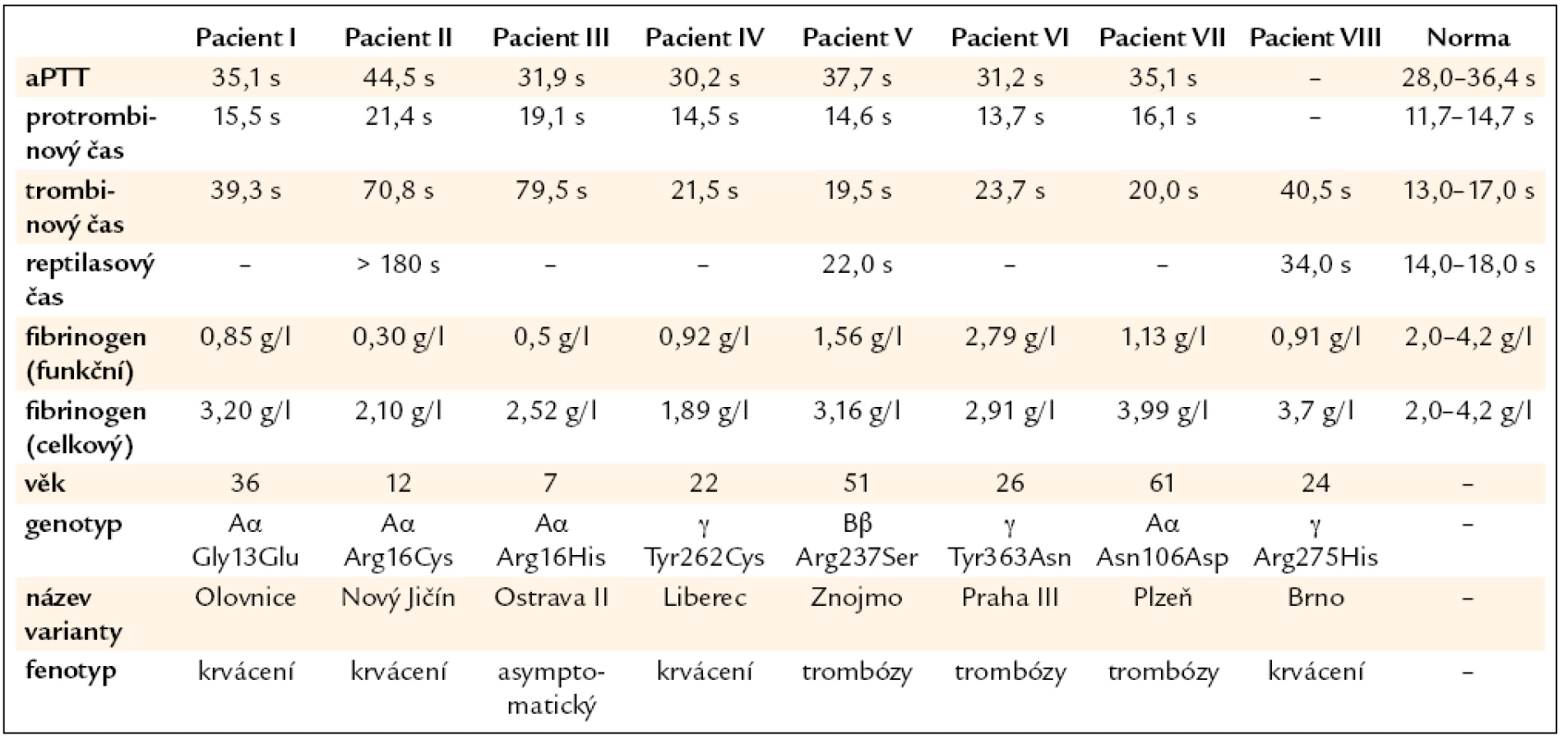
Pacient II: 12letá dívka, která udávala častou epistaxi, krvácení z dásní a snadnou tvorbu hematomů. Koagulační vyšetření odhalilo výrazně prodloužený trombinový čas a sníženou hladinou funkčního fibrinogenu (tab. 1). Výsledky koagulačního vyšetření její matky byly normální.
Pacient III: 7letý asymptomatický chlapec s výrazně prodlouženým trombinovým časem a sníženou hladinou funkčního fibrinogenu (tab. 1). Podobné výsledky koagulačního vyšetření byly nalezeny i u jeho 1ročního bratra a 30letého otce. Jeho 31letá matka měla normální výsledky koagulačních vyšetření.
Pacient IV: 22letá žena, která udávala snadnou tvorbu modřin. Koagulační vyšetření odhalilo prodloužený trombinový čas a sníženou hladinu funkčního fibrinogenu (tab. 1).
Pacient V: 55letý muž s hlubokou žilní trombózou v pravém třísle a plicní embolií v pravé plíci. Pacient dále uváděl zvýšenou tvorbu hematomů po venepunkci. Výskyt běžných rizikových faktorů pro trombózu nebyl u pacienta potvrzen. U pacienta byla zjištěna snížená hladina funkčního fibrinogenu a prodloužený trombinový čas (tab. 1).
Pacient VI: 26letá žena, která v roce 2003 prodělala plicní embolii s akutním cor pulmonale a hlubokou žilní trombózu v ileofemorální oblasti. Plicní embolie se objevila u pacientky znovu v roce 2009. Výskyt běžných rizikových faktorů pro trombózu nebyl u pacientky potvrzen. Její 59letý otec prodělal plicní embolii v roce 2008. U pacientky byl zjištěn prodloužený trombinový čas a normální hladina fibrinogenu (tab. 1).
Pacient VII: 61letý muž, který v roce 2005 prodělal hlubokou žilní trombózu ve femoropopliteální oblasti vpravo, plicní embolii, tromboflebitis v lýtku a tromboflebitis v. saphena magna. Po 5 týdnech po trombotické příhodě se objevilo závažné subarachnoidální krvácení (aneurysma a. cerebri media), které si vyžádalo neurochirurgický zákrok. Koagulační vyšetření u pacienta odhalilo mírně prodloužený trombinový čas a sníženou hladinu funkčního fibrinogenu (tab. 1). Otec pacienta zemřel ve věku 37 let na plicní embolii.
Pacient VIII: 24letá žena s menoragií. U pacientky byla zjištěna snížená hladina funkčního fibrinogenu a prodloužený trombinový čas (tab. 1). Její 45letá matka měla podobné výsledky koagulačního vyšetření a byla asymptomatická. Její 12letý bratr udával epistaxi a snadnou tvorbu modřin. Výsledky koagulačního vyšetření ukázaly podobně jako u jeho sestry a matky prodloužení trombinového času a snížení funkční hladiny fibrinogenu.
Genetická analýza
Provedená genetická analýza odhalila u pacientů následující heterozygotní mutace ve fibrinogenu.
U pacienta I byla nalezena bodová mutace Aα Gly13Glu a tato varianta fibrinogenu byla nazvána Olovnice [38]. U pacienta II byla nalezena bodová mutace Aα Arg16Cys a tato varianta fibrinogenu byla nazvána Nový Jičín [39]. U pacienta III byla nalezena bodová mutace Aα Arg16His a tato varianta fibrinogenu byla nazvána Ostrava II [40]. U pacienta IV byla nalezena bodová mutace γ Tyr262Cys a tato varianta fibrinogenu byla nazvána Liberec [41]. U pacienta V byla nalezena bodová mutace Bβ Arg237Ser a tato varianta fibrinogenu byla nazvána Znojmo [42]. U pacienta VI byla nalezena bodová mutace γ Tyr363Asn a tato varianta fibrinogenu byla nazvána Praha III [43]. U pacienta VII byla nalezena bodová mutace Aα Asn106Asp a tato varianta fibrinogenu byla nazvána Plzeň [43]. U pacienta VIII byla nalezena bodová mutace γ Arg275His a tato varianta fibrinogenu byla nazvána Brno [44].
Polymerace fibrinu
Polymerace fibrinu byla ovlivněna ve všech 8 případech. U všech pacientů byla pozorována snížená výsledná turbidita. U případů Aα Gly13Glu, Aα Arg16Cys a Aα Arg16His byla silně ovlivněna i lag fáze. Prodloužená lag fáze je v těchto případech způsobena ovlivněním odštěpování fibrinopeptidů.
Fibrinolýza
Fibrinolýza byla ovlivněna v případě trombotických mutací Bβ Arg237Ser, γ Tyr363Asn a Aα Asn106Asp (obr. 1). Ve všech 3 případech byla ovlivněna lytická fáze. V případě mutace Bβ Arg237Ser byla lytická fáze zhruba 2,5krát delší než u kontroly, v případě mutace γ Tyr363Asn byla lytická fáze 1,7krát delší než u kontroly a v případě mutace Aα Asn106Asp byla lytická fáze zhruba 2,8krát delší než u kontroly.

Kinetika odštěpování fibrinopeptidů
Abnormální kinetika odštěpování fibrinopeptidů byla pozorována u mutací Aα Gly13Glu, Aα Arg16Cys a Aα Arg16His. Ve všech 3 případech bylo zjištěno pomalejší odštěpování fibrinopeptidů A s menším celkovým podílem z odštěpených fibrinopeptidů (obr. 2). U ostatních případů byla kinetika odštěpování fibrinopeptidů normální.

Skenovací elektronová mikroskopie
Vliv mutace na strukturu fibrinové sítě se uplatňoval u různých mutací různě (obr. 3). V případě trombotických mutací Bβ Arg237Ser, γ Tyr363Asn a Aα Asn106Asp byla fibrinová síť spíše rigidnější, s menšími póry a signifikantně užšími vlákny. Mutace Aα Gly13Glu, γ Arg275His, Aα Arg16Cys a Aα Arg16His neměly zásadní vliv na morfologii sítě. Mutace γ Tyr262Cys naopak způsobovala vznik signifikantně širších vláken. Průměrné šířky vláken jsou shrnuty v tab. 2.


Modifikace fibrinogenu
Stanovení karbonylů
Všechny zmíněné modifikační systémy vnesly do molekuly fibrinogenu nové karbonylové skupiny. Nejvíce jich bylo způsobeno malondialdehydem. O něco méně jich bylo způsobeno modifikací chlornanem, avšak za 1/6 doby působení MDA (tab. 3).

U fibrinogenu modifikovaného GSNO byly karbonyly detekovány pomocí SDS-PAGE. Více karbonylů v molekule bylo zjištěno u modifikovaného fibrinogenu než u kontrolního (data neuvedena).
Stanovení dityrosylů
K detekci vzniklých radikálů jsme využili SDS-PAGE s následnou imunochemickou detekcí podle Vlies et al [33]. Tyrosylové radikály byly detekovány u fibrinogenu modifikovaného systémy: Fe3+/ASC; Fe3+/ASC/H2O2; MetMB/H2O2; NaOCl a SIN-1. Pro kvantifikaci u fibrinogenu modifikovaného Fe3+/ASC a NaOCl bylo využito stanovení fluorescence. Bylo zjištěno, že modifikovaný fibrinogen má přibližně 6krát vyšší fluorescenci než kontrolní. U fibrinogenu modifikovaného NaOCl byla zjištěna přímá úměra mezi dobou působení modifikačního činidla a hodnotou fluorescence (obr. 4).

Reaktivita oxidovaného fibrinogenu
U fibrinogenu modifikovaného NaOCl a SIN-1 byla stanovena reaktivita oxidovaného fibrinogenu. Tato metoda může být použita k monitoringu oxidačního stresu pacientů v kritickém stavu [13]. Vyšší hodnoty konečné absorbance dosáhl fibrinogen modifikovaný NaOCl (obr. 5).

Turbidimetrie
Vznik fibrinové sítě z modifikovaného a kontrolního fibrinogenu byl sledován pomocí zákalových křivek. Ve většině případů s délkou působení modifikačních činidel klesala schopnost fibrinogenu tvořit fibrinovou síť. Výjimkou byla modifikace MetMb/H2O2, kdy s vzrůstající dobou modifikace roste i maximální hodnota absorbance. U modifikace GSNO je signifikantně nižší turbidita u fibrinogenu modifikovaného 500 μM GSNO než u kontrolního fibrinogenu (obr. 6).

Odštěpování FPP
Odštěpování FPP bylo sledováno pomocí HPLC. Ve většině případů strukturní změny vyvolané působením modifikačních činidel vedly k ovlivnění odštěpování fibrinopeptidů (FPP). Odchylka v kinetice odštěpování FPP nebyla zjištěna u fibrinogenu modifikovaného MDA a GSNO. Z fibrinogenu modifikovaného MetMb/H2O2 se fibrinopeptidy A a B lépe odštěpovaly než z kontrolního fibrinogenu (o 1/4, resp. o 1/3). U ostatních modifikací došlo k snížení odštěpování fibrinopeptidů z modifikovaného fibrinogenu v porovnání s kontrolním (tab. 4).

Skenovací elektronová mikroskopie
U fibrinogenu modifikovaného NaOCl, MDA a SIN-1 byly pomocí skenovací mikroskopie pořízeny fotografie vzniklé sítě. Architektura sítí vzniklých z modifikovaného fibrinogenu se odlišovala od normální (obr. 7). U všech sítí byla vzniklá vlákna slabší v porovnání s kontrolní sítí. Sítě zformované z fibrinogenu modifikovaného Sin-1a NaOCl se liší i hustotou vláken. Zatímco v 1. případě byl počet vláken na jednotku plochy vyšší, v 2. případě zase mnohem nižší.

Statická adheze
Modifikace fibrinogenu SIN-1 a MetMB/H2O2 výrazně neovlivnila schopnost fibrinogenu se vázat na krevní destičky. U fibrinogenu modifikovaného Fe3+/ASC/(H2O2), GSNO a MDA došlo k snížení počtu adherovaných destiček na sorbovaný modifikovaný fibrinogen v porovnání s kontrolním o 15–20 %. U fibrinogenu modifikovaného NaOCl došlo k zvýšení počtu adherovaných destiček o 10 % v porovnání s kontrolním fibrinogenem (tab. 5).

Dynamická adheze
U modifikací SIN-1, NaOCl, MDA a GSNO byla provedena dynamická adheze, která zahrnuje nejen adhezi krevních destiček, ale i jejich agregaci. Smykové tření v tomto provedení nahrazuje průtok krve cévami. Jako hodnoticí ukazatel bylo vybráno procento pokrytí povrchu. Bylo zjištěno, že modifikace vyvolané MDA a GSNO znesnadňují interakci s krevními destičkami, kdežto změny vyvolané NaOCl a SIN-1 ji usnadňují (obr. 8).

Diskuze
Jednotlivé nalezené mutace se vyskytují v různých částech molekuly fibrinogenu a podle toho ovlivňují jeho funkci.
Mutace Aα Gly13Glu se nachází ve fibrinopeptidu A. Tato mutace byla již dříve popsána. Mutace byla popsána u 4členné rodiny, ve které byli 3 nositelé mutace asymptomatičtí a 1 projevoval sklon k trombózám [45]. Asselta et al [46] popsali mutaci Aα Gly13Stop. Tento případ se projevoval jako hypofibrinogenemie, neboť bylo zjištěno pomocí COS-1 buněk, že tato mutace způsobuje poruchu sekrece molekul fibrinogenu. Mutace Aα Gly12Glu se projevovala podobně jako mutace Aα Gly13Glu pomalejším odštěpováním fibrinopeptidu A [47]. Aminokyselinové zbytky Aα 12GGG14 jsou důležité pro správnou orientaci zbytků Aα Phe8 a Aα Arg16. Aminokyselinový zbytek Aα Gly13 je v těsném kontaktu s aminokyselinovým zbytkem 217Glu trombinu během katalýzy hydrolytického štěpení vazby mezi Aα 16 Arg a Aα 17 Gly. Aminokyselinové zbytky Aα 6–18 zapadají do aktivního místa trombinu [48], a správná konformace fibrinopeptidu A je tedy velmi důležitá pro správné odštěpování fibrinopeptidu A. Záměnou malého zbytku glycinu za polární postranní řetězec glutaminu a funkční aminoskupinou zřejmě ovlivňuje vzájemné interakce trombinu s mutantním Aα řetězcem, což vede k poruše v odštěpování fibrinopeptidu A.
Mutace Aα Arg16Cys a Aα Arg-16His se nacházejí v místě hydrolytického štěpení vazby Aα Arg16-Gly17. Jedná se o celosvětově nejvíce frekventované mutace. Měření kinetiky odštěpování fibrinopeptidů ukázalo abnormální odštěpování fibrinopeptidu A v obou případech. Kinetika odštěpování fibrinopeptidů z obou fibrinogenových variant odpovídala kinetice odštěpování fibrinopeptidu z fibrinogenu vázaného na povrchu [49]. Stejné výsledky publikovali dříve Lane et al [50], Lee et al [51], Stücki et al [52] a Reber et al [53]. V případě záměny Aα Arg16Cys není trombin schopen katalyzovat odštěpování fibrinopeptidu A, v případě záměny Aα Arg16His dochází k výraznému ovlivnění enzymatické kinetiky trombinu [54]. Pacienti s těmito mutacemi se většinou klinicky projevují asymptomaticky nebo mají krvácivé projevy.
Mutace Aα Asn106Asp je situována v coiled-coil konektoru, který spojuje centrální region s distálními regiony ve fibrinogenu. Coiled-coil konektor je tvořen převážně α-helikálními strukturami, které dodávají molekule fibrinogenu vysokou pružnost a výrazně ovlivňují jeho viskoelastické vlastnosti [55]. Mutace je přibližně uprostřed coiled-coil konektoru v těsné blízkosti segmentu senzitivního na plazmin, kolem kterého fibrinogen zaujímá různé konformace [56], a blízko místa vazby plazminogenu (Aα 148–160) [57]. Mutace měla signifikantní efekt na fibrinolýzu, kdy kompletní lýza klotu vyžadovala více než 40 min v porovnání s 15 min potřebnými pro lýzu kontrolního klotu. Mutace také ovlivňovala morfologii fibrinové sítě, kdy vznikající síť měla užší vlákna, byla rigidnější a byla pozorována přítomnost volných nepolymerovaných konců. Abnormální morfologie sítě a fibrinolýza zřejmě vedly k trombotickým komplikacím u pacienta.
Mutace Bβ Arg237Ser je lokalizována v β skládaném listu Bβ Tyr236-Asp241, který tvoří β vlásenku s β skládaným listem Bβ Trp249-Arg255 (třída 7 : 7), v centrální B-doméně β nodulu [55]. Záměna pozitivně nabitého zbytku asparaginu za neutrální seryl mění konformaci β skládaného listu Bβ Tyr236-Asp241 a celé β vlásenky. Tyto konformační změny pak ovlivňují jak polymeraci fibrinu a jeho morfologii, tak i fibrinolýzu. Fibrin připravený ze vzorků plazmy pacienta měl méně vláken a mnoho volných nepolymerovaných konců. Tato abnormální morfologie sítě následně ovlivnila i fibrinolýzu, která byla přibližně 3krát delší než u kontrolních vzorků. Kombinace těchto faktorů byla zřejmě příčinou trombotických komplikací u pacienta.
Mutace γ Tyr262Cys se nachází v β skládaném listu γ 257-264 (sekvence TSTADYAM) blízko polymeračního místa a místa vazby vápenatých iontů. Tyrosyl 262 je situován uvnitř γC domény. Záměna aromatického postranního řetězce za postranní řetězec cysteinu působí konformační změny v γC doméně, které následně vedou k poruše polymerace a krvácivým obtížím u pacienta. Aromatický kruh tyrosylu je také schopen zprostředkovávat π-π interakce s dalšími aromatickými postranními řetězci. Postranní řetězec cysteinu není schopen tyto interakce zprostředkovávat, což přispívá k ovlivnění funkčních vlastností fibrinogenu pacienta. V okolí γ Tyr262 bylo popsáno několik mutací, které způsobují dysfibrinogenemii spojenou s abnormální polymerací fibrinu podobnou jako v tomto případě [58,59].
Mutace γ Arg275His se nachází na povrchu γC domény a hraje klíčovou roli při D:D interakcích během tvorby protofibril. Jedná se o nejčastější mutaci v g řetězci. Arg275 zprostředkovává vodíkové vazby mezi 2 D-fragmenty fibrinového mono-meru [55]. Jeho záměna za histidyl negativně ovlivňuje interakci distálních regionů, neboť postranní řetězec histidinu vodíkové vazby s dalším distálním regionem netvoří, a tím výrazně brzdí polymeraci a ovlivňuje tvorbu protofibril [60]. To vede následně ke krvácivým klinickým projevům u pacienta. Byly popsány záměny γ 275Arg za cystein [60,61], serin [62] i histidin [61], všechny ovlivňující polymeraci fibrinu a tvorbu protofibril.
Mutace γ Tyr363Asn se nachází v polymerační prohlubni „a“, do které komplementárně zapadá polymerační hrbolek „A“ z centrálního regionu další molekuly fibrinu. Polymerační prohlubeň „a“ se nachází v P-doméně γ nodulu, který hraje klíčovou úlohu během polymerace fibrinu, při D:D interakcích, při vazbě vápenatých iontů či během ligace fibrinových molekul faktorem XIIIa. Tyrosyl 363 je součástí inverzního γ ohybu γ 363–365 (sekvence YDN) [63] ve smyčce III P-domény a je silně konzervován. Záměna velkého aromatického postranního řetězce tyrozinu za malý polární postranní řetězec asparaginu vede ke konformačním změnám inverzního γ ohybu, které působí konformační změny v polymeračním místě, což vede k porušení polymerace fibrinových monomerů. Molekulárním modelováním bylo zjištěno, že přítomnost postranního řetězce asparaginu v polymerační prohlubni vyvolává tvorbu nových vodíkových vazeb s γ glutaminylem 329 a postranním řetězcem asparagylu z polymeračního hrbolku „A“ [43]. To zřejmě vede k ovlivnění laterální agregace protofibril a morfologie vznikající fibrinové sítě.
Každá z popsaných mutací se funkčně manifestuje jiným způsobem. Zatímco mutace ve fibrinopeptidu A vedou k prodloužení doby polymerace, ovlivňují kinetiku odštěpování fibrinopeptidů a vedou spíše ke krvácivým projevům, mutace Bβ Arg237Ser, γ Tyr363Asn a Aα Asn106Asp se manifestují tromboticky. Mutace Bβ Arg237Ser, γ Tyr363Asn, γ Tyr262Cys a Aα Asn106Asp nebyly dříve popsány. Zdá se, že v některých případech trombózy bez určené příčiny mohou být příčinou právě mutace ve fibrinogenu, které nebyly u pacientů dosud diagnostikovány. Znalost mutace pomáhá lékařům lépe předvídat možné klinické projevy dysfibrinogenemie.
Modifikovaný fibrinogen byl objeven v plazmě pacientů s různými onemocněními. Mnoho prací je zaměřeno na důsledky těchto změn na funkci fibrinogenu a další rozvoj onemocnění. V naší laboratoři byl kladen důraz na možné příčiny vzniku odlišností v molekule fibrinogenu a jejich důsledky. Nebyly tak zkoumány jen vzniklé strukturní změny, ale i jejich vliv na fyziologickou funkci fibrinogenu. Byly vybrány takové modifikační systémy, které se mohou běžně vyskytovat při (pato)fyziologických procesech organizmu, a mohou tak ovlivňovat další molekuly.
Vznik nových karbonylových skupin, ale nejen těch, je výsledkem působení látek oxidačního stresu na molekuly proteinů. Zvýšené markery oxidačního stresu byly zaznamenány u mnoha onemocnění. Jedná se např. o kardiovaskulární onemocnění [7–10,12,64], neurodegenerativní poruchy [65], zánětlivé stavy [66] nebo diabetes mellitus [10]. Při všech těchto onemocněních v důsledku oxidačního stresu může docházet k vzniku modifikovaných plazmatických proteinů. Mezi nejnáchylnější bílkoviny v plazmě patří fibrinogen [67].
V modifikovaném fibrinogenu byl zaznamenán nárůst obsahu karbonylových skupin a vznik tyrosylových radikálů. Ačkoli dosažené hodnoty karbonylů byly nižší než při podobných in vitro modifikacích [15], byly ve stejném rozsahu, jenž byl zjištěn u pacientů s MI [7].
Jedním z markerů oxidačního stresu v plazmě diabetiků je hladina MDA [16,68] a právě tato látka vyvolala nejvyšší nárůst nových karbonylů. Po 2 hod působení narostl obsah karbonylových skupin 20krát v porovnání s kontrolním fibrinogenem. Reaktivnější než MDA byl NaOCl, jehož vlivem vzrostl obsah karbonylů po 20 min více než 6krát (tab. 3).
Ve fibrinogenu modifikovaném Fe3+/ASC, Fe3+/ASC/H2O2, MetMB/H2O2, NaOCl a SIN-1 byl zjištěn vznik tyrosylových radikálů. Z tyrosylových radikálů vznikají dityrosyly, které mohou prokřížit řetězce fibrinogenu mezi sebou. Pokud dojde k prokřížení v oblasti Aα řetězců, může být negativně ovlivněna fibrinolýza vzniklé sítě [68].
Názory na vliv modifikace na schopnost fibrinogenu interagovat s trombinem a tvořit fibrinovou síť se různí. Některé studie tvrdí, že modifikace fibrinogenu vede k snížení tvorby sítě (Shacter et al [15], Piryazev et al [69], Tetik et al [70,71]), jiné zase předkládají důkazy o protrombogenním působení modifikace fibrinogenu jeho větší ochotou tvořit fibrinovou síť (Vadseth et al [19], Paton et al [7] nebo Upchurch et al [8]). V této práci byly zjištěny obě varianty v závislosti na použitém modifikačním systému. Při modifikaci MetMB/H2O2 dochází k rychlejší tvorbě fibrinové sítě a vyšší celkové turbiditě ve srovnání s kontrolním fibrinogenem. To potvrzuje i o přibližně 1/3 více uvolňování fibrinopeptidů z modifikovaného fibrinogenu oproti kontrolnímu. U ostatních modifikací dochází k celkovému snížení schopnosti tvorby fibrinové sítě. Odštěpování FPP z fibrinogenu modifikovaného MDA a GSNO nebylo, na rozdíl od ostatních modifikačních systémů, signifikantně ovlivněno. Vzhledem k tomu, že i tyto modifikace vykazovaly podle turbidimetrie omezenou schopnost tvořit síť, není nejspíše fibrinogen modifikován v místě ovlivňujícím interakci s trombinem, ale spíše v polymeračním místě. Pro porovnání, při tvorbě sítě z fibrinogenu modifikovaného v reálných podmínkách oxidačního stresu byla popsána kratší lag fáze a výsledná vyšší turbidita v porovnání s kontrolou [7–9,12].
Paton et al [7] dávají do přímé souvislosti obsah karbonylových skupin a rychlost polymerace. S přibývajícím obsahem karbonylů roste i rychlost polymerace. Jen u fibrinogenu modifikovaného MetMB/H2O2 byl potvrzen tento trend. Avšak u ostatních činidel s délkou modifikace, a tím i nárůstu karbonylů, polymerace klesá.
Rychlost tvorby sítě ovlivňuje její výsledný vzhled a architekturu. Kratší lag fáze svědčí o rychlejší laterální agregaci, a tím i vzniku silnějších vláken. V případě fibrinových sítí vzniklých z plazem pacientů dochází k rychlému vzniku fibrinové sítě se silnějšími vlákny a nižší permeabilitou v porovnání s kontrolními vzorky [7–10]. Síť s tenčími vlákny byla získána z plazem kuřáků. Tato síť byla v porovnání se sítěmi nekuřáků hustší a více organizovaná [72].
V této práci jsme analyzovali sítě vzniklé z fibrinogenu modifikovaného MDA, NaOCl a SIN-1. Ve všech případech byla vzniklá vlákna signifikantně tenčí v porovnání s kontrolou, což odpovídá datům získaným ze sledování tvorby sítě a odštěpování fibrinopeptidů. Sítě vytvořené z fibrinogenu modifikovaného MDA a SIN-1 v porovnání s kontrolou obsahují více vláken na plochu než kontrolní, na rozdíl od sítě z fibrinogenu modifikovaného NaOCl, která obsahuje mnohem méně vláken v porovnání s kontrolou.
Při statické adhezi je sledována schopnost krevních destiček interagovat s fibrinogenem sorbovaným na povrch. V této práci se nepodařilo prokázat signifikantní vliv modifikací fibrinogenu způsobených SIN-1 a MetMB/H2O2 na jeho interakci s krevními destičkami. Na fibrinogen modifikovaný NaOCl se destičky vázaly ochotněji než na kontrolní. U ostatních modifikačních činidel došlo k snížení počtu adherovaných krevních destiček. Dynamická adheze sleduje nejen adhezi, ale i agregaci krevních destiček za podmínek toku krve v cévách.
Dynamická adheze krevních destiček byla provedena v přítomnosti fibrinogenu modifikovaného GSNO, MDA, NaOCl a SIN-1. Modifikace SIN-1 signifikantně ovlivnila interakci fibrinogenu s krevními destičkami, na rozdíl od statické adheze, kde nebyl rozdíl mezi kontrolním a modifikovaným fibrinogenem průkazný. Dynamická adheze potvrdila výsledky získané z adheze statické. Fibrinogen modifikovaný in vivo více agreguje s krevními destičkami [8,73].
Každá modifikace způsobuje odlišné změny v molekule proteinu co do rozsahu modifikací a rychlosti jejich vzniku. Celkové vyznění vzniklých změn, zda se jedná spíše o trombogenní, nebo protitrombogenní charakter, je kombinací všech faktorů. U většiny modifikací dochází jak k ovlivnění tvorby fibrinové sítě, tak i k interakci s krevními destičkami. Modifikace způsobené MDA, GSNO a Fe3+/ASC/(H2O2) mají spíše protitrombotické vyznění. Změny vyvolané jejich působením negativně ovlivnily tvorbu fibrinové sítě i adhezi krevních destiček. V případě systémů obsahujících Fe3+ a ASC dochází při působení po dobu delší než 8 hod k tak zásadním změnám ve struktuře molekuly, že v podstatě nedochází k tvorbě sítě (data neuvedena). Podobné výsledky publikovali i Schacter et al. Modifikace způsobené působením NaOCl sice negativně ovlivnily tvorbu sítě a její architekturu, ale je to vyváženo mnohem ochotnější adhezí a agregací krevních destiček v přítomnosti takto modifikovaného fibrinogenu. Modifikace MetMB/H2O2 nijak neovlivnila adhezi krevních destiček. Tvorba sítě vzniklé z takto modifikovaného fibrinogenu odpovídá vzniku sítí vzniklých z plazem pacientů s kardiologickou diagnózou [7–10].
Fibrinogen byl v této práci modifikován vždy pouze jedním činidlem a za definovaných podmínek, což se diametrálně odlišuje od oxidačního stresu v organizmu, kdy může na jednu molekulu fibrinogenu působit několik různých činidel. Faktorem ovlivňujícím celkové vyznění vzniklých modifikací je i jejich rychlost vzniku. Důsledkem může být potlačení změn způsobených slabšími modifikačními činidly (např. MDA nebo MetMB/H2O2) ve prospěch rychleji působících činidel (NaOCl, GSNO).
Při porovnávání dat získaných z in vitro modifikací se studiemi pracujícími s plazmami pacientů je nutné brát v úvahu působení i dalších modifikovaných nebo nemodifikovaných složek plazmy. Některé námi zachycené změny fibrinogenu mohou být u pacientů překryty, nebo naopak umocněny působením dalších složek krevní plazmy. Ve studii provedené Pietersem et al [74] byl porovnáván vliv glykemické kontroly na glykozylaci fibrinogenu a dále na charakteristiky vzniklé sítě. Byly zde uvedeny výsledky jak ve fibrinogenu izolovaného z plazmy pacientů, tak i pokusy přímo s jejich plazmou. Zatímco fibrinová síť vzniklá z plazmy pacientů se v ničem neodlišovala od kontrolní, tak síť vzniklá z fibrinogenu izolovaného z plazem stejných pacientů se statisticky významně odlišovala permeabilitou a rychlostí svého vzniku a rozpouštění v porovnání s kontrolou.
V této práci byl prokázán vliv látek oxidačního stresu na molekulu fibrinogenu. Vzniklé strukturní změny signifikantně ovlivňují rychlost vzniku, architekturu fibrinové sítě a interakci fibrinogenu s krevními destičkami. Rozsah a rychlost modifikace jsou závislé na použitém modifikačním činidle. Celkové funkční vyznění vzniklých změn závisí na charakteru a intenzitě oxidačního činidla a, což je velmi důležité, pohybuje se od protitrombogenního až po výrazně trombogenní. Výhodou takto navržené studie je přesnější popis změn způsobených jednotlivými modifikačními činidly a identifikace pozměněných aminokyselinových zbytků v molekule pro jednotlivá činidla.
Poděkování
Tato práce vznikla za podpory grantu P205/12/G118 Grantové agentury České republiky a grantu Evropské unie ERDF OPPK 24001.
prof. Ing. Jan E. Dyr, DrSc.
www.uhkt.cz
e-mail: jan.dyr@uhkt.cz
Doručeno do redakce: 12. 5. 2012
Zdroje
1. Weisel JW. Fibrinogen and fibrin. Adv Protein Chem 2005; 70 : 247–299.
2. Brennan SO, Fellowes AP, George PM. Molecular mechanisms of hypo - and afibrinogenemia. Ann NY Acad Sci 2001; 936 : 91–100.
3. Hanss M, Biot F. A Database For Human Fibrinogen Variants. Ann N Y Acad Sci 2001; 936 : 89–90.
4. Sies H. Strategies of antioxidant defense. Eur J Biochem 1993; 215 : 213–219.
5. Griendling KK, FitzGerald GA. Oxidative Stress and Cardiovascular Injury Part I: Basic Mechanisms and In Vivo Monitoring of ROS. Circulation 2003; 108 : 1912–1916.
6. Ashi N, Hayes KC, Bao F. The peroxynitrite donor 3-morpholinosydnonimine induces reversible changes in electrophysiological properties of neurons of the gunea-pig spinal cord. Neuroscience 2008; 156 : 107–117.
7. Paton LN, Mocatta TJ, Richards AM et al. Increased thrombin-induced polymerization of fibrinogen associated with high protein carbonyl levels in plasma from patients post myocardial infarction. Free Radic Biol Med 2010; 48 : 223–229.
8. Upchurch GR, Ramdev N, Wash MT et al. Prothrombotic Consequences of the Oxidation of Fibrinogen and their Inhibition by Aspirin. J Thromb Thrombolysis 1998; 5 : 9–14.
9. Mill JD, Ariëns RAS, Mansfield MW et al. Altered Fibrin clot Structure in the Healthy Relatives of Patients With Premature Coronary Artery Disease. Circulation 2002; 106 : 1938–1942.
10. Scott EM, Ariëns RAS, Grant PJ. Genetic and Enviromental Determinants of Fibrin Structure and Function. Arterioscler Tromb Vasc Biol 2004; 24 : 1558–1566.
11. Alexandru N, Constantin A, Popov D. Carbonylation of platelet proteins occurs as consequence of oxidative stress and thrombin activation, and is stimulated by ageing and type 2 diabetes. Clin Chem Lab Med 2008; 46 : 528–536.
12. Undas A, Szułdrzynski K, Stepien E et al. Reduced clot permeability and susceptibility to lysis in patients with acute coronory syndrome: Effects of inflammation and oxidative stress. Atherosclerosis 2007; 196 : 551–557.
13. Selmeci L, Seres L, Székely M et al. Assay of oxidized fibrinogen reactivity (OFR) as a biomarker of oxidative stress in human plasma: the role of lysina analogs. Clin Chem Lab Med 2010; 48 : 379–382.
14. Schmidt D, Brennan SO. Modified form of the fibrinogen Bb chain (des-Gln Bb), potential long-lived marker of pancreatitis. Clin Chem 2007; 53 : 2105–2111.
15. Shacter E, Williams JA, Levine RL. Oxidative modification of fibrinogen inhibits thrombin-catalyzed clot formation. Free Radic Biol Med 1995; 18 : 815–821.
16. Nielsen F, Mikkelsen BB, Nielsen JB et al. Plasma malodiadehyde as biomarker for oxidative stress: reference interval and effects of life-style factors. Clin Chem 1997; 43 : 1209–1214.
17. Hawkins CL, Davies MJ. Hypochlorite-induced damage to proteins: formation of nitrogen-centred radicals from lysine residues and their role in protein fragmentation. Biochem J 1998; 332 : 617–625.
18. Hazell LJ, Stocker R. Oxidation of low-density lipoprotein with hypochlorite causes transformation of the lipoprotein into a high-uptake form for macrophages. Biochem J 1993; 290 : 165–172.
19. Vadseth C, Souza JM, Thomson L et al. Pro--trombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. J Biol Chem 2004; 279 : 8820–8826.
20. Nowak P, Wachowicz B. Peroxinitrite-mediated modification of fibrinogen affects platelet agregation and adhesion. Platelets 2002; 13 : 293–299.
21. Nowak P, Zbikowska HM, Ponczek M et al. Different vulnerability of fibrinogen subunits to oxidative/nitrative modifications induced by peroxynitrite: functional consenqueces. Thromb Res 2002; 121 : 163–174.
22. Akhter S, Vignini A, Wen Z et al. Evidence for S-nitrosothiol-dependent changes in fibrinogen that do not involve transnitrosation or thiolation. PNAS 2002; 99 : 1972–1977.
23. Miller SA, Dykes PD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acid Res 1988; 16 : 1215.
24. Kotlín R, Sobotková A, Riedel T et al. Acquired Dysfibrinogenemia Secondary to Multiple Myeloma. Acta Haematol 2008; 120 : 75–81.
25. Kotlín R, Chytilová M, Suttnar J et al. A novel fibrinogen variant – Praha I: hypofibrinogenemia associated with γ 351 Gly → Ser substitution. Eur J Haematol 2007; 78 : 410–416.
26. Kotlín R, Suttnar J, Čápová I et al. Fibrinogen Šumperk II: dysfibrinogenemia in an individual with two coding mutations. Am J Hematol 2012; 87 : 555–557.
27. Kotlín R, Reicheltová Z, Suttnar J et al. Two novel fibrinogen variants in the C-terminus of the Bβ-chain: Fibrinogen Rokycany and fibrinogen Znojmo. J Thromb Thrombolysis 2010; 30 : 311–318.
28. Suttnar J, Dyr JE, Fořtová H et al. Determination of fibrinopeptides by high performance liquid chromatography. Biochem Clin Bohemoslov 1989; 18 : 17–25.
29. Libondi T, Ragone R, Vincenti D et al. In vitro cross-linking of calf lens a-crystallin by malondialdehyde. Int J Pept Protein Res 1994; 44 : 342–347.
30. Morris JS. The acid ionization constant of HOCl from 5 to 35. J Phys Chem 1966; 70 : 3798–3805.
31. Levine RL, Garland D, Oliver CN et al. Determination of carbonyl content in oxidatively modified proteins. Meth Enzymol 1990; 186 : 464–478.
32. Robinson CE, Keshavarzian A, Pasco DS et al. Determination of Protein Carbonyl Groups by Immunoblotting. Anal Biochem 1999; 266 : 48–57.
33. Vlies DV, Wirtz KW, Pap EH. Detection of protein oxidation in rat-1 fibroblasts by fluorescently labeled tyramine. Biochemistry 2001; 40 : 7783–7788.
34. Bekman EM, Baranova OA, Gubareva EV et al. Evaluation of the Resistance of Blood Plasma to Oxidative Stress by Oxidizability of Proteins and Lipids during Metal-Catalyzed Oxidation Bull. Exp Biol Med 2006; 142 : 268–272.
35. Vaníčková M, Suttnar J, Dyr JE. The adhesion of blood platelets on fibrinogen surface: Comparison of two biochemical microplate assays. Platelets 2006; 17 : 470–476.
36. Bellavite P, Andrioli G, Guzzo P et al. A colorimetric method for the measurement of platelet adhesion in microtiter platelets. Anal Biochem 1994; 216 : 445–450.
37. Sobotková A, Mášová-Chrastinová L, Suttnar J et al. Antioxidant Change platelet responses to various stimulating events. Free Radic Biol Med 2009; 47 : 1707–1714.
38. Kotlín R, Zichová K, Suttnar J et al. Congenital dysfibrinogenemia Aα Gly13Glu associated with bleeding during pregnancy. Thromb Res 2011; 127 : 277–278.
39. Kotlín R, Chytilová M, Suttnar J et al. Fibrinogen Nový Jičín and Praha II: Cases of hereditary Aα 16 Arg→Cys and Aα 16 Arg→His dysfibrinogenemia. Thromb Res 2007; 121 : 75–84.
40. Kotlín R, Blažek B, Suttnar J et al. Dysfibrinogenemia in childhood: two cases of congenital dysfibrinogens. Blood Coagul Fibrinolysis 2010; 21 : 640–648.
41. Kotlín R, Sobotková A, Suttnar J et al. A novel fibrinogen variant – Liberec: dysfibrinogenaemia associated with γ Tyr262Cys substitution. Eur J Haematol 2008; 81 : 123–129.
42. Kotlín R, Reicheltová Z, Suttnar J et al. Two novel fibrinogen variants in the C-terminus of the Bβ-chain: fibrinogen Rokycany and fibrinogen Znojmo. J Thromb Thrombolysis 2010; 30 : 311–318.
43. Kotlín R, Reicheltová Z, Malý M et al. Two cases of congenital dysfibrinogenemia associated with thrombosis – Fibrinogen Praha III and Fibrinogen Plzeň. Thromb Haemost 2009; 102 : 479–486.
44. Kotlín R, Reicheltová Z, Sobotková A et al. Three cases of abnormal fibrinogens: Šumperk (BβHis67Leu), Uničov (BβGly414Ser), and Brno (γ Arg275His). Thromb Haemost 2008; 100 : 1199–1200.
45. Gaja A, Terasawa F, Okumuna N. Hereditární dysfibrinogenemie s Aa13Gly®Glu mutací. In: Malý M, Pecka J (eds). Trombóza a hemostáza. Hradec Králové: HK Credit 2001 : 114.
46. Asselta R, Duga S, Spena S et al. Congenital afibrinogenemia: mutations leading to premature termination codons in fibrinogen Aα-chain gene are not associated with the decay of the mutant mRNAs. Blood 2001; 98 : 3685–3692.
47. Ménaché D. Congenital fibrinogen abnormalities. Ann NY Acad Sci 1983; 408 : 121–129.
48. Rose T, Di Cera E. Three-dimensional modeling of thrombin-fibrinogen interaction. J Biol Chem 2002; 277 : 18875–18880.
49. Riedel T, Suttnar J, Brynda E et al. Fibrinopeptides A and B release in the process of surface fibrin formation. Blood 2011; 117 : 1700–1706.
50. Lane DA, Ireland H, Thompson E et al. Two more fibrinogens (London III and Sydney) with impaired fibrinopeptide release. Thromb Res 1982; 28 : 821–824.
51. Lee MH, Kaczmarek E, Chin DT et al. Fibrinogen Ledyard (AαArg16→Cys): Biochemical and physiologic characterization. Blood 1991; 78 : 1744–1752.
52. Stucki B, Zenhäusern R, Biedermann B et al. Fibrinogens Bern IV, Bern V and Milano XI: three dysfunctional variants with amino acid substitutions in the thrombin cleavage site of the Aα – chain. Blood Coagul Fibrinolysis 1999; 10 : 93–99.
53. Reber P, Furlan M, Beck EA et al. Fibrinogen Bergamo I (Aα16Arg→Cys): Susceptibility Towards Thrombin Following Aminoethylation, Methylation or Carboxyamidomethylation of Cysteine Residues. Thromb Haemost 1985; 54 : 390–393.
54. Soria J, Soria C, Samama M et al. Fibrinogen Troyes – Fibrinogen Metz. Two cases of congenital dysfibrinogenemia. Thromb Diath Haemorrh 1972; 27 : 619–633.
55. Spraggon G, Everse SJ, Doolittle RF. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature 1997; 389 : 455–462.
56. Doolittle RF, Goldbaum DM, Doolittle LR. Designation of sequence involved in the coiled-coil interdomainal connections in fibrinogen: construction of an atomic scale model. J Mol Biol 1978; 120 : 311–325.
57. Voskuilen M, Vermond A, Veeneman GH et al. Fibrinogen lysine residue A alpha 157 plays a crucial role in the fibrin-induced acceleration of plasminogen activation, catalyzed by tissue-type plasminogen activator. J Biol Chem 1987; 262 : 5944–5946.
58. Niwa K, Takebe M, Sugo T et al. A γ Gly-268 to Glu substitution is responsible for impaired fibrin assembly in a homozygous dysfibrinogen Kurashiki I. Blood 1996; 87 : 4686–4694.
59. Castaman G, Ghiotto R, Duga S et al. A novel fibrinogen γ chain mutation (γ 239 Gln.His) is the cause of dysfibrinogenemia Vicenza. J Thromb Haemost 2005; 3 : 600–601.
60. Imafuku Y, Tanaka K, Takahashi K et al. Identification of a dysfibrinogenemia of γR275C (Fibrinogen Fukushima). Clin Chim Acta 2002; 325 : 151–156.
61. Borrell M, Garí M. Coll I et al. Abnormal polymerization and normal binding of plasminogen and t-PA in three new dysfibrinogenaemias: Barcelona III and IV (gamma Arg 275→His) and Villajoyosa (gamma Arg 275→Cys). Blood Coagul Fibrinolysis 1995; 6 : 198–206.
62. Mimuro J, Kawata Y, Niwa K et al. A new type of Ser substitution for γ Arg-275 in fibrinogen Kamogawa I characterized by impaired fibrin assembly. Thromb Haemost 1999; 81 : 940–944.
63. Medved L, Weisel JW. Recommendations for nomenclature on fibrinogen and fibrin. J Thromb Haemost 2009; 7 : 355–359.
64. Banfi C, Brioschi M, Barcella S et al. Oxidazed proteins in plasma of patients with heart failure: Role in endothelial damage. Eur J Heart Fail 2008; 10 : 244–251.
65. Lee JR, Kim JK, Lee SJ et al. Role of Protein Tyrosine Nitration in Neurodegenerative Diseases and Atherosclerosis. Arch Pharm Res 2009; 32 : 1109–1118.
66. Heffron SP, Parastatidis I, Cuhel M et al. Inflammation induces fibrinogen nitration in experimental human endotoxemia. Free Radic Biol Med 2009; 47 : 1140–1146.
67. Shacter E, Williams JA, Lim M et al. Differential susceptibility of plasma proteins to oxidative modification – examination by western blot immunoassay. Free Radic Biol Med 1994; 17 : 429–437.
68. Lipinski B. Pathophysiology of oxidativestress in diabetes mellitus. J Diabetes Complications 2001; 5 : 203–210.
69. Piryazev AP, Aseichev AV, Azizova OA. Effect of Oxidation-Modified Fibrinogen on the Formation and Lysis of Fibrin Clot in the Plasma. Bull Exp Biol Med 2009; 148 : 881–883.
70. Tetik S, Kaya K, Demir M et al. Oxidative modification of fibrinogrn affects its binding activity to glycoprotein (GP) IIb/IIIa. Clin Appl Thromb Hemost 2010; 16 : 51–59.
71. Tetik S, Kaya K, Yardimci TK. Effect of Oxidized Fibrinogen on Hemostatic System: In Vitro Study. Clin Appl Thromb Hemost 2011; 17 : 259–263.
72. Barua RS, Sy F, Srikanth S et al. Effects of cigarette smoke exposure on clot dynamics and fibrin structure: an ex vivo investigation. Artherioscler Thromb Vasc Biol 2010; 30 : 75–79.
73. Azizova OA, Aseychev AV, Piryazev AP et al. Effects of oxidized fibrinogen on the functions of blood cells, blood clotting, and rheology. Bull Exp Biol Med 2007; 144 : 397–407.
74. Pieters M, Covic N, van der Westhuizen FH et al. Glycaemic control improves fibrin network characteristics in type 2 diabetes – A purified fibrinogen model. Thromb Haemost 2008; 99 : 691–700.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2012 Číslo Suppl 2
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Omeprazol a mechanismus hojení krvácení z peptického vředu
- Porovnání farmakologických vlastností mikronizovaného diosminu a hesperidinu v terapii chronické žilní insuficience a hemoroidů
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
Nejčtenější v tomto čísle
- Imunohematologie – historie, současný stav poznání a role ÚHKT
- Naléhavé stavy v hematologii
- Metabolizmus železa a jeho regulace
- Hemaferéza – vysoce účinná technika v terapii nemocných
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy