
Klinické obrazy histiocytózy z Langerhansových buněk v dospělosti
Clinical presentations of Langerhans cell histiocytosis in adults
Langerhans cell histiocytosis (LCH) is a rare disease affecting both genders, which may occur at any age. It often involves recurrent flare-ups and its severity varies from benign forms that do not require treatment to life threatening disease. Some patients have serious functional impairment with psychological and social consequences and prolonged disability. LCH may affect a single organ, with uni - or multifocal involvement or it can involve multiple organs -multisystemic disease. The organs most frequently involved are bones, lung, skin and the endocrine system. Isolated pulmonary LCH is related to smoking, but this is not true in the case of multisystemic disease. Some patients have mixed histiocytosis combining LCH and other histiocytic disorders. The diagnosis is based on histological examination of tissue samples that demonstrates tissue infiltration with cells staining for CD1a and Langerin (CD207) on immunohistochemistry. The BRAFV600E mutation is observed in tissue samples in approximately one half of patients. Treatment must be adapted to the severity of the disease and ranges from conservative observation to systemic chemotherapy. Therapies targeting the RAS-RAF-MEK-ERK pathway are promising treatments for progressive disease in patients treated with 2-chlorodeoxyadenosine or vinca alkaloids and glucocorticoids.
Keywords:
Langerhans cell histiocytosis
Autoři:
Z. Adam 1; Marta Ježová 2; T. Nebeský 3; Z. Řehák 4; A. Fassman 5; P. Smilek 6; M. Krejčí 1; L. Pour 1; Z. Král
Působiště autorů:
Interní hematoonkologická klinika LF MU a FN Brno
1; Patologický ústav LF MU a FN BRNO
2; Radiologická klinika LF MU a FN Brno
3; PET CT Oddělení Masarykův onkologický ústav
4; Stomatologická klinika LF MU a FN Brno
5; ORL klinika LF MU a FN Brno
6
Vyšlo v časopise:
Transfuze Hematol. dnes,25, 2019, No. 3, p. 219-228.
Kategorie:
Souhrnné/edukační práce
Souhrn
Histocytóza z Langerhansových buněk (Langerhans cell histiocytosis – LCH) je velmi vzácné onemocnění postihující jedince obou pohlaví jakéhokoliv věku. Často má formu recidivujícího onemocnění a závažnost tohoto onemocnění je individuální, od benigních forem, které nevyžadují léčbu až po agresivní, život ohrožující formy. U některých pacientů nemoc způsobuje závažné funkční poruchy s odpovídajícími psychologickými a sociálními důsledky a dlouhodobou pracovní neschopností. LCH může postihnout pouze jeden orgán ve formě unifokální či multifokální, anebo může postihovat více systémů a orgánů. Nejčastěji postiženými orgány jsou kosti, plíce, kůže a endokrinní systém. Izolované plicní formy LCH jsou asociovány s kouřením, v případech s mimoplicním postižením však není souvislost s kouřením prokázána. U některých pacientů se kombinuje LCH s jinými histiocytárními malignitami. Stanovení diagnózy je založeno na histologickém vyšetření s imunohistochemickým průkazem CD1a a langerinu (CD207). Mutace BRAF V600E se nachází v tkáňových vzorcích asi u poloviny pacientů s LCH. Léčba LCH musí být vždy upravena podle agresivity nemoci a kolísá od konzervativní observace po systémovou chemoterapii. Léčebné strategie cílené na RAS-RAF-MEK-ERK signální cesty jsou určeny pro progredující nemocné na klasické léčbě 2-chlorodexyadenosinem nebo vinka-alkaloidy a kortikoidy.
Klíčová slova:
histiocytóza z Langerhansových buněk
ÚVOD
Histiocytóza z Langerhansových buněk je nejčastější nemocí ze všech chorob ze skupiny histiocytárních a dendritických malignit. Věnujeme jí proto samostatný článek. Histiocytóza z Langerhansových buněk (LCH) je nozologická jednotka tvořená velmi širokým spektrem forem s různou závažností klinických projevů. Lze říci, že tato nemoc představuje chameleona mezi krevními nemocemi. Projevy LCH dospělých počínají náhodným rentgenologickým nálezem jednoho osteolytického ložiska, ale mohou mít podobu i generalizované systémové choroby. Dříve, vzhledem ke své tajuplnosti, nejasné etiologii a rozpakům, zdali toto onemocnění řadit k nádorovým nebo infekčním onemocněním či lipidovým tezaurismózám, bylo nazýváno histiocytosis X a nemoc byla podle svých forem pojmenována po autorech, kteří ji popsali, jako nemoc Lettererova-Siweho a nemoc Handova-Schüllerova--Christianova. Tato označení jsou historická a dnes se již nepoužívají!
Incidence LCH v ČR zcela přesně neznáme. Nicholson (1998) uvádí incidenci 5/1 milión všech osob s tím, že u dětí je tato nemoc podstatně častější [1].
V České republice je dětskými hematology a onkology ročně registrováno cca 15 dětí s nově diagnostikovanou LCH, počet dětí s izolovanou kostní formou LCH, která nevyžaduje jinou než chirurgickou léčbu, není znám [2, 3, 4].
Němečtí autoři uvádějí incidenci LCH u dospělých 1–2 případy/1 milion obyvatel [5]. Díky nízké incidenci je nutné získávat informace o této nemoci z popisů jednoho či více případů, klinické studie se u takto vzácné nemoci organizují velmi obtížně.
Pokud se nemoc projeví až v dospělosti, dominuje kostní manifestace. Méně častý je souběh kostního a plicního postižení. Podle analýzy, provedené Baumgartnerem (1997), je postižení hypofýzy, lymfatických uzlin a jater v dospělém věku vzácné, ale vyskytuje se. Při analýze projevů této nemoci u dospělých byly získány následující údaje: v 80 % bývají postiženy kosti, v 60 % kůže, ve 33 % játra, slezina a uzliny, ve 30 % kostní dřeň, ve 25 % plíce, ve 25 % orbita, ve 20 % ucho a orodentální oblast, kde nemoc může zapříčinit uvolňování zubů [6].
Při pohledu do Medline na přehled publikovaných případů lze říci, že nemoc může i u dospělého postihnout skoro každý orgán. Z orgánů, které jsou postihovány u dospělých nejčastěji, lze jmenovat kosti, kůži a plíce. Z ostatních méně častých lokalizací se pak jedná o játra a žlučové cesty, lymfatické uzliny a slezinu, orbitu, orodentální oblast a sluchové ústrojí. Některé histiocytární nemoci včetně LCH mají nevysvětlenou afinitu k hypotalamu. Diabetes insipidus bývá nejčastějším projevem poškození hypothalamo-hypofyzární osy.
Průběh je u dospělých velmi různorodý. U některých postihne jenom jedno ložisko a po léčbě se již neobjeví, u jiných má recidivující charakter, objevují se stále nová a nová ložiska a choroba může být příčinou omezené hybnosti či může dokonce přivodit smrt.
V dalším textu probereme jednotlivé manifestace.Formy LCH u dospělých shrnuje tabulka 1.
![Klasifikace Langerhansovy histiocytózy u dospělých [6]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/79fd9666f8e638c0ec58208289baaca2.png)
Zásadním vyšetřením, nutným pro stanovení diagnózy, je samozřejmě bioptická excize. Pro lékaře, který není odborníkem na histologii, je dobré vědět, že v době akutní choroby jsou v ložisku přítomny četné velké Langerhansovy buňky a eozinofily. Později v ložisku ubývá Langerhansových buněk, přibývá makrofágů střádajících lipidy, vícejaderných buněk a fibroblastů/myofibroblastů. V pozdním stadiu (vyhořelé léze) odpovídá histologický obraz nespecifické fibróze. Histologický vývoj ložiska Langerhansovy histiocytózy lze tedy rozdělit do následujících fází:
- Proliferativní stadium (převažují Langerhansovy buňky).
- Granulomatózní stadium (pestrá cytologie).
- Xantomatózní stadium a tvorba jizev.
Někdy může být obtížné rozlišení osteomyelitidy od kostních ložisek histiocytózy, stejně jako odlišení plicní formy histiocytózy od jiných intersticiálních plicních procesů. V kůži a uzlinách může být problematické odlišení reaktivní proliferace Langerhansových buněk (např. dermatoplastická lymfadenopatie, dermatitida) od proliferace nádorové. K potvrzení diagnózy Langerhansovy histiocytózy se proto používají další znaky, jejichž přítomnost je typická.
- Cytoplazmatická ATP-áza a D-manosidáza (v praxi pro technickou náročnost nepoužíváno).
- Průkaz Birbeckových granulí v cytoplazmě elektronovou mikroskopií (v dnešní době již není standardem, prováděno výjimečně, časově náročné, vyžaduje speciální fixaci).
- Exprese proteinu S-100 (imunohistochemicky, cytoplazmaticky a jaderně pozitivní).
- Exprese antigenu CD1a (imunohistochemicky, membránově pozitivní).
- Exprese langerinu (imunohistochemicky, granulárně pozitivní v cytoplazmě).
- Antigen CD68 a CD45 se vyskytuje nepravidelně, většinou je negativní.
- Jaderná exprese cyklinu D1 pomůže rozlišit nádorovou a reaktivní proliferaci Langerhansových buněk [7].
- Pro Langerhansovu histiocytózu je patognomonický nález antigenů II. HLA třídy, přítomnost CD1a antigenu a často i přítomnost antigenů fyziologicky se nacházejících na fagocytujících histiocytech: CD11c a CD14. Dále jsou uváděny následující znaky CD1a++, CD 11c++, CD44++, CD54++, CD58++. Znaky CD2, CD11a, CD11b jsou na těchto patologických buňkách výjimečně a ve slabé intenzitě. V běžné histopatologické praxi se diagnóza potvrzuje kombinací tří protilátek – S100, CD1a a langerinu. Langerin (CD207), který je součástí Birbeckových granulí, je nejspecifičtější, ale míra jeho pozitivity kolísá od fokální po difuzní [8, 9, 10].
WHO klasifikace uvádí dále termín Langerhans cell sarcoma, který je vyhrazen pro maligní proliferaci Langerhansových buněk, které jsou morfologicky charakterizovány dediferenciací a obvykle také agresivnějším sarkomatózním růstem, vysokou mitotickou aktivitou a imunofenotypem Langerhansových buněk.
Od roku 2012 se při histologickém průkazu této nemoci pátrá po aktivační bodové mutaci genu BRAF v exonu 15, kodonu 600 (BRAF V600E), protože průkaz této mutace signalizuje léčitelnost nemoci preparátem vemurafenib [12, 13]. Mutaci je možné prokazovat jak imunohistochemicky, tak molekulárně-genetickými metodami (PCR). Monoklonální protilátka anti-BRAFV600E je vhodná pro vyšetřování vzorků fixovaných formolem a zalitých do parafinu. Vyšetření je možné doplnit i dodatečně z archivovaných bloků. Molekulárně-genetickými metodami je možné analyzovat kromě periferní krve, likvoru a kostní dřeně i tkáň tumoru v parafinových bločcích. Nejvhodnější metoda je zvolena podle množství a kvality extrahované DNA a podle zastoupení nádorových buněk v analyzovaném vzorku. Patolog je zodpovědný za výběr bločku s maximálním zastoupením vitální nádorové tkáně, jejíž podíl uvede v procentech [11, 12]. Histologickou charakteristiku této nemoci ilustrují obrázky 1–3.
Langerhansovy buňky mají světlou cytoplazmu a bledá ledvinovitá
jádra se zářezy. Příměs tvoří zejména eozinofily.


Pacient je léčen BRAF inhibitorem s výborným výsledkem.

KOSTNÍ PROJEVY LCH
Histiocytóza z Langerhansových buněk v dospělosti postihuje dominantně skelet, vytváří osteolytická ložiska podobná osteolýze při mnohočetném myelomu. Nejčastěji je uváděno postižení kalvy (obr. 4), následuje pak osteolytické postižení žeber. S menší frekvencí bývají postiženy další části skeletu, například dlouhé kosti jako na obr. 5 [13, 14].
Kostní ložiska LCH mají osteolytický charakter, na rozdíl od podobných
osteolytických ložisek u mnohočetného myelomu, u LCH tumorózní
hmoty častěji expandují do okolí kosti a způsobují hmatné zduření,
pokud expandují zevně, anebo neurologické či oční příznaky, pokud
tumorózní hmoty komprimují mozek i obsah očnice. Na obrázku je
zřetelný osteolytický proces okcipitálně, který expandoval jak vně kosti,
takže pacient udával v tom místě hmatné zduření, tak také expandoval
do nitra kalvy a komprimoval šedou koru mozkovou v oblasti zrakového
centra, což se projevilo poruchou zraku. Po léčbě kladribinem se vše
upravilo a pacient je více než 10 let v remisi. K podobné osteolýze
s expanzí zevně i dovnitř může dojít v kterékoliv lebeční kosti.

Snímek zapůjčil pro tuto publikaci prof. Claus Doberaurer
z Gelsenkirchenu, Německo.

U některých pacientů jsou kostní ložiska různého stáří, hojící se místa mají sklerotický lem. Všechna ložiska nemusí bolet. Zduření tkání, přiléhajících ke kosti, signalizuje, že choroba prorůstá do okolí a mnohdy až zduření nad kostí upozorní na přítomnost kostního ložiska. Kostní ložiska často způsobují zvýšený kostní obrat, a proto jsou znázornitelná metodou scintigrafie skeletu pomocí technecium-pyrofosfátu. Novou metodou, která detekuje ložiska LCH v kostech i jinde je FDG--PET/CT. Tato metoda vypovídá o aktivitě či neaktivitě ložiska a může pomoci při plánování radioterapie a vyhodnocování léčebné odpovědi. Velmi citlivou metodou zobrazení patologického děje v kosti je však magnetická rezonance (MR), která je optimální zobrazovací metodou pro plánování rozsahu radioterapie. Zobrazovací vyšetření však pouze detekuje obecně patologické změny v kosti. Teprve histologické vyšetření materiálu z ložiska přinese diagnózu. V případě kostních defektů jsou ložiska patology často popisována jako eozinofilní granulomy [15]. Je tomu tak proto, že ložiska obsahují kromě Langerhansových buněk i velké množství eozinofilních leukocytů ve shlucích (tzv. eozinofilní abscesy).
Izolované kostní ložisko lze léčit lokálně, kyretáží zasahující do zdravé tkáně, nebo radioterapií, případně instilací depotních glukokortikoidů do ložiska. V případě vícečetných ložisek se doporučuje po určitou dobu podávat bisfosfonáty [15].
Za zvláště rizikové kostní lokalizace jsou považovány kostní defekty v oblasti orbitální se supraorbitálními infiltráty a obecně postižení obličejových kostí. Uvedené postižení bývá s vyšší pravděpodobností provázeno pozdějším postižením CNS [7, 8].
KOŽNÍ PROJEVY LCH
Kožní manifestace jsou u Langerhansovy histiocytózy velmi časté a mohou být vůbec prvními zachytitelnými projevy nemoci. Infiltrace kůže buňkami Langerhansovy histiocytózy často postihuje intertriginózní oblasti (perianální oblast, vulvu, třísla, pupek) – obrázek 6. Typickou morfou je hnědorůžová papula velikosti 1–5 mm, při tendenci ke splývání, zvláště v oblasti kůže kštice, se objevuje i šupení (obr. 7). Popisovány jsou i vezikuly a pustuly. Papulózní projevy jsou často hodnoceny jako nespecifické či ekzémové. Šupící plošky jsou zaměňovány se seboroickou dermatitidou, zvláště u kojenců a malých dětí při postižení vlasaté části hlavy. Vezikulózní projevy mohou napodobovat varicelu i ekzematizovaný scabies. U nejbenignější formy choroby, Hashimoto--Pritzkerovy nemoci, se již krátce po narození dítěte objevují mnohočetné nebo solitární červenohnědé uzlíky. Kožní projevy se mohou sdružovat s kostním či viscerálním postižením, v případě závažnějšího průběhu splývají, exulcerují a stav se může komplikovat bakteriální či mykotickou superinfekcí. To, že jde o kožní projev LCH, nelze obvykle rozpoznat při makroskopickém pohledu, ale pro rozpoznání je většinou potřeba provést excizi a histologické hodnocení vzorku [16]. Kožní projevy se mohou sdružovat s kostním či viscerálním postižením, může však jít také o izolovanou morfu, která často spontánně regreduje [17].
Prvním příznakem onemocnění byla bolest a svědění anální krajiny po
jízdě na horském kole. LCH zde mělo formu připomínající kondylomata,
ale histologické vyšetření v těchto morfách prokázalo LCH. Anus byl
ozářen 20 Gy a proces zcela vymizel.

Makroskopicky může připomínat folikulitidu a jedině histologické
vyšetření excise kůže může správně stanovit diagnózu. U špatně
se hojících či nehojících se kožních morf je proto na místě vždy
histologická verifikace kožního onemocnění.

PLICNÍ PROJEVY LCH
Respirační cesty jsou postiženy častěji u dospělých než u dětí, 60–100 % pacientů s plicní formou jsou kuřáci. Incidence plicního postižení mezi všemi pacienty s LCH se udává kolem 20 %. Pacienti přicházejí s anamnestickým údajem nejen dušnosti, ale i bolesti na hrudníku, neproduktivním kašlem, někdy udávají teploty a úbytek hmotnosti.
Infiltrace plic vyvolává restrikční změny, které předcházejí rentgenologickým změnám. Radiografický nález je tvořen cystami a intersticiálními nodulárními opacitami uloženými obvykle blíže hilům. Optimálním prostředkem pro diagnostiku plicní formy histiocytózy je HRCT, s jehož pomocí je možno rozpoznat tenkostěnné cysty, mikronoduly, opacity a zesílení intersticiálních prostorů. Cysty jsou častěji v horních lalocích, méně jich je ve středních lalocích a vynechávají kostodiafragmatický úhel. Zesílení intersticia je při HRCT vyšetření zřetelné hlavně bazálně (obr. 8). Prasknutí cyst a jejich komunikace s pleurální dutinou způsobují spontánní pneumothorax (obr. 9).
Na snímku plic není zřetelná patologie. Pro dušnost však provedeno
HRCT i s trojrozměrnou rekonstrunkcí. HRCT prokázalo cystické změny
plicního parenchymu. Proto máme-li podezření na plicní formu LCH, je
nutno provést HRCT zobrazení i při nomálním RTG snímku plic.

Nodularity přecházejí v kavitované nodularity, později v cysty, které
mají tendenci ke splývání. A velké plicní cysty pak mohou být příčinou
spontánního pneumothoraxu. U tohoto pacienta byla diagnóza
stanovena až po třetím spontánním pneumothoraxu, kdy byla
provedena thorakoskopie a odběr vzorku na histologii.

Postižení plic histocytózou bývá často komplikováno nasedající oportunní infekcí. Odlišit infekci od LCH může být problém, neboť teplota a hmotnostní úbytek mohou být jak prvními projevy plicní histiocytózy, tak mohou mít i jiné, například infekční příčiny. V diferenciální diagnostice pomůže buď bronchoalveolární laváž, nebo thorakoskopie s excizí materiálu na histologické vyšetření.
Bronchoalveolární laváž je přínosná jedině v tom případě, když se provedou odpovídající imunohistochemická vyšetření. Samotná bronchoskopie s biopsií bronchiální sliznice je pro průkaz LCH zcela nepřínosná. Langerhansovy buňky lze identifikovat průtokovou cytometrií flowcytometricky anebo v sedimentu z bronchoalveolární laváže imunohistochemicky průkazem Langerhansových buněk s pozitivitou znaku CD1a proteinu S-100 [18, 19]. Stanovení diagnózy pomocí průtokové cytometrie vyžaduje určitou zkušenost, kterou však je při vzácnosti této nemoci obtížné získat. V našem souboru pacientů s plicní formou LCH máme jednoho nemocného, u něhož byla diagnóza stanovena s pomocí bronchoalveolární laváže. U dalších byly plicní formy postižení verifikovány thorakoskopicky s odběrem vzorku k histologickému vyšetření. Odborná literatura udává, že v případě plicní formy histiocytózy bývá v laváži více než 5 % CD1a+ buněk, zatímco u zdravých pacientů je počet CD1a+ menší než l %. Dále bývá zvýšen počet buněk v aspirované tekutině nad 106/ml, s prevalencí alveolárních makrofágů. Makrofágová alveolitida je totiž přítomna u kuřáků s LCH a chybí u vzácných případů plicní LCH nekuřáků [20].
Podle nálezů z konce 90. let minulého století se plicní LCH dospělých liší od ostatních forem tím, že proliferující Langerhansovy buňky jsou polyklonální, zatímco u ostatních forem jsou Langerhansovy buňky spíše monoklonální. Plicní forma Langerhansovy histiocytózy se považuje za reaktivní proces na kouření či jiné stimuly. U této formy bylo také potvrzeno, že v případech pacientů se silnou vůlí, kteří dokázali přestat kouřit, došlo podle zobrazovacích a funkčních vyšetřené k spontánní regresi nemoci. Plicní forma se považuje za relativně příznivou, pokud jde o izolovanou formu u kuřáka. Pokud však jde o plicní formu navazující na generalizované postižení skeletu a dalších orgánů, tak je průběh relativně nepříznivý. V plicním parenchymu však může vzniknout i Erdheimova--Chesterova choroba [20, 21]. Erdheimova-Chesterova choroba (Erdheim-Chester disease) je histocytární onemocnění, patřící do skupiny juvenilního xantogranulomu. Choroba se projevuje symetrickou osteosklerózou, postihující diafýzu i metafýzu dlouhých kostí, šetřící epifýzy, fibrózou v retroperitoneu a také zesílením stěny velkých cév. Radiologický nález je pro tuto nemoc patognomický.
Pro sledování vývoje plicní formy LCH se používají jak funkční testy, tak HRCT a pro měření aktivity plicní formy lze použít i PET/CT [22, 23].
POSTIŽENÍ ENDOKRINNÍHO SYSTÉMU U LCH
Diabetes insipidus je typickým projevem dětské formy LCH, výjimečně může vzniknout i v dospělosti. V literatuře se popisuje při multifokálním kostním postižení. Zvláště v dětském věku však deficit nepostihuje pouze sekreci adiuretinu, ale občas dochází v důsledku nemoci k deficitu i dalších hormonů včetně poškození tvorby somatotropinu [24].
Defekt tvorby somatotropinu vzniklý v dětství má za následek nejen malý vzrůst, ale nedostatek somatotropinu může být příčinou i neúspěchu in vitro fertilizace, protože přiměřená tvorba somatotropinu či jeho substituce jsou podmínkou k otěhotnění a úspěšnému donošení plodu [25].
Ale také u dospělých může být nově vzniklý diabetes insipidus prvním projevem LCH [25]. Vizualizovat lze postižení CNS klasicky s pomocí MR vyšetření (obr. 10), ale také s pomocí FDG-PET/CT [25].
Řez v koronární rovině, T1 vážený MR obraz po aplikaci kontrastní látky
Tato infiltrace byla příčinou diabetu insipidu.

MOZEK A HYPOFÝZA
Diabetes insipidus je dobře známou komplikací LCH, postihuje 10–50 % dětských pacientů, ale může se objevit i v dospělosti jako první příznak této nemoci. Při MR zobrazení CNS nacházíme u těchto pacientů patologické infiltráty ve stopce hypofýzy nebo v oblasti hypotalamu.
Přibližně u poloviny z nich se diabetes insipidus projeví jako první příznak nemoci a u dalších se objeví do 4–5 let od prvních symptomů LCH. Postižení přední části hypofýzy je méně časté, asi u 10 % pacientů. Deficit růstového hormonu bývá méně častý než malý vzrůst pacientů. Příčina nedostatečného vzrůstu je asi komplexní. Přispívá k němu jak léčba kortikoidy, tak nediagnostikované postižení střeva a malabsorpce. Někdy může vzniknout až úplný panhypopituitarismus. Hypotalamický syndrom s hyperfagií je vzácný, provází jej porucha termoregulace a nezvyklá náladovost.
Metodou detekce těchto změn v CNS jsou funkční endokrinologické testy a magnetická resonance nebo CT s aplikací kontrastní látky. Expanzivní ložiska jsou těmito metodami prokazatelná u 50 % pacientů s poruchou funkce hypofýzy. Disperzní infiltraci prokáže jedině autopsie.
Velmi vzácnou manifestací LCH v dospělosti je extrahypothalamické postižení CNS. Podezření na tuto formu je nutno mít u všech nemocných s LCH, u nichž se objeví jakékoliv neurologické příznaky. Infiltráty lze znázornit pomocí MR a CT s aplikací kontrastní látky. Není jasné, proč Langerhansovy buňky mají takovou afinitu k hypotalamu a jeho stopce [26, 27] (viz obr. 10).
Vzhledem k nemožnosti zkoumat průběh těchto komplikací opakovanými biopsiemi, musí poznání vycházet ze zobrazovacích metod, nemnohých výsledků biopsií a autoptických studií a případně analýz cerebrospinálního moku.
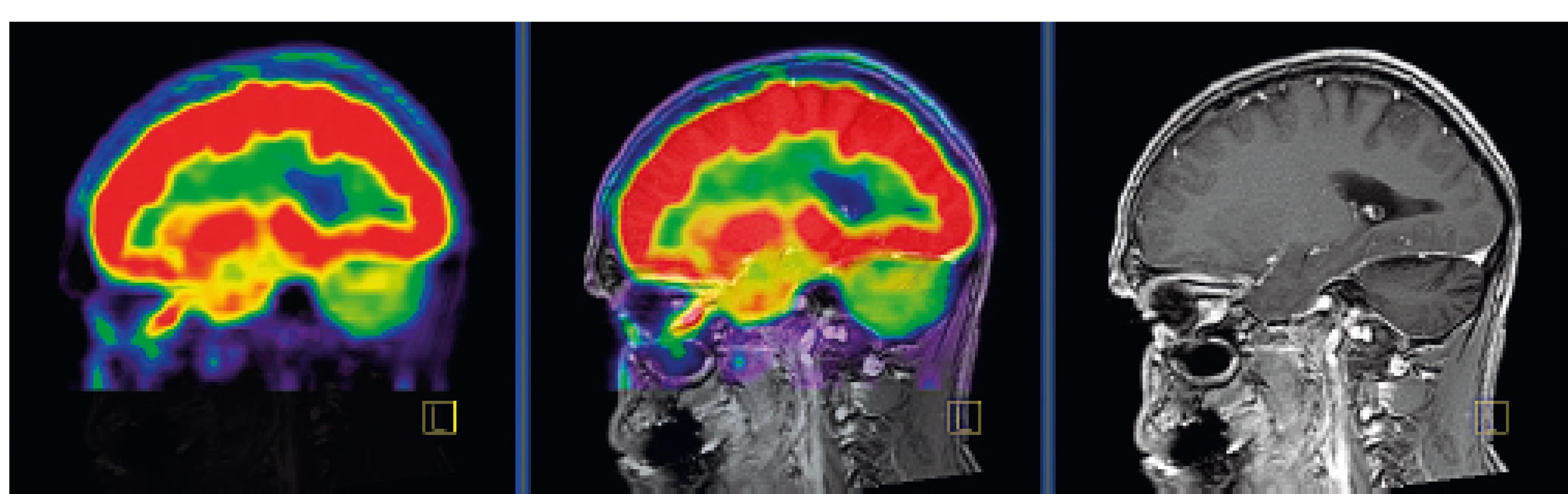
Po dlouhém průběhu nemoci se objevují neurodegenerativní ložiska, postrádající CD1a+ buňky. Nejčastěji bývá postiženo cerebellum, nukleus dentatus, cerebellární bílá hmota a mozkový kmen, s výraznou zánětlivou infiltrací, obsahující CD8 lymfocyty. Tento proces vede k degeneraci a glióze nervové tkáně. Neurodegenerativní proces se objevuje na základě T-buněčného zánětlivého procesu. Je provázen destrukcí neuronů a axonů se sekundární demyelinizací, připomínající paraneoplastickou encefalitidu. Tento typ postižení se objevuje až za dlouhou dobu od stanovení diagnózy, je progredující a nevratný (obr. 11). Klinicky se projevuje hyporeflexií, ataxií, závratěmi, dysartrií, nystagmem, tremorem, diplopií, psychomotorickou retardací a neuropsychologickými defekty [26, 27].
V oblasti cerebella, které je postiženo neurodegenerativními změnami,
je zřetelné snížené vychytávání FDG a neurodegenerativní změny byly
u tohoto pacienta zřetelné i na MR zobrazení.
LYMFADENOPATIE
Histiocytóza obvykle nedělá výraznou lymfadenopatii. Pokud ano, jde spíše o ložiskové než generalizované postižení [28]. U našich pacientů jsme se setkali jak s případem lokalizované lymfadenopatie, kterou vyléčil operační výkon, tak s případem generalizované lymfadenopatie, která měla stejný obraz při PET/CT zobrazení jako generalizovaný nehodgkinský lymfom (obr 12).
PET-CT znázorňujě postižení lymfatických
uzlin od krku až po inguiny. LCH z tohoto
pacienta měla neobvykle vysoký proliferační
index a vysoký počet mitóz, onemocnění
se chovalo velmi agresivně, takže po remisi
navozené lenalidomidem byla provedena
alogenní transplantace krvetvorné tkáně
a pacient je nyní 8 let od transplantace
v remisi LCH.

UŠI
Histiocytóza z Langerhansových buněk (LCH) někdy postihuje zevní zvukovod.
Způsobí zánět makroskopicky neodlišitelný od klasického zánětu zevního zvukovodu. Pouze histologie postižení kůže by mohla prokázat, že se jedná o LCH. Ta se ale obvykle neprovede, takže infiltrace přestoupí na spánkovou kost a vytvoří v ní osteolytická ložiska. Ta se pak obvykle přihlásí narušením sluchu či přímo hluchotou. To pak vede konečně k CT vyšetření, které prokáže osteolytický proces ve spánkové kosti, operační revizi a konečně i histologické vyšetření, které prokáže LCH. Bylo by proto vhodné u dlouhodobých nehojících se zánětů zevního zvukovodu provést histologické vyšetření.
Poruchy sluchu mohou nastat jak postižením zevního sluchového kanálu, tak poruchou středního či vnitřního ucha propagací choroby z processus mastoideus. Infiltrace je nebolestivá a postupně vede k hluchotě. Časté jsou sekundární infekce, které jsou příčinou záměny za chronickou otitidu. U každé dlouhodobé afekce, připomínající zánět zevního zvukovodu, by proto mělo být histologicky ověřeno, zda se nejedná o první projev LCH. Nemoc totiž dále progreduje do vnitřního ucha a bohužel je často diagnostikovaná až při operaci pro proces, který destruuje celé vnitřní ucho, jak je ukázáno na obrázku 13 [29].
Způsobí zánět makroskopicky neodlišitelný od klasického zánětu
zevního zvukovodu. Pouze histologie postižené kůže by mohla
prokázat, že se jedná o LCH. Toto vyšetření se ale obvykle neprovede,
takže infiltrace přestoupí na spánkovou kost a vytvoří v ní osteolytická
ložiska.

V některých případech však postihuje i kost a vede k destrukci čelisti.
Tyto snímky zapůjčil prof. Fassmann ze svého archivu.

OČI
Intraokulární postižení je vzácné, zatímco infiltrace orbitálního prostoru je relativně častá. Dětští lékaři se s ní setkávají u 20–30 % nemocných Langerhansovou histiocytózou. Projevuje se ptózou víčka, edémem papily a poruchou funkce VII. nervu. Může být poškozen i optický nerv, což si někdy kromě systémové léčby vynutí i akutní léčbu nitroložiskovou aplikací kortikosteroidů a radioterapii [30]. Sami jsme se setkali pouze s případy postižení zevní stěny orbity, které vyřešil operační výkon.
JÁTRA A SLEZINA
Játra i slezina mohou být touto chorobou také postižena, což se projeví jejich zvětšením. Infiltrace jater může vyvolat příznaky jaterního selhání (pokles koncentrace albuminu, snížení aktivity koagulačních faktorů, žloutenka bez výrazného zvýšení jaterních enzymů). U chronických forem může vzniknout periportální fibrotizace s příznaky shodnými se sklerotizující cholangoitidou a obstrukční biliární žloutenkou, kterou je nutno na základně biopsie odlišit od primární sklerotizující cholangitidy a adekvátně léčit [31].
DUTINA ÚSTNÍ
Počínající infiltrace se v dutině ústní projevuje zduřením dásní a sliznice patra. Může dojít i k postižení kostí a uvolňování zubů či hypertrofii dásní. Progrese infiltrátů pak vytváří ulcerace v ústech. Někdy je projev LCH v ústech bez histologie nerozeznatelný od paradentózy, projevu se zánětem dásní, a někdy proces přechází i na kostní strukturu čelisti (obr. 14). Toto postižení ilustruje snímek dutiny ústní dospělého člověka [32, 33]. Diagnostické je až histologické vyšetření.
TRÁVICÍ TRAKT
Sliznice střevního traktu je postižena jen zřídka. Prvními příznaky je celkové neprospívání a hubnutí. Klasické projevy malabsorpce se objevují až při rozsáhlejším postižení trávicího traktu. Anální kanál a perianální oblast jsou infiltrovány často, a tvoří tak součást kožního postižení. Infiltrace kůže perianálně je makroskopicky nerozeznatelná od ekzému, pouze histologické vyšetření kůže může identifikovat LCH. Někdy má nemoc v této oblasti podobu verukovitých výrůstků podobných kondylomatům [34, 35].
LÉČBA LCH
Cílem uvedeného textu je podat stručné informace o klinických projevech a problémech diagnostiky, a proto léčbu zmíníme jen heslovitě a budeme se jí věnovat v dalším článku.
Historicky první léčebnou alternativou byly a stále jsou vinka alkaloidy, konkrétně vinblastin v kombinaci s glukokortikoidy. Tato léčba je používána u dětí a používá se i u dospělých. U dospělých nemocných je tato léčba provázena neuropatií. Proto před léčbou vinblastinem a prednisonem preferujeme léčbu kladribinem.
Kladribin (2-chlorodeoxyadenosin) je u této nemoci excelentně účinný. U pediatrických pacientů se často kombinoval 2-chlorodexyadenosin s vysokými dávkami cytosinarabinosidu.
U dospělých pacientů používáme podkožní aplikaci kladribinu, preparát Litak, v dávce 5 mg/m2 s. c. 5 dní po sobě, ve čtyřtýdenních intervalech. Celkem podáváme 4 cykly této léčby. Kladribin je v této dávce výborně tolerován. Léčba této nemoci kladribinem není provázena cytopenií. Pouze v případě, když ke kladribinu v uvedené dávce jsme přidali cyklofosfamid, tak jsme pozorovali přechodné cytopenie.
Z klasických cytostatik mezi účinné léky pro tuto nemoc patří etoposid. Etoposid lze použít v rámci kombinace CHOEP (cyklofosfamid, vinkristin, adria-mycin, etoposid a prednison), anebo jej lze použít v monoterapii.
Z novějších léků byla účinnost prokázána u thalidomidu a také u lenalidomidu. Na našem pracovišti jsme lenalidomid použili u dvou případů a v obou navodil remisi nemoci.
Objev mutace BRAF v roce 2012 u pacientů s LCH vedl k používání inhibitorů této signální cesty pro léčbu této nemoci. Prvním z úspěšně použitých preparátů byl vemurafenib. Takže i u LCH lze dnes použít cílenou terapii [36].
V případech, kdy nemoc špatně reaguje na výše popsanou léčbu, je možné použít vysokodávkovanou chemoterapii s autologní transplantací.
V případně značně agresivního onemocnění je možné přistoupit k vysokodávkované chemoterapii s alogenní transplantací. V našem souboru nemocných máme jednoho pacienta, který měl velmi agresivní onemocnění s vysokou proliferací. Předchozí léčebné linie sice navodily vždy kompletní remisi, nemoc však brzy recidivovala. A proto jsme přistoupili k alogenní transplantaci, po níž je tento pacient již 9 let v kompletní remisi a v dobrém stavu.
V případně nově diagnostikového multiložiskového onemocnění volíme kladribin jako léčbu první volby. V případně nedosažení kompletní remise zvažujeme léky ze skupiny imidů.
SLEDOVÁNÍ NEMOCNÝCH
Zatím nemáme v praxi dostupný žádný laboratorní parametr, takže nezbývá nic jiného než používat odpovídající zobrazovací metody a klinická vyšetření. Použití sérové hladiny proteinu S100 se zatím nedostalo do rutinní praxe. LCH výrazně vychytává FDG, a proto je pro sledování velmi užitečným vyšetřením FDG-PET/CT [36].
Podíl autorů na přípravě rukopisu
ZA, LP, MK – odpovídá za úhel pohledu klinického lékaře
MJ – odpovídá za úhel pohledu patologa
TN – odpovídá za uvedená zobrazovací vyšetření (CT a MR)
ZŘ – odpovídá za uvedené zobrazovací vyšetření pomocí radioizotopů
AF – autor stomatologických obrázků
PS – autor obrázků poškožení ORL oblasti
Čestné prohlášení
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Doručeno do redakce dne 16. 5. 2018.
Přijato po recenzi dne 11. 12. 2018.
prim. MUDr. Zdeněk Král, CSc.
Interní hematologická a onkologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno-Bohunice
e-mail: kral.zdenek@fnbrno.cz
Zdroje
1. Nicholson SH, Egeler M, Nesbit ME. The epidemiology of Langerhans cell histiocytosis. Hematol Oncol Clin North Amer 1998;12 : 379–348.
2. Bubanská E, Stančoková T, Dluholucký S. Histiocytóza z Langerhansových buněk. Čes-slov Pediat 1998;53(1):18–19.
3. Mottl H. Histiocytóza z Langerhansových buněk u dětí a dospívajících. Vnitřní Lék 2010; 56(Supl 2):64–73.
4. Mottl H, Mráček J, Kabelka Z, et al. Histiocytóza z Langerhansových buněk u dětí. Čes-slov Pediat 1992;47(9):530–533.
5. Baumgartner I, Hochstetter A, Baumert B, et al. Langerhans cell histiocytosis in adults. Med Pediatric Oncol 1997;28 : 9–14.
6. Stockschlaeder M, Sucker C. Adult Langerhans cell histiocytosis. Eur J Haematol 2006; 76(5):363–368.
7. Shanmugan V. Cyclin D1 is expressed in neoplastic cells of Langerhans cell histiocytosis but not reactive Langerhans cell proliferation. Am J Surg Path 2017;41 : 1390–1396.
8. Kodet R, Mrhalová M. Histiocytóza z Langerhansových buněk z pohledu patologa. Vnitřní Lék 2010;56(Supl 2):27–23.
9. Plank L. Diagnostická patológia non-Langerhansových histiocytóz. Vnitřní Lék 2010; 56(Supl 2):29–63.
10. Nakamine H, Yamakawa M, Yoshino T, et al. Langerhans cell histiocytosis and Langerhans cell sarcoma: current understanding and differential diagnosis. J Clin Exp Hematop 2016;56(2):109–118.
11. Sahm F, Capper D, Preusser M, et al. BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood 2012;120(12):e28–34.
12. El Demellawy D, Young JL, de Nanassy J, et al. Langerhans cell histiocytosis: a comprehensive review. Pathology 2015;47(4):294–301.
13. Smilek P, Krejčová B, Čada K, et al. Histiocytóza z Langerhansových buněk, případ postižení spánkové kosti. Otorinolaryng Foniat 1994;43(4):263–265.
14. Angelini A, Mavrogenis AF, Rimondi E, et al. Current concepts for the diagnosis and management of eosinophilic granuloma of bone. J Orthop Traumatol 2017;18(2):83–90.
15. Chellapandian D, Makras P, Kaltsas G, et al. Bisphosphonates in Langerhans cell histiocytosis: an international retrospective case series. Mediterr J Hematol Infect Dis 2016; 8(1):e2016033.
16. Ferreli C, Aste N, Pinna LA, et al. Langerhans cell histiocytosis in an adult. J Eur Acad Dermatol Venereol 1997;9 : 253–255.
17. Vašků V. Histiocytóza z Langerhansových buněk – kožní aspekty onemocnění. Vnitřní Lék 2010;56(Supl 2):91–93.
18. Radzikowska E. Pulmonary Langerhans‘ cell histiocytosis in adults. Adv Respir Med 2017;85(5):277–289.
19. Cooley J, Lee YCG, Gupta N. Spontaneous pneumothorax in diffuse cystic lung diseases. Curr Opin Pulm Med 2017;23(4):323–333.
20. Tazi A, de Margerie C, Naccache JM, et al. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis 2015;10 : 30. Publikováno elektronicky 14. března 2015. DOI: 10.1186/s13023-015-0249-2.
21. Tazi A, Marc K, Dominique S, et al. Serial computed tomography and lung function testing in pulmonary Langerhans‘ cell histiocytosis. Eur Respir J 2012;40(4):905–912.
22. Adam Z, Nebeský T, Szturz P, et al. Postižení plic u pacientů s multiorgánovou formou histiocytózy z Langerhansových buněk, popis 8 pacientů a přehled literatury. Vnitřní Lék 2010;56(Supl 2):105–122.
23. Adam Z, Řehák Z, Koukalová R, et al. Přínos PET-CT pro diagnostiku a sledování plicní formy histiocytózy z Langerhansových buněk. Vnitřní Lék 2010;56(Supl 2):123–130.
24. Makras P, Kaltsas G. Langerhans cell histiocytosis and pituitary function. Endocrine 2015;48(3):728–729.
25. Makras P, Alexandraki KI, Chrousos GP, et al. Endocrine manifes-tations in Langerhans cell histiocytosis. Trends Endocrinol Metab 2007;18(6):252–257.
26. Shamim SA, Tripathy S, Mukherjee A, et al. (18-)F-FDG PET/CT in localizing additional CNS lesion in a case of Langerhans cell histiocytosis: determining accurate extent of the disease. Indian J Nucl Med 2017;32(2):162–163.
27. Grois N, Prayer D, Prosch H, et al Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005;128 : 829–838.
28. Edelweiss M, Medeiros LJ, Suster S, et al. Lymph node involvement by Langerhans cell histiocytosis: a clinicopathologic and immunohistochemical study of 20 cases. Hum Pathol 2007;38(10):1463–1469.
29. Smilek P, Pažourková M. Projevy histiocytózy z Langerhansových buněk v ORL oblasti. Vnitřní Lék 2010;56(Supl 2):76–84.
30. Esmaili N, Harris GJ. Langerhans cell histiocytosis of the orbit: spectrum of disease and risk of central nervous system sequelae in unifocal cases. Ophthal Plast Reconstr Surg 2016; 32(1):28–34.
31. Tang Y, Zhang Z, Chen M, et al. Severe sclerosing cholangitis after Langerhans cell histiocytosis treated by liver transplantation: An adult case report. Medicine (Baltimore) 2017;96(9):e5994.
32. Fassmann A, Izakovičová-Hollá L, Augustín P, et al. Projevy histiocytózy z Langerhansových buněk v orofaciální oblasti Vnitřní Lék 2010;56(Supl 2):85–90.
33. Bansal M, Srivastava VK, Bansal R, et al. Severe periodontal disease manifested in chronic disseminated type of Langerhans cell histiocytosis in a 3-year old child. Int J Clin Pediatr Dent 2014;7(3):217–219.
34. Doberauer C. Langerhans cell histiocytosis in adults. Vnitřní Lék 2010;56(Supl 2):22–26.
35. Nanduri VR, Kelly K, Malone M, et al. Colon involvement in Langerhans‘ cell histiocytosis. J Pediatr Gastroenterol Nutr 1999;29(4):462–466.
36. Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis 2013;8 : 72. Publikováno elektronicky 14. května 2013. DOI:10.1186/1750-1172-8-72.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2019 Číslo 3
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Diagnostika von Willebrandovy choroby krok za krokem
- Těhotenství a porod u ženy s VWD − kazuistika
Nejčtenější v tomto čísle
- Osteonekróza čelisti, atypické fraktury kostí a další méně časté nežádoucí účinky bisfosfonátů
- Revakcinace dospělých pacientů po alogenní transplantaci krvetvorných buněk: doporučení České leukemické skupiny – pro život
- Profesor MUDr. Jaroslav Čermák, CSc., 65letý
- Farmakokinetika v léčbě hemofilie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy