
IL-33-Mediated Protection against Experimental Cerebral Malaria Is Linked to Induction of Type 2 Innate Lymphoid Cells, M2 Macrophages and Regulatory T Cells
Cerebral malaria (CM) caused by the parasite Plasmodium sp. is a fatal disease, especially in children. Currently there is no effective treatment. We report here our investigation on the role of a recently discovered cytokine IL-33, in treating experimental cerebral malaria (ECM) in the susceptible C57BL/6 mice. IL-33 protects the mice against ECM. The protection is accompanied by a reduction of Th1 response and the enhancement of type 2 cytokine response. We also found that IL-33 mediates its protective effect by inducing a population of type 2 innate lymphoid cells (ILC2), which then polarize macrophages to alternatively-activated phenotypes (M2). M2 in turn expand regulatory T cells (Tregs) which suppress the deleterious Th1 response. Our report therefore reveals hitherto unrecognised mechanisms of the regulation of ECM and provide a novel function of IL-33.
Published in the journal:
. PLoS Pathog 11(2): e32767. doi:10.1371/journal.ppat.1004607
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004607
Summary
Cerebral malaria (CM) caused by the parasite Plasmodium sp. is a fatal disease, especially in children. Currently there is no effective treatment. We report here our investigation on the role of a recently discovered cytokine IL-33, in treating experimental cerebral malaria (ECM) in the susceptible C57BL/6 mice. IL-33 protects the mice against ECM. The protection is accompanied by a reduction of Th1 response and the enhancement of type 2 cytokine response. We also found that IL-33 mediates its protective effect by inducing a population of type 2 innate lymphoid cells (ILC2), which then polarize macrophages to alternatively-activated phenotypes (M2). M2 in turn expand regulatory T cells (Tregs) which suppress the deleterious Th1 response. Our report therefore reveals hitherto unrecognised mechanisms of the regulation of ECM and provide a novel function of IL-33.
Introduction
Malaria remains a major health problem for humans infected with Plasmodium species. Cerebral malaria (CM) is a severe and potentially fatal neurological manifestation of infection and accounts for approximately one million deaths annually of children in sub-Saharan Africa alone [1]. CM is characterized by a strong Th1 immune response, with a robust and uncontrolled production of proinflammatory cytokines (IFN-γ and TNF-α) and chemokines (IP-10/CXCL10, KC/CXCL1 and MCP-1/CCL2) [2,3] that contribute to vascular leakage and sequestration of parasitized red blood cells (pRBCs) and leukocytes within the brain blood vessels [4,5]. In malaria, the balance between pro - and anti-inflammatory cytokines is critical in determining the outcome of infection, and recent evidences suggest that helminth co-infection may dampen immunopathological responses to malaria parasite by inducing a protective type-2 response [6,7]. Studies using murine models of malaria have established that genetic background of the host affects the development and outcome of ECM. Infection of C57BL/6 mice, which present a Th1-biased phenotype, with the rodent parasite Plasmodium berghei ANKA (PbA) induces a fatal cerebral disease characterized by neurological disorders including paralysia, convulsion and coma. In contrast, BALB/c mice, that present a Th2-biased phenotype, do not develop neurological complications and die at later stages from high parasitemia and anaemia [8,9].
IL-33, the latest member of the IL-1 cytokine family [10], plays an important role in Th2-associated immune responses [11,12]. IL-33 has been linked to a number of inflammatory disorders including allergic asthma, rheumatoid arthritis, allergic rhinitis and ulcerative colitis [13]. IL-33 binds to a heterodimer receptor composed of ST2 (IL-33R) and IL-1R accessory protein, leading to the production of IL-4, IL-5, IL-10 and IL-13 from mast cells, eosinophils, Th2 lymphocytes and the newly discovered population of type 2 innate lymphoid cells (ILC2) [12]. In vitro, IL-33 has also been shown to synergize with IL-4 to drive the polarization of alternatively-activated macrophages (M2) [14], that secrete high levels of IL-10 and TGF-β. We hypothesized that IL-33 could divert the deleterious Th1-immune response during infection and therefore protects mice from ECM.
Results reported here demonstrate that PbA-infected C57BL/6 mice treated with recombinant IL-33 presented no signs of neurological pathology associated with CM and had reduced production of pro-inflammatory cytokines and chemokines. This IL-33-protective effect was mediated by the activation of ILC2 that produced type 2 cytokines which in turn polarized anti-inflammatory M2 macrophages. Furthermore, M2 macrophages expanded Tregs, the depletion of which abrogated the protective effect of IL-33. We therefore present a previously unrecognised role of IL-33 in ECM, and provide evidence that induction of type 2 immunity by an exogenous cytokine treatment was sufficient to down-regulate the inflammatory Th1 response and ECM induced by PbA.
Results
IL-33 treatment protects mice from cerebral malaria induced by PbA
To determine whether IL-33 could modulate malaria pathogenesis, we first infected C57BL/6 mice with Plasmodium berghei ANKA (PbA) parasites (104 parasitized red blood cells, pRBC) and treated the mice with recombinant murine IL-33 (0.2 μg/mouse/day, intraperitoneally) starting from day 0. Body weight loss, parasitemia, clinical score and survival were monitored daily. PbA-infected control mice developed parasitemia, body weight loss and CM symptoms (head deviation, ataxia and paraplegia) from day 5 post infection, and all mice succumbed to CM by day 7–8 (Fig. 1A-D). In contrast, mice treated with IL-33 displayed reduced body weight loss, clinical score and survived up to day 20 post-infection when they were euthanised due to development of hyperparasitemia (up to 40% parasitemia) (Fig. 1D). This indicates that IL-33 administration protects mice from ECM but not from malaria-induced hyperparasitemia and death. Similar results were obtained when the IL-33 treatment began one day after infection (day +1). Similar results were also obtained with 100× higher PbA infective dose (S1A-S1B Fig.).

Adherence of parasited red blood cells (pRBC) to the vascular endothelium of organs plays a key role in the pathogenesis of Plasmodium species allowing the parasite to escape clearance in the spleen [15]. In vivo imaging using luciferase-expressing PbA confirmed that the parasite biomass was significantly reduced in IL-33-treated mice indicating that the reduction in blood parasitemia was not due to an increase of parasite sequestration in the peripheric organs (Fig. 1E, F).
CM is associated with parasite sequestration into the brain microvasculature and cerebral hemorrhage that result from excessive systemic inflammation, which involves pro-inflammatory cytokine production leading to endothelial cell activation and vascular permeability [16]. Using the luciferase-expressing PbA, we found a strong increase in parasite biomass in the brain of PBS-treated mice on day 7 post-infection (Fig. 1G, H). In contrast, IL-33-treated animals showed a significant reduction of luciferase activity in the brain, indicating diminished pRBC accumulation. Histopathological analysis of brains from PBS-treated mice showed microhemorrhages and cytoadhesion of erythrocytes and leucocytes to the brain vasculature on day 7. Mice treated with IL-33 displayed markedly fewer hemorrhages and less vessel obstruction compared to the PBS-treated mice (Fig. 1I, J). Finally, quantitative PCR analysis showed an upregulation of Icam-1 expression in the brain tissues of PbA-infected PBS-treated mice, that was absent in IL-33-treated mice (Fig. 1K), consistent with the reduction of cytoadhesion in IL-33-treated mice compared to the controls.
IL-33 reduces pro-inflammatory cytokine and chemokine production in PbA-infected mice
To investigate how IL-33 might interfer with the host immune response, we measured the levels of the key Th1 cytokines (IFN-γ, IL-12 and TNF-α), key Th2 cytokines (IL-4, IL-5 and IL-13), and the regulatory cytokine IL-10 in the serum at various time-points after infection with PbA. Levels of IFN-γ, IL-12 and TNF-α rose progressively to day 5 in the serum of PBS-treated mice. In contrast, the levels of these cytokines were significantly reduced in IL-33-treated mice (Fig. 2A-C). The levels of serum IL-5 were low in the control mice but were strongly enhanced in IL-33-treated mice, (Fig. 2D). The levels of serum IL-4 and IL-13 were barely detectable in all groups of mice. The concentrations of serum IL-10 also increased in PBS control mice but were reduced in IL-33-treated mice (Fig. 2E). Early production of the pro-inflammatory chemokines IP-10/CXCL10, KC/CXCL1 and MCP-1/CCL2 was also reduced in the serum of IL-33-treated mice compared to PBS-control mice (Fig. 2F-H). We then assessed the expression of lineage-specific transcription factors in CD4+ T cells purified from the spleen of PbA-infected mice. The Th1-specific transcription factor Tbet peaked at day 3 of infection in PBS control mice but was reduced on day 3 and unchanged on day 5 in IL-33-treated mice compared to PBS-treated mice (Fig. 2I). IL-33 did not affect Gata3 expression in purified CD4+ T cells (Fig. 2J), suggesting that Th2 cells are unlikely involved in IL-33-mediated protection. T cell-derived Granzyme B (GrmB) is known to drive cytotoxic T cell-mediated cerebral pathology [17]. We therefore determined GrmB expression in splenic CD8+ and CD4+ T cells. Percentage and frequency of GrmB+CD8+ and GrmB+CD4+ T cells in PbA-infected mice was markedly increased compared to non-infected mice (Fig. 2K, L). Granzyme B positive CD8+ and CD4+ T cells were significantly reduced in percentage and number in IL-33-treated mice compared to PBS-treated control mice (Fig. 2K, L). Together, our results suggest that IL-33-mediated protection against ECM is likely associated with reduction in the early pro-inflammatory type-1 response.

IL-33 induces ILC2 expansion and type 2 cytokine production in PbA-infected mice
Recently, IL-33 has been found to directly induce ILC2 expansion and cytokine production in vitro and in vivo [18–20].We therefore analysed the effect of IL-33 on ILC2 in the ECM model. The frequency and number of ILC2 in the spleen of non-infected (NI) or PbA-infected mice after treatment with PBS or IL-33 were analysed by FACS. A small but consistent percentage of lineage negative CD45+ST2+ICOS+ cells, corresponding to ILC2 [21], was found in the spleen of NI mice in the absence of IL-33-treatment. However IL-33-treatment significantly increased the percentage and number of ILC2 in the spleen in NI and PbA-infected mice compared to PBS-treated mice (Fig. 3A-C). These cells were negative for lineage markers (CD4, CD11b, CD11c, NK1.1, CD3e, Ter119, FcεRI, Siglec F, Gr1, CD49b, CD5, F4/80) and positive for the innate lymphoid cell markers CD127, CD44, Sca-1, IL-1R1 and CD25 (S1C-S1D Fig.). The proportion of Ki67+ cells among splenic ILC2 was higher in IL-33-treated mice, suggesting that at least some of these cells proliferated in situ (Fig. 3D). Intracellular staining revealed that ILC2 from IL-33-treated mice, but not from PBS-treated mice, expressed substantial levels of IL-4, IL-5 and IL-13 after ex vivo PMA-ionomycin stimulation (Fig. 3E-F). We were unable to detect IL-4, IL-5 or IL-13 production by CD4+ T cells or FcεR1+ cells. These data demonstrated that IL-33 not only induced the recruitment and proliferation of ILC2 but further activated these cells to produce Type-2 cytokines in vivo during PbA infection.

To investigate the potential role of ILC2 in IL-33-driven protection from CM, we sorted ILC2 and adoptively transferred them into naïve WT mice. One day after ILC2 transfer, mice were infected with PbA and monitored daily for parasitemia, body weight loss and neurological symptoms. Two injections of IL-33 (0.2 μg/mouse, i.p.) were given to the recipient mice 30 min and 24 h after cell transfer. An earlier study has shown that the provision of IL-33 boosts the survival and cytokine production of the transferred ILC2 cells [22]. A control group infected with PbA and similarly treated with IL-33 confirmed that IL-33 at this dose and schedule of treatment was suboptimal and not protective (Fig. 3G, H). While the two control groups developed severe body weight loss and succumbed to CM by day 7, all the recipient mice given ILC2 exhibited limited clinical disease and survived beyond day 14 (Fig. 3G, H). The parasitemia of ILC2-transferred mice was reduced within the first week of infection compared to PbA control group (Fig. 3I). Mice that received only 2 injections of IL-33, exhibited a slight reduction of parasitemia but nevertheless succumbed to CM by day 7. Histopathology analysis of the brain on day 7 revealed a significant reduction of microhemorrhages and vessel obstruction in the ILC2 recipients compared to the control mice (Fig. 3J, K). The cells producing IL-5 and IL-13 in the ILC2 recipient mice are CD4− T cells and not CD4+ T cells (S2A-S2C Fig.), indicating that they are unlikely to be Th2 cells. These results therefore demonstrate that ILC2 play an important role in the IL-33-mediated protection against ECM.
IL-33 polarizes M2 macrophages
We then investigated the mechanism by which ILC2 protects mice against ECM. Macrophages can be divided into specific subsets according to their polarization environment, phenotype, and function. M1 (classically-activated macrophages) typically produce pro-inflammatory cytokines, including TNF-α and IL-12, whereas M2 (alternatively-activated macrophages) have been implicated in immune regulation, phagocytosis, and tissue remodeling [23,24]. As M2 macrophages are polarized by IL-4 and IL-13, and that these cytokines are produced by ILC2, we therefore assessed the profile of macrophage polarization in mice infected with PbA with or without IL-33 treatment.
The percentage and number of CD11b+F4/80+CD11c− cells in the spleen of mice administered with IL-33 were significantly elevated compared to that treated with PBS (Fig. 4A-C). Expansion of CD11b+F4/80+CD11c− cells was accompanied by increased expression of the key M2 marker (CD206, mannose receptor) and the reduction of the M1 markers (CD86, MHC-II and CD40) on macrophages recovered from IL-33-treated mice compared to that of the PBS control mice (Fig. 4D and S3A Fig.). The expression of other M2 markers (Arginase-1, Ym1/chitinase 3–like 3 and Fizz1/resistin-like α) were also elevated in the spleen whereas the expression of the key M1 marker, Nos2, was reduced in IL-33-treated mice compared to that of the PBS control mice (Fig. 4E). Interestingly, mRNA expression of heme oxygenase-1 (Hmox-1), an enzyme that converts heme into carbon monoxide, was significantly increased in the spleen of IL-33-treated mice compared to PBS-control mice (Fig. 4E). QPCR analysis on sorted CD11b+F4/80+CD11c− cells confirmed that splenic macrophages from IL-33-treated mice, but not from PBS-treated mice, exhibited an M2-polarization status (S3B Fig.). Overall, our data indicate that IL-33 increases macrophage number in the spleen and promotes their polarization towards M2 phenotype.

ILC2 promote M2 macrophage polarization
Since IL-33 alone is not sufficient to fully differenciate M2 macrophages [14], we investigated the potential role of ILC2 in M2 polarization in vitro. Purified ILC2 were co-cultured in a transwell culture with bone marrow-derived macrophages (BMDM) in culture medium alone (M0) or supplemented with IL-4 (M2-polarizing conditions). After 24 h, BMDM were harvested and analysed by qPCR for M2 markers. Under the M0 conditions, BMDM alone did not expressed detectable M2 markers. However, when co-cultured with ILC2, a low level of Arginase-1, Ym1 and Fizz1 RNA became detectable (Fig. 5A). Under the M2-polarizing conditions, these markers were clearly detected and markedly enhanced by the presence of ILC2 (Fig. 5A). Functionally, the polarized M2 macrophages displayed enhanced capability to uptake dextran-FITC or pRBC compared to unpolarized macrophages (S3C-S3D Fig.).

Since ILC2 proliferation and cytokine production are IL-7 - and IL-33-dependent [25], we determined the effect of activated-ILC2 on BMDM. To avoid any direct effect of IL-33 on BMDM, we used ST2-deficient BMDM. When cultured alone, ST2-deficient BMDM did not express any M2 markers even when IL-33 or IL-33 + IL-7 were added to the culture. In the presence of ILC2, the expression of Arginase-1 and Ym1 in the ST2-deficient BMDM was increased, and the expression of these markers were further enhanced by the addition of IL-33 alone or in combination with IL-7 (Fig. 5B), suggesting that activated-ILC2 can produce cytokines involved in M2 polarization. The level of Fizz1 expression was high in the presence of ILC2 alone and was reduced in the presence of IL-33/IL-7. The reason of this reduction is not clear but could be due to over-stimulation of Fizz1 expression. We then analysed the production of Th2 cytokines by ILC2 in vitro. ILC2 alone (without BMDM) were able to produce low levels of IL-4, IL-5 and IL-13 in the culture supernatants. This production was markedly increased after stimulation by IL-33 and IL-7 (Fig. 5C). Flow cytometry analysis of ILC2 confirmed that ILC2 in the culture produced IL-4 and IL-13 and the production was further enhanced by the presence of IL-33 + IL-7 (Fig. 5D, E). The polarized M2 did not produce detectable amount of IL-33.
We next investigated the role of ILC2 in the polarization of M2 in vivo. Sorted ILC2 were adoptively transferred to naïve C57BL/6 mice which were infected with PbA and treated with suboptimal doses of IL-33, as described in Fig. 3F. The protected ILC2 recipients had increased expression of Arginase-1, Ym1 and Fizz1 in their spleen cells (Fig. 5F), indicating that ILC2 are involved in M2 polarization in vivo. Together, our data showed that IL-33-induced ILC2 can effectively drive M2 polarization in vitro and in vivo.
The role of ILC2, M2 and Tregs in IL-33-mediated protection against ECM
Previous studies have implicated Tregs to limit disease and immunopathology in the PbA-induced models of ECM [26–28] and IL-33 has been shown to induce Tregs in vivo [20,29–31]. We therefore explored the impact of IL-33, ILC2 and M2 macrophages on Tregs in the ECM model. First, we noted that the level of Foxp3 message in sorted splenic CD4+ T cells was increased in IL-33-treated mice during PbA-infection compared to PBS-treated mice. The percentage and total number of Foxp3+ splenic CD4+ T cells in the mice infected with PbA were significantly increased by the treatment with IL-33 (Fig. 6A-B). We then determined the effect of adoptively transferred ILC2 in the induction of Tregs in vivo (as described in Fig. 3F). Suboptimal doses of IL-33 led to increased frequency of Foxp3+ cells among splenic CD4+ suggesting that IL-33 alone could induce Treg polarization which was however not sufficient to protect the mice from ECM (see Survival curve and Clinical Score in Fig. 3F). However, the spleens of IL-33-treated PbA-infected mice given ILC2 contained significantly higher frequency of Foxp3+ among CD4+ T cells compared to those not given ILC2 (Fig. 6C-D). To demonstrate a direct link between M2 and Tregs, we co-cultured purified CD4+CD25+ T cells with M2 in the presence of soluble anti-CD3. M2 significantly expanded the Foxp3+ Tregs (Fig. 6E). We also cultured CD4+CD25− T cells under the inducible Treg (iTregs) conditions (plate-bound anti-CD3, soluble anti-CD28 + TGF-β, anti-IL-4 and anti-IFN-γ) in the presence or absence of M2. M2 markedly enhanced the development of iTregs as determined by the frequency of Foxp3+CD4+ T cells (Fig. 6F).

We then investigated whether the Treg population was involved in IL-33-mediated protection from ECM. For this purpose, we used DEREG mice, in which administration of diphtheria toxin (DT) leads to specific-depletion of Tregs due to expression of DT receptor-enhanced Gfp under the control of the Foxp3 promoter [32]. DEREG mice were infected with PbA and treated daily with PBS or IL-33 from the start of infection. DT was administered intraperitoneally every second day from day 1. Treg depletion in IL-33-treated DEREG mice was confirmed in the peripheral blood by flow cytometry (Fig. 7A). As previously reported [33], Treg depletion in PBS-treated DEREG mice has no effect on the parasitemia and survival. PbA-infected DEREG mice treated with PBS died on day 7 with severe ECM (Fig. 7B, C). IL-33-treated infected DEREG mice did not develop CM and died at later stages from hyperparasitemia. In contrast, IL-33-treated PbA-infected DEREG mice that received DT developed cerebral disease and died by day 7. IFN-γ and Granzyme B production by splenic CD8+ T cells, which were significantly reduced in the IL-33-treated mice, was partly reversed in IL-33-treated mice after DT administration (Fig. 7D-E). In addition, serum levels of IFN-γ and IL-12, which were markedly reduced in IL-33-treated mice, were also restored when Tregs were depleted (Fig. 7F).
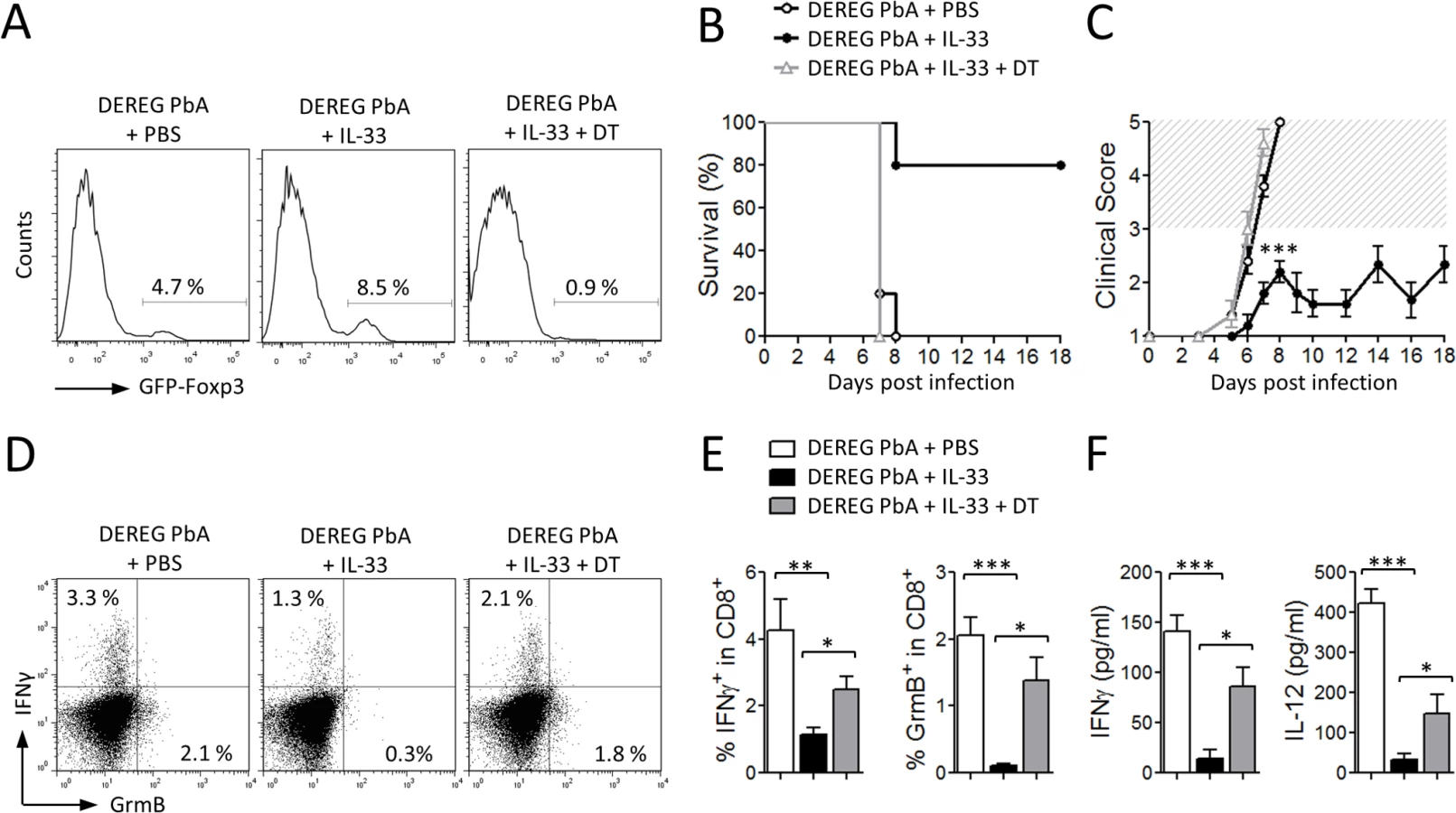
Together these results indicate that IL-33 induces ILC2 which in turn polarize M2 macrophages. M2 can expand Tregs which mediate the suppression of the Th1 response, which is critical to ECM pathogenesis (Fig. 8).

Discussion
Data reported here reveal a previously unrecognised role of IL-33 in the protection against cerebral malaria by reducing pro-inflammatory cytokines and chemokines production and inhibiting vascular sequestration of infected erythrocytes and inflammatory cells in the brain. Furthermore, we provide a plausible mechanistic pathway by which IL-33 induces the expansion of ILC2 which in turn promote the polarization of M2 macrophages and Tregs that are critical for the protection against ECM.
ILC2 have emerged as key players in experimental and clinical diseases [34]. They expand strongly in vivo in response to IL-25 and IL-33, and represent the predominant early source of IL-5 and IL-13 during allergic inflammation and worm infection [25,35]. In PbA-infected mice, exogenous IL-33 induces a robust expansion and mobilization of ILC2 which have the potential to produce IL-4, IL-5 and IL-13. Adoptive transfer of ILC2 markedly ameliorated ECM. IL-33 administration following ILC transfer was necessary to induce the protection, likely because ILC2 require IL-33 stimulation to expand and produce sufficient amount of type 2 cytokines [22]. However, the possibility that IL-33 may also act on other cell types that participate in the protection against ECM cannot be excluded. In vitro, we confirmed that ILC2 can polarize BMDM into M2 macrophages in a cell-cell contact independant manner. IL-33 increased production of IL-4 and IL-13 by ILC2, which can synergize to polarize M2 macrophages [14,36]. This is supported by our data showing that adoptively transferred ILC2 can collaborate with IL-33 to polarize M2 in vivo.
M2 macrophages exhibit potent anti-inflammatory properties and play important roles in parasite clearance, tissue repair and remodeling [37]. One of the proposed mechanism for the immunomodulatory role of M2 macrophages is the competition between Arginase-1 (expressed by M2) and iNOS (expressed by M1) for the subtrate L-Arginine [37]. Another mechanism is the production of carbon monoxide (CO) by heme-oxygenase-1 (HO-1), an enzyme which has been shown to be preferentially expressed in CD206+ M2 macrophages [38]. HO-1 catalyzes the degradation of heme into biliverdin, iron and CO [39]. Here, we found that IL-33-treated mice expressed higher levels of HO-1 in the spleen. Free heme release during Plasmodium spp. infection contributes to blood brain barrier disruption and ECM pathogenesis [40]. CO production by HO-1 has been shown to suppress PbA-induced ECM by inhibiting blood brain barrier disruption, reducing adhesion molecule expression in the brain microvasculature and CD8+ T cell sequestration in the brain [40]. The role of IL-33 in the induction of CO via HO-1 merits further investigation.
Tregs expansion by IL-2/anti-IL-2 complexes in vivo has been implicated to protect mice against T cell-mediated immune pathology in PbA-induced ECM [28], although direct evidence for a role of Tregs in ECM remains elusive. We and others have shown earlier that IL-33 administration leads to Treg induction [20,29–31,41]. Here, we provide data supporting that IL-33-mediated induction of Tregs in PbA-infected mice involves the activity of ILC2 and M2 macrophages. We also show that M2 expand natural Tregs and inducible Tregs in vitro. Importantly, Tregs depletion abrogated the protective effect of IL-33 in ECM by reducing the Th1 cell response. These results therefore demonstrate a cascade of events leading to the protection of ECM by IL-33 (Fig. 8). The detailed mechanism by which Tregs suppress effector T cells, the major immunopathological mediators of ECM, remains to be explored.
It is important to note that IL-33-treated mice produced minimal amount of IL-10 during PbA-infection (Fig. 2E). Furthermore, administration of anti-IL-10 monoclonal antibody in IL-33-treated mice did not affect the protection conferred by IL-33 (S4A-S4B Fig.). Therefore, it is unlikely that M2 - or Tregs-produced-IL-10 participates to ECM protection by IL-33. This is consistent with an earlier report which shows CTLA4-dependent but IL-10-independent protection against ECM [28].
IL-33-treated mice, though consistently showed significant reduction in parasitemia at the early stage of infection (day 5–7) compared to untreated mice, were unable to clear the parasite and eventually died at later stage from hyperparasitemia. IL-33-mediated protection was achieved when the cytokine was given relatively early after infection and delaying treatment by 48 h failed to control the disease. This observation suggests a fine temporal interplay between the protective T cell response against the parasite and the anti-inflammatory response during PbA infection. Spleen is a key site for removal of pRBC during malaria through production of reactive oxygen species and phagocytosis by activated macrophages [42]. Moreover, it has been shown that Flt3L-induced CD11bint F4/80+ red pulp macrophages, which ressemble the macrophages induced by IL-33, displayed higher phagocytic activity and contributed to parasite clearance in PbA-infected mice [43]. Here, we observed that polarized M2 macrophages showed enhanced capacity in dextran-FITC or pRBC uptake compared to unpolarized macrophages (S3C-S3D Fig.). Therefore, the initial reduced parasitemia observed in our model could be explained by local activation and proliferation of red pulp macrophages, which might contribute to the parasite killing. Although it is possible that the low parasitemia contributed to IL-33-mediated protection by reducing PbA antigenic stimulation, it is unlikely an influential mechanism since we found that IL-33 treatment was still equally protective in mice infected with a 100× higher dose of pRBC which displayed a parasitemia > 5% (S1A-S1B Fig.).
IL-33 can directly stimulate eosinophil differentiation and survival [44]. Eosinophil granules contain cytotoxic, highly basic proteins, including the eosinophilic cationic protein that has been shown to inhibit P. falciparum in culture [45]. In our model, although the percentage and number of splenic eosinophils were augmented by 6 folds in IL-33-treated mice compared to untreated mice, eosinophil depletion using anti-Siglec-F antibody did not affect IL-33-mediated protection during PbA infection (S4C-S4G Fig.) suggesting that eosinophils are unlikely an influential mechanism in IL-33-mediated protection against ECM.
Increased number of CD4+CD25+Foxp3+ Tregs have been observed in humans infected with Plasmodium falciparum [27,46,47]. It would be of considerable interest to investigate if the observation reported here is also applicable to clinical cerebral malaria.
Materials and Methods
Animals
Female C57BL/6 mice (8–10 weeks old) were obtained from Charles River UK Ltd. ST2−/− female mice (on the C57BL/6 genetic background) were originally provided by Dr. Andrew McKenzie (Medical Research Council Laboratory of Molecular Biology, Cambridge,U.K.) and bred in-house in a pathogen-free facility at University of Glasgow. DEREG mice (on the C57BL/6 genetic background, originally provided by Dr. Tim Sparwasser, Hannover Medical School, Germany) were bred at Transgenose Institute animal facility (UPS44 CNRS, Orleans, France). Mice under procedure were kept in polyethylene boxes with free access to food and water, and subjected to 12 h light-dark cycles. All experiments were performed in accordance with the UK Home Office guidelines and within the terms of the Project License (PPL 70/7293) granted for this work under the Animals (Scientific Procedures) Act 1986. All efforts were made to minimize the number of animals used and their suffering.
Experimental infection
C57BL/6 red blood cells infected with Plasmodium berghei ANKA parasites expressing a green fluorescent protein (PbA GFPcon 259cl2, MRA-865, deposited by CJ Janse and AP Waters) were stored in liquid nitrogen and thawed and passed into wild-type mice that served as parasite donor. All mice, unless otherwise stated, were inoculated intravenously (i.v.) into the tail vein with 1×104 parasitized red blood cells (pRBC). Parasitemia was monitored daily (from day 4) by flow cytometry using the FL3 channel (PbA-GFP) and TER-119 APC (erythrocytes) in a BD FACScalibur cytometer (BD Biosciences). Clinical score was assessed using the following clinical scale: 1 = no signs; 2 = ruffled fur and/or abnormal posture; 3 = lethargy; 4 = reduced responsiveness to stimulation and/or ataxia and/or respiratory distress/hyperventilation; and 5 = prostration and/or paralysis and/or convulsions. All animal that reachs stage 4 developped ECM. Recombinant murine IL-33 (Biolegend) was injected intraperitoneally (0.2 μg/mouse/200 μl) daily, routinely from the beginning of infection (day 0). As a control for IL-33 effects, non-infected (NI) mice also received IL-33 for 5 consecutive days. For some experiments, mice were administered with anti-Siglec F (MAB17061, R&D Systems, 50 μg daily) or anti-IL-10 (MAB417, R&D Systems, 40 μg daily) or appropriate isotype control Abs.
Bioluminescence imaging with IVIS
Mice infected with the transgenic PbA strain that constitutively expresses luciferase (PbA-luc, gift of Dr. AP Waters, Glasgow, UK) were imaged using an IVIS Imaging 100 system (Xenogen Corp.) Mice were injected intraperitoneally with D-luciferin (PerkinElmer, 150 mg/kg in DPBS) and anesthetized in 5% isoflurane/1L O2.min−1 atmosphere. The animals were then placed in the imaging chamber of the IVIS and anesthesia was maintained using 2% isoflurane/0.2L O2 per mouse min−1 atmosphere. Bioluminescence (photons per second per square centimeter per steridian) was monitored over a 20 min period in previously defined regions of interest (ROI). Exposure times varied between 0.5 and 1 min, depending on signal intensity. To standardize imaging and to allow comparison between mice, the images presented in the figures were taken once luminescence plateaued. For brain bioluminescence imaging, mice were sacrificed on day 7, perfused with 20 ml ice-cold PBS and the whole brains were excised and imaged ex vivo as described previously [48]. To enhance the signal and avoid desiccation, 100 μl D-luciferin (150 μg/ml) was pipetted onto the surface of each brain 5–10 min prior to imaging.
Histology
After intracardiac perfusion with 20 ml ice-cold PBS the brain was removed, fixed with 4% neutral phosphate-buffered formalin (Merck) and embedded in paraffin. The tissue were cut into 4 μm sections and stained with hematoxylin-eosin (H&E) following standard procedures. Brain microvascular obstruction in coronal brain sections was scored by two independent observers blinded to the experimental groups using a Nikon Eclipse E400 microscope at ×400. For each brain, fields containing vessels were scored using a semi-quantitative scale (0–5) according to the severity of obstruction and the presence of microhaemorrhages: 0, no obstruction; 1, only small vessels obstructed; 2, presence of leukocytes attached to the endothelium; 3, partial obstruction, presence of leukocytes and RBC; 4, total obstruction, without haemorrhages; 5, total obstruction with haemorhages. Data are presented as the average score for each brain.
Real-time PCR
Brains and spleens were excised at indicated time-points and preserved in RNAlater (Qiagen). CD4+ T cells were purified from total splenocytes at indicated time-point by negative selection (AutoMACS, Miltenyi Biotec) with 85–90% purity. After homogenization in TRIzol (Sigma-Aldrich), total RNA was extracted with an RNeasy Mini kit (Qiagen). cDNA was synthesized using M-MLV Reverse Transcriptase (Promega). The quantitative RT-PCR (qPCR) assays were performed using TaqMan Real-Time PCR Master Mix in an ABI PRISM 7500 Fast Sequence Detection System (Applied Biosystems). Relative expression levels were calculated as ΔCt values by normalizing Ct values of target genes to Ct values of hypoxanthine phosphoribosyl transferase-1 (Hprt1). Data are represented as relative % of Hprt1 expression. All primers were purchased from Applied Biosystems (TaqMan Gene Expression Assay).
Flow cytometry analysis
Cells were first blocked with FcγR blocker and stained with fluorochrome labeled Abs or their corresponding isotype controls. Abs were purchased from BD Bioscience, Biolegend or eBioscience. The following Abs were used: anti-ST2 (DJ8), anti-CD45 (30-F11), anti-ICOS (C398.4A), anti-CD11b (M1/70), anti-F4/80 (BM8), anti-CD11c (N418), anti-CD40 (1C10), anti-CD206 (C068C2), anti-CD86 (GL1), anti-MHC-II (M5/114.15.2), anti-IL-4 (11B11), anti-IL-5 (TRFK5), anti-IL-13 (eBio13A), anti-Granzyme B (NGZB). For intracellular cytokine staining, cells were incubated for 4 h with phorbol-12-myristate-13-acetate (50 ng/ml; Sigma-Aldrich), ionomycin (750 ng/ml; Sigma-Aldrich) and GolgiStop (1 μl/ml; BD Biosciences). After surface staining, cells were fixed and permeabilized with BD Fixation/permeabilization kit (BD Biosciences) and stained for intracellular cytokines. For all experiments, cells were stained with a Live/Dead Fixable dye (Molecular Probes) to allow gating on viable cells. Data were acquired using a Beckman Coulter CyAn ADP (Beckman Coulter, USA). Gating strategy and analysis were performed using the FlowJo software (treeStar Software, USA) and shown in S1C Fig.
ILC2 sorting and adoptive transfer
To induce ILC2 in vivo, naive C57BL/6 mice were inoculated intranasally with 1 μg recombinant IL-33 (BioLegend) on five consecutive days. Lung tissue was digested with Liberase TL (Roche, 0.2 mg/ml) and DNAse I (Sigma, 0.5 mg/ml) for 45 min at 37°C under rotation. Total lung cells were stained with lineage cocktail Abs (anti-CD3ε, anti-CD11b, anti-CD11c, anti-NK1.1, anti-siglec F, anti-FcεRI, anti-B220), anti-CD45, anti-ST2 and anti-ICOS Abs for 30 min at 4°C. ILC2 were sorted by FACSAria (BD Biosciences) (purity >98%). ILC2 were defined as CD45+ICOS+ST2+ lymphoid cells negative for lineage markers as described previously [18]. For adoptive transfer, 2×106 freshly purified ILC2 were injected i.v. to naive C57BL/6 mice, which were infected i.v. with 104 pRBC 24 h later. Mice were then treated with IL-33 (0.2 μg, i.p.) 30 min and 24 h after cell transfer.
Bone Marrow Derived Macrophages/ILC2 co-culture and phagocytosis assay
Bone marrow cells were harvested from femur bones of C57BL/6 WT or ST2-deficient mice and cultured in petri dishes in complete medium [RPMI-1640 supplemented with 10% (vol/vol) FCS (LONZA), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin] containing 25% L929 cell-conditioning medium as a source of macrophage colony-stimulating factor (M-CSF) to differentiate into bone marrow-derived macrophages (BMDM). BMDM were harvested on day 6 and co-cultured (106 cells) in the lower chamber of a 24-well transwell plate (0.4 μM porous membrane, Corning) under M0 polarizing conditions (complete medium only) or M2 polarizing conditions (+ 10 ng/ml IL-4). In some experiments, 2×105 freshly sorted ILC2 were added in the upper chamber of the transwell. Cells from the lower chamber were collected for RNA analysis after 24 h co-culture.
For phagocytosis assay, polarized M0 or M2 (from BMDM) were plated at 106 cells/ml and incubated overnight in complete medium. FITC-labeled Dextran (Sigma, 1 mg/ml) or PbA-GFP-parasitized RBC (ratio macrophage:pRBC, 1 : 100) were then added and the cells were incubated at 4°C (controls) or 37°C for 30 min. Cells were harvested, washed and analyzed by FACS (Beckman Coulter CyAn ADP). At least 20,000 events were collected and data were analyzed by FlowJo software, and changes were presented as percentage of FITC+ or GFP+ cells.
Tregs/macrophages co-cultures
CD4+CD25+ T cells were purified (AutoMACS, Miltenyi) from the spleen and lymph nodes of naïve C57BL/6 mice and cultured (5×105 cells/ml) with equal number of BMDM-derived M2 for 2 days. Foxp3 expression was determined by FACS gated on live CD4+ cells. In some experiments, CD4+CD25− T cells from naïve C57BL/6 mice were cultured (5×105 cells/ml) for 2 days under iTreg polarizing conditions (3 μg plate-bound anti-CD3, 1.5 μg soluble anti-CD28 + 10 ng/ml TGF-β, 10 μg/ml anti-IL-4 and anti-IFN-γ) in the presence or absence of equal number of M2. Foxp3 expression was determined by FACS gated on live CD4+ cells.
ELISA and multiplex
ELISA for serum IL-13 (Ebioscience), IL-4, IL-5, CXCL1, CXCL10, CCL2 (all from R&D Systems) were performed following the manufacturer’s instructions. Sensitivity of the assays was between 20 and 40 pg/ml. Concentrations of serum IFN-γ, IL-4, IL-5, IL-10, IL-12 and TNF-α were determined using a multiplex mouse cytokine assay (Invitrogen) according to the manufacturer’s instructions using a Luminex 200 reader (Luminex Corp.).
Statistical analysis
Comparisons between 2 groups were performed using a 2-tailed unpaired Student’s t test. Multiple groups were compared using a 2-way ANOVA followed by a Bonferroni’s post-test. Values for all measurements are expressed as mean ± SEM. P<0.05 was considered statistically significant. Data are representative of at least 3 separate experiments unless otherwise stated in the legend. Statistical analysis were performed using GraphPad Prism 5.0. (GraphPad Software).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. World Health Organization (2012) World Malaria Report 2012.
2. Belnoue E, Potter SM, Rosa DS, Mauduit M, Gruner AC, et al. (2008) Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol 30 : 544–553. doi: 10.1111/j.1365-3024.2008.01053.x 18665903
3. Campanella GS, Tager AM, El Khoury JK, Thomas SY, Abrazinski TA, et al. (2008) Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc Natl Acad Sci U S A 105 : 4814–4819. doi: 10.1073/pnas.0801544105 18347328
4. van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE (2006) A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol 22 : 503–508. doi: 10.1016/j.pt.2006.09.002 16979941
5. Shikani HJ, Freeman BD, Lisanti MP, Weiss LM, Tanowitz HB, et al. (2012) Cerebral malaria: we have come a long way. Am J Pathol 181 : 1484–1492. doi: 10.1016/j.ajpath.2012.08.010 23021981
6. Angulo I, Fresno M (2002) Cytokines in the pathogenesis of and protection against malaria. Clin Diagn Lab Immunol 9 : 1145–1152. 12414742
7. Knowles SC (2011) The effect of helminth co-infection on malaria in mice: a meta-analysis. Int J Parasitol 41 : 1041–1051. doi: 10.1016/j.ijpara.2011.05.009 21777589
8. de Kossodo S, Grau GE (1993) Profiles of cytokine production in relation with susceptibility to cerebral malaria. J Immunol 151 : 4811–4820. 8409439
9. Bagot S, Campino S, Penha-Goncalves C, Pied S, Cazenave PA, et al. (2002) Identification of two cerebral malaria resistance loci using an inbred wild-derived mouse strain. Proc Natl Acad Sci U S A 99 : 9919–9923. doi: 10.1073/pnas.152215199 12114535
10. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, et al. (2005) IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23 : 479–490. doi: 10.1016/j.immuni.2005.09.015 16286016
11. Liew FY, Pitman NI, McInnes IB (2010) Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol 10 : 103–110. doi: 10.1038/nri2692 20081870
12. Mirchandani AS, Salmond RJ, Liew FY (2012) Interleukin-33 and the function of innate lymphoid cells. Trends Immunol 33 : 389–396. doi: 10.1016/j.it.2012.04.005 22609147
13. Liew FY (2012) IL-33: a Janus cytokine. Ann Rheum Dis 71 Suppl 2: i101–104. doi: 10.1136/annrheumdis-2011-200589 22460136
14. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, et al. (2009) IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol 183 : 6469–6477. doi: 10.4049/jimmunol.0901575 19841166
15. Franke-Fayard B, Fonager J, Braks A, Khan SM, Janse CJ (2010) Sequestration and tissue accumulation of human malaria parasites: can we learn anything from rodent models of malaria? PLoS Pathog 6: e1001032. doi: 10.1371/journal.ppat.1001032 20941396
16. Gimenez F, Barraud de Lagerie S, Fernandez C, Pino P, Mazier D (2003) Tumor necrosis factor alpha in the pathogenesis of cerebral malaria. Cell Mol Life Sci 60 : 1623–1635. doi: 10.1007/s00018-003-2347-x 14504653
17. Haque A, Best SE, Unosson K, Amante FH, de Labastida F, et al. (2011) Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J Immunol 186 : 6148–6156. doi: 10.4049/jimmunol.1003955 21525386
18. Salmond RJ, Mirchandani AS, Besnard AG, Bain CC, Thomson NC, et al. (2012) IL-33 induces innate lymphoid cell-mediated airway inflammation by activating mammalian target of rapamycin. J Allergy Clin Immunol 130 : 1159–1166 e1156. doi: 10.1016/j.jaci.2012.05.018 22738676
19. Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, et al. (2012) Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci U S A 109 : 3451–3456. doi: 10.1073/pnas.1201042109 22331917
20. Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, et al. (2013) Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 210 : 535–549. doi: 10.1084/jem.20121964 23420878
21. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, et al. (2012) Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol 129 : 191–198 e191–194. doi: 10.1016/j.jaci.2011.09.041 22079492
22. Barlow JL, Peel S, Fox J, Panova V, Hardman CS, et al. (2013) IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol 132 : 933–941. doi: 10.1016/j.jaci.2013.05.012 23810766
23. Gordon S, Taylor PR (2005) Monocyte and macrophage heterogeneity. Nat Rev Immunol 5 : 953–964. doi: 10.1038/nri1733 16322748
24. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M (2013) Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol 229 : 176–185. doi: 10.1002/path.4133 23096265
25. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, et al. (2010) Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464 : 1367–1370. doi: 10.1038/nature08900 20200518
26. Amante FH, Stanley AC, Randall LM, Zhou Y, Haque A, et al. (2007) A role for natural regulatory T cells in the pathogenesis of experimental cerebral malaria. Am J Pathol 171 : 548–559. doi: 10.2353/ajpath.2007.061033 17600128
27. Vigario AM, Gorgette O, Dujardin HC, Cruz T, Cazenave PA, et al. (2007) Regulatory CD4+ CD25+ Foxp3+ T cells expand during experimental Plasmodium infection but do not prevent cerebral malaria. Int J Parasitol 37 : 963–973. doi: 10.1016/j.ijpara.2007.01.004 17350019
28. Haque A, Best SE, Amante FH, Mustafah S, Desbarrieres L, et al. (2010) CD4+ natural regulatory T cells prevent experimental cerebral malaria via CTLA-4 when expanded in vivo. PLoS Pathog 6: e1001221. doi: 10.1371/journal.ppat.1001221 21170302
29. Brunner SM, Schiechl G, Falk W, Schlitt HJ, Geissler EK, et al. (2011) Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int 24 : 1027–1039. doi: 10.1111/j.1432-2277.2011.01306.x 21797940
30. Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, et al. (2011) IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol 187 : 4598–4610. doi: 10.4049/jimmunol.1100519 21949025
31. Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, et al. (2012) IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur J Immunol 42 : 1804–1814. doi: 10.1002/eji.201141947 22585447
32. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, et al. (2007) Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med 204 : 57–63. doi: 10.1084/jem.20061852 17200412
33. Steeg C, Adler G, Sparwasser T, Fleischer B, Jacobs T (2009) Limited role of CD4+Foxp3+ regulatory T cells in the control of experimental cerebral malaria. J Immunol 183 : 7014–7022. doi: 10.4049/jimmunol.0901422 19890049
34. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, et al. (2013) Innate lymphoid cells—a proposal for uniform nomenclature. Nat Rev Immunol 13 : 145–149. doi: 10.1038/nri3365 23348417
35. Monticelli LA, Sonnenberg GF, Artis D (2012) Innate lymphoid cells: critical regulators of allergic inflammation and tissue repair in the lung. Curr Opin Immunol 24 : 284–289. doi: 10.1016/j.coi.2012.03.012 22521139
36. Lawrence T, Natoli G (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11 : 750–761. doi: 10.1038/nri3088 22025054
37. Biswas SK, Mantovani A (2010) Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11 : 889–896. doi: 10.1038/ni.1937 20856220
38. Choi KM, Kashyap PC, Dutta N, Stoltz GJ, Ordog T, et al. (2010) CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology 138 : 2399–2409, 2409 e2391. doi: 10.1053/j.gastro.2010.02.014 20178793
39. Cairo G, Recalcati S, Mantovani A, Locati M (2011) Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol 32 : 241–247. doi: 10.1016/j.it.2011.03.007 21514223
40. Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, et al. (2007) Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med 13 : 703–710. doi: 10.1038/nm1586 17496899
41. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, et al. (2014) The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513 : 564–568. doi: 10.1038/nature13577 25043027
42. Buffet PA, Safeukui I, Deplaine G, Brousse V, Prendki V, et al. (2011) The pathogenesis of Plasmodium falciparum malaria in humans: insights from splenic physiology. Blood 117 : 381–392. doi: 10.1182/blood-2010-04-202911 20852127
43. Tamura T, Akbari M, Kimura K, Kimura D, Yui K (2014) Flt3 ligand treatment modulates parasitemia during infection with rodent malaria parasites via MyD88 - and IFN-gamma-dependent mechanisms. Parasite Immunol 36 : 87–99. doi: 10.1111/pim.12085 24400637
44. Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY (2010) IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol 185 : 3472–3480. doi: 10.4049/jimmunol.1000730 20693421
45. Waters LS, Taverne J, Tai PC, Spry CJ, Targett GA, et al. (1987) Killing of Plasmodium falciparum by eosinophil secretory products. Infect Immun 55 : 877–881. 3549562
46. Couper KN, Blount DG, Wilson MS, Hafalla JC, Belkaid Y, et al. (2008) IL-10 from CD4CD25Foxp3CD127 adaptive regulatory T cells modulates parasite clearance and pathology during malaria infection. PLoS Pathog 4: e1000004. doi: 10.1371/journal.ppat.1000004 18401464
47. Walther M, Jeffries D, Finney OC, Njie M, Ebonyi A, et al. (2009) Distinct roles for FOXP3 and FOXP3 CD4 T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS Pathog 5: e1000364. doi: 10.1371/journal.ppat.1000364 19343213
48. Fauconnier M, Bourigault ML, Meme S, Szeremeta F, Palomo J, et al. (2011) Protein kinase C-theta is required for development of experimental cerebral malaria. Am J Pathol 178 : 212–221. doi: 10.1016/j.ajpath.2010.11.008 21224058
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Control of Murine Cytomegalovirus Infection by γδ T Cells
- Dimorphism in Fungal Pathogens of Mammals, Plants, and Insects
- ATPaseTb2, a Unique Membrane-bound FoF1-ATPase Component, Is Essential in Bloodstream and Dyskinetoplastic Trypanosomes
- Rational Development of an Attenuated Recombinant Cyprinid Herpesvirus 3 Vaccine Using Prokaryotic Mutagenesis and In Vivo Bioluminescent Imaging
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy