
A Membrane-bound eIF2 Alpha Kinase Located in Endosomes Is Regulated by Heme and Controls Differentiation and ROS Levels in
Trypanosoma cruzi proliferates as epimastigotes in the midgut of the insect vector filled with blood meal. There, it accumulates nutrients in specific endosomal organelles. The parasite moves towards the hindgut and when the blood is completely digested, these organelles are consumed. At this moment, the insect is ready for a new feeding cycle that promotes the release of infective metacyclic-trypomastigote forms. We have previously found that such differentiation involves protein synthesis arrest through the phosphorylation of the eukaryotic translation initiation factor 2α (eIF2α). Now, we show that one of the kinases (TCK2) that phosphorylate eIF2α is localized in these endosomes. TcK2 binds and is specifically inhibited by heme derived from blood hemoglobin. We also found that heme inhibits differentiation, suggesting that it is an important signal for differentiation. By generating knockouts of TcK2, we observed an increased accumulation of heme in the cytosol, which induced cellular damage by affecting the reactive oxygen metabolism in the parasite. We conclude that this eIF2α kinase senses cytosolic heme obtained from the blood meal, promoting its storage in the cytosolic organelles. When heme levels are decreased in the cytosol, TcK2 activation can then arrest protein synthesis that is followed by the induction of the differentiation of proliferative epimastigote forms to infective metacyclic-trypomastigotes.
Published in the journal:
. PLoS Pathog 11(2): e32767. doi:10.1371/journal.ppat.1004618
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004618
Summary
Trypanosoma cruzi proliferates as epimastigotes in the midgut of the insect vector filled with blood meal. There, it accumulates nutrients in specific endosomal organelles. The parasite moves towards the hindgut and when the blood is completely digested, these organelles are consumed. At this moment, the insect is ready for a new feeding cycle that promotes the release of infective metacyclic-trypomastigote forms. We have previously found that such differentiation involves protein synthesis arrest through the phosphorylation of the eukaryotic translation initiation factor 2α (eIF2α). Now, we show that one of the kinases (TCK2) that phosphorylate eIF2α is localized in these endosomes. TcK2 binds and is specifically inhibited by heme derived from blood hemoglobin. We also found that heme inhibits differentiation, suggesting that it is an important signal for differentiation. By generating knockouts of TcK2, we observed an increased accumulation of heme in the cytosol, which induced cellular damage by affecting the reactive oxygen metabolism in the parasite. We conclude that this eIF2α kinase senses cytosolic heme obtained from the blood meal, promoting its storage in the cytosolic organelles. When heme levels are decreased in the cytosol, TcK2 activation can then arrest protein synthesis that is followed by the induction of the differentiation of proliferative epimastigote forms to infective metacyclic-trypomastigotes.
Introduction
The phosphorylation of the alpha subunit of the trimeric eukaryotic initiation factor 2 (eIF2) complex is a key event in the cellular stress response [1]. This phosphorylation inhibits the conversion of GDP to GTP in eIF2 by the eIF2B, a guanosine exchange factor, thus decreasing the levels of the ternary complex eIF2-GTP-Met-tRNAi available for new rounds of translation initiation. At the same time that global translation is inhibited, there is preferential translation of specific messages that promote the remediation from stresses in several eukaryotes [2]. The phosphorylation of eIF2α is catalyzed by serine-threonine kinases specifically activated by the different stress conditions [3]. Mammals have four kinases that phosphorylate eIF2α: a) The general control nonderepressible 2 (GCN2) kinase (KEGG K16196, also known as eIF2AK4), which is activated by amino acid deprivation through binding to uncharged tRNAs [4]. b) The RNA-activated protein kinase (PKR) (KEGG 16195, also known as eIF2AK2), which is activated by double strand RNA in cells infected with viruses [5, 6, 7]. c) The PKR-like endoplasmic reticulum kinase (PERK) (KEGG K08860, also known as eIF2AK3), which is activated by unfolded proteins in the endoplasmic reticulum [8]. d) The heme-regulated eIF2α kinase (HRI) (KEGG K16194, also known as eIF2AK1), which is activated by heme deficiency [9]. Different stress conditions result in conformational changes within the catalytic domain of these kinases leading to autophosphorylation and activation, allowing for the binding and subsequent phosphorylation of the eIF2α substrate.
Similar kinases of eIF2α are present in other organisms and are involved in stress sensing and remediation [10]. While GCN2, or a GCN2-like kinase, is present in virtually all eukaryotic organisms, other eIF2α kinases are more scattered through different organisms. For example, HRI is present in some fungi including Schizosaccharomyces pombe. PERK orthologues have been identified in worms, insects and vertebrates. Specific phosphorylation of eIF2α by unique kinases is also directly involved in responses to environmental changes throughout the lifecycle of protozoan parasites (reviewed in [11, 12]). For example, the eIF2α kinase eIK2 controls the latency of Plasmodium sporozoites in insect salivary glands through eIF2α phosphorylation [13]. The eIK1 is homologous to GCN2, as it is activated by amino acid starvation, regulating the erythrocyte cycle of Plasmodium [14]. The PK4 has a topology similar to PERK and controls the growth of schizonts and gametocytes of Plasmodium [15]. In Toxoplasma gondii eIF2α phosphorylation takes place during the differentiation to bradyzoites, which remain latent in the vertebrate host, or during parasite passage through the extracellular medium, when the parasite is subjected to lower amino acid levels [16, 17, 18].
The phosphorylation of eIF2α is also involved in the control of trypanosome differentiation in response to variations in nutrients and temperature. In these organisms, there are three putative eIF2α kinases: a) K1, which is similar to GCN2, b) K2 with a topology similar to PERK, and c) K3, with little similarity to the known eIF2 kinases [19]. The physiological significance of these kinases is mostly unknown. In Leishmania, the K2 ortholog (LinPERK) is localized in the endoplasmic reticulum and regulates the differentiation of promastigotes into amastigotes by unknown stimulus [20, 21]. Interestingly, in Trypanosoma brucei the homologous kinase (TbeIF2αK2) is localized in the flagellar pocket membrane, site of intense membrane internalization, and early endocytic vesicles [19]. TbeIF2αK3 has been recently shown to migrate from the endoplasmic reticulum to the nucleus upon persistent stress, abrogating transcription events [22]. In Trypanosoma cruzi, we have demonstrated that eIF2α phosphorylation is required for the differentiation of the non-infective epimastigotes into infective metacyclic trypomastigotes (metacyclogenesis) stimulated by starvation [23], mimicking what happens in the insect hindgut [24, 25]. Metacyclogenesis takes about 72 h to be accomplished when the parasites finally cease translation [23]. As nutrients stored in endosome-like organelles, also called reservosomes, are consumed during metacyclogenesis [26], we presumed that TcK2, the kinase homologous to the T. brucei flagellar pocket eIF2 kinase (TbeIF2αK2) might be involved in T. cruzi differentiation. Herein, we studied the expression and the localization of TcK2 kinase in different life stages of the parasite and generated knockout parasites of this kinase. We provide evidence that in T. cruzi, TcK2 is located in the endosomal compartment and it is regulated by heme, controlling the parasite’s growth and differentiation.
Results
TcK2 is a membrane bound eIF2α kinase that undergoes autophosphorylation and phosphorylates the parasite eIF2α
The predicted topology of T. cruzi K2 was found similar to mammalian PERK. TcK2 contains a kinase domain at its C-terminal, a transmembrane domain in the middle, and a signal peptide at its N-terminal (Fig. 1A). Polyclonal antibodies generated against the kinase domain of TbeIF2αK2 [19] and a monoclonal antibody (mAb) 5D10 obtained from mice immunized with the TcK2 N-terminal region recognized specifically two bands (140–160 kDa) in Western blots of total parasite lysates (Fig. 1B). The other bands seen in the Western blot were either degradation products, or non-specific antibody labeling, as they were not seen in all experiments. The protein is associated with parasite membranes as it could be solubilized only in the presence of detergents, in contrast to the soluble cytosolic heat shock protein (Hsp) 70 (Fig. 1C). Moreover, TcK2 was only solubilized in high concentrations of digitonin, differently from BiP, present in the endoplasmic reticulum lumen or the Hsp70, located in the cell cytosol (Fig. 1D).

The kinase domain of TcK2 fused with the glutathione S-transferase (GST) was expressed in E. coli. The recombinant GST-TcK2, but not GST, obtained from E. coli was able to phosphorylate the purified His6x-tagged-TceIF2α in vitro, as shown in Fig. 1E. In addition, it became phosphorylated, as indicated by the presence of the second labeled band in the autoradiogram with the expected size of GST-TcK2 (68 kDa). Phosphorylation of TcK2 was also suggested to occur in vivo, as treatment of cell lysates with calf intestine alkaline phosphatase (CIAP) decreased the relative intensity of the slow migrating band (Fig. 1F). In addition, pre-treatment of live parasites with calyculin A, a general phosphatase inhibitor, increased the levels of the slow migrating band. These observations indicated that this slow migrating band might correspond to the autophosphorylated and active form of the TcK2 kinase.
T. cruzi K2 is localized in endosomes and the upper phosphorylated band predominates in non-proliferative stages
Immunofluorescence showed that TcK2 is located in cellular organelles in the epimastigote form of the parasite (Fig. 2A). This form corresponds to the stage that proliferates in the gut of the insect vector. The same result was obtained with a C-terminal HA-tagged TcK2 expressed in epimastigotes (Fig. 2B). TcK2 was found expressed in both replicative and infective forms (Fig. 2A and 2C). In epimastigotes and amastigotes, respectively the forms that proliferate in the insect gut and in the cytosol of mammalian cells, the enzyme showed poor colocalization with BiP, an ER-resident protein (Fig. 2A). Increased, but incomplete colocalization with BiP was observed in trypomastigote stages, which are the infective and non-proliferative forms. Interestingly, in proliferating epimastigotes and amastigotes, the lower band, which may correspond to the non-phosphorylated form of TcK2, predominated while in the infective forms (metacyclic-trypomastigotes and cell-derived trypomastigotes) the upper phosphorylated band was predominant (Fig. 2C), suggesting that the kinase could be more active in the infective forms.

Hemin starvation causes an increase in the slow migrating TcK2 form
Epimastigotes proliferate in the insect gut, a compartment full of blood meal. The parasite stores different nutrients, including heme [27] and lipids, such as cholesterol [28], in organelles derived from endosomes called reservosomes [29]. As nutrients become scarce, differentiation of epimastigotes into metacyclic-trypomastigotes is triggered and concomitantly these endosomes are consumed [26]. As heme is largely present in the blood meal and is required for epimastigote growth [30], we tested the effect of adding heme in the form of hemin into the culture medium and checked parasite growth, TcK2 localization and the enzyme migration by gel electrophoresis. As shown in Fig. 3A, parasites maintained in heme-rich medium (LIT with hemin) propagated faster than parasites cultivated in medium without heme supplementation, as shown previously [31]. In the heme-rich medium, TcK2 was detected mainly in endosomes, co-localizing with cruzipain (Fig. 3B) a protease present in these organelles [32]. Both proteins were found more diffuse in the parasites maintained in media with reduced amounts of hemin (Fig. 3B). This is compatible with the different solubilization of TcK2 by digitonin in trypomastigote forms when compared to epimastigotes (S1 Fig. and Fig. 1D), or when cells were maintained in reduced amounts of hemin (Fig. 2A), suggesting a different localization of TcK2 in the various parasite stages.

In parasites cultivated in medium supplemented with hemin, electron microscopy showed more organelles appearing as electron-dense vesicles, while in hemin poor medium electron-lucent structures predominate (Fig. 3C). These results are in agreement with the proposal that heme causes the elevated electron density of these T. cruzi structures, located in the posterior end of the cell, possibly reservosomes, based on incorporation of tracers added externally to parasites [32].
More importantly, in the absence of hemin, the upper band of TcK2 predominated in total parasite lysates (Fig. 3D). Similarly, when parasites were transferred to saline buffer (PBS), the upper band of TcK2 prevailed and this was inhibited by addition of hemin (Fig. 3E). These results indicate that heme accumulation in the endosomes could be related to TcK2 inactivation.
Hemin binds to and specifically inhibits TcK2
Two putative heme binding sites in the catalytic domain of TcK2 could be predicted based on sequence analysis [33], as shown in Fig. 4A. These sites are represented in more detail as horizontal bars in the alignment of the kinase domain of HRI, PERK and the two kinases of T. cruzi (S2 Fig.). Indeed, a GST-TcK2 recombinant protein displayed a typical Soret spectral band, with a peak from 410 to 420 nm in the presence of heme, not seen in the GST-TcK2 alone, as detected by spectroscopy analysis (Fig. 4B). This spectral band is formed in proteins that bind to heme. Quantitative assays using different amounts of hemin revealed that the increase in absorbance at 410 nm was saturable, suggesting specific binding and allowing us to estimate an apparent heme binding constant (Kdapp) of 2.8 ± 0.6 μM (Fig. 4C).

To test the effect of heme in the regulation of TcK2 activity, we assessed the in vitro phosphorylation of TceIF2α by GST-TcK2 in the presence of heme and several analogs. Heme (Fe-Protoporphyrin IX) was able to inhibit the activity of GST-TcK2, while protoporphyrin IX, mesoporphyrin IX, and Sn protoporphyrin had no effect (Fig. 4D). Importantly, this inhibitory effect was specific, as the PK4 kinase from P. falciparum did not bind to heme and heme did not inhibit eIF2α phosphorylation by PK4 (S3B Fig.) [15].
When we analyzed the effect of heme and its analogs, we also found that only heme inhibited the differentiation of epimastigotes into metacyclic trypomastigotes (Fig. 4E). As previously reported, the differentiation depends on eIF2α phosphorylation and protein synthesis attenuation [23]. Taken together, these data suggest that heme binding inactivates TcK2, thus inhibiting T. cruzi differentiation.
TcK2 null cells show decreased differentiation and proliferation rates
To better understand the TcK2 function in the parasite, we generated TcK2-null cell lines by a two-step gene replacement through homologous recombination and antibiotic selection as illustrated in Fig. 5A. The integration of resistance genes (Neo and Hygro, or Blast) in the TcK2 locus of the two homologous chromosomes was confirmed by PCR using specific primers (Fig. 5B). The TcK2 gene was not amplified from the double KOs (−/−) (Fig. 5C) and the cells did not express the kinase as shown by Western blot (Fig. 5D) and immunofluorescence assays (Fig. 5E). TcK2 null cells still expressed cruzipain in organelles (Fig. 5E), showing that the kinase is not necessary for cruzipain incorporation into endosomes. As expected, epimastigotes with deletion of one TcK2 allele (−/+) and the TcK2 null cell line (−/−) showed a progressive inhibition in differentiation to metacyclic-trypomastigotes (Fig. 5F), compatible with the heme starvation followed by eIF2α phosphorylation being a factor inducing differentiation. Strickling, in hemin-containing medium, only the TcK2 null cells showed a reduced growth rate (Fig. 5G). The same phenotypes were obtained in four independent clones, two of them using blasticidin instead of hygromycin resistance genes (see below).
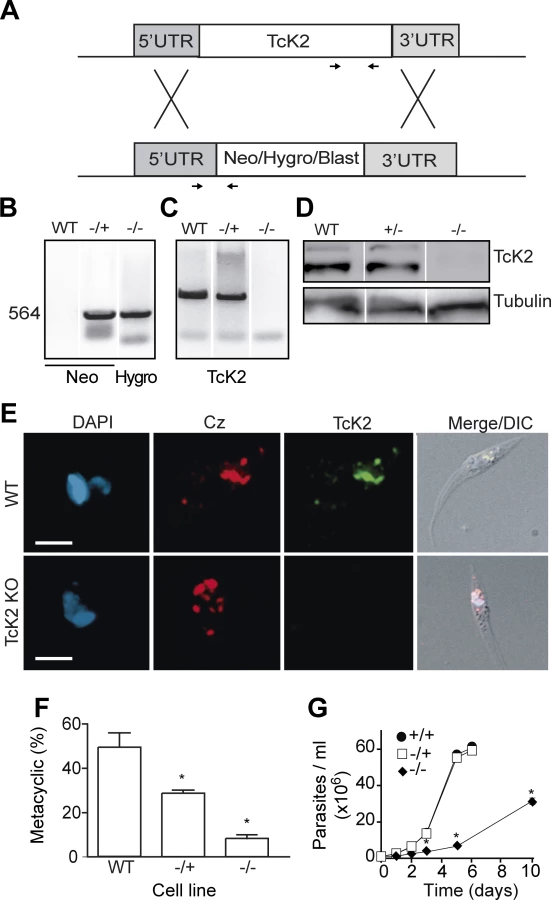
Ascorbate restores the growth rate and cell viability but not the full differentiation capacity
The slow growth phenotype of the TcK2 null cells could be due to oxidative damage generated by the hemin in parasites lacking this kinase. Therefore, we examined the effect of antioxidant agents in parasite growth. As shown in Fig. 6A, ascorbate restored normal growth when used at concentration of 10 μM; with 1 mM of ascorbate, growth was initially restored but the proliferation rate diminished after a few days. At the concentration of 10 mM, ascorbate was not effective. The effect of ascorbate was not because it restored the levels of sulfhydryl groups, as N-acetyl-cysteine, a sulfhydryl reducing agent, did not restore growth. Moreover, the effect of ascorbate disappeared after three days when cells were washed and maintained in medium without ascorbate (Fig. 6B), suggesting that a factor that prevents growth progressively accumulates in the medium of TcK2-KO cells.

On the other hand, 10 μM ascorbate only partially reverted the poor differentiation capacity of TcK2 null epimastigotes to metacyclic-trypomastigotes (Fig. 6C). This partial reversion of differentiation is suggestive that TcK2 affects differently the growth and the metacyclogenesis. The generated TcK2 null metacyclics also showed a reduced capacity to invade mammalian cells (Fig. 6D). Moreover, after infection, the TcK2 null parasites were able to differentiate into amastigotes but did not grow (Fig. 6E), or produce trypomastigotes, even after long incubations. Also, when metacyclics-trypomastigotes from TcK2 null cells were produced in the presence of ascorbate, the invasion was largely increased (Fig. 6D), without intracellular multiplication (Fig. 6E).
TcK2 null cells display more electron lucent structures
The TcK2 null cells still showed organelles that resemble reservosomes, as revealed by the cruzipain signal in the immunofluorescence analysis (Fig. 5E). Indeed, TcK2 null parasites examined by transmission electron microscopy showed cytoplasmic organelles that were more electron-lucent than in wild type parasites (Fig. 7). The empty structures were also found in parasites cultivated in medium without hemin supplementation (Fig. 3C), suggesting that heme does not accumulate in these organelles in cells lacking TcK2. These structures were clearly distinguishable from the mitochondrion, which was identified by the double membranes. Remarkably, in TcK2 null parasites treated with 10 μM ascorbate, which restored epimastigotes growth, these organelles remained electron-lucent, probably not accumulating heme. This observation suggests that the reduced proliferation in the absence of ascorbate was not because of the lack of heme in the endosomal compartment.

Heme levels and superoxide dismutase activity increase and peroxidase decreases in TcK2 null parasites
Next, we quantified the total amount of heme in the parasites after gentle lysis, expecting to measure mainly molecules present in the cytosol. The TcK2 null parasites had larger amounts of heme than wild type parasites, and this increase was not reversed by ascorbate, as shown in Fig. 8A. We also observed a larger uptake of the fluorescent heme analogue [Zn(II) Mesoporphyrin IX] by the TcK2 null cells when compared to the wild type cells. This uptake was strongly inhibited by heme in TcK2 null parasites as compared to wild type parasites, ruling out a non-specific binding to the cell (Fig. 8B). These results suggest that TcK2 could act in sensing and/or regulating the amount of heme that is present in the cells. Together with the electron-microscopy results, such data indicate that in parasites lacking TcK2, heme taken up by the cell is not directed to the lumen of endosomes, remaining in the cytosol.

The absence of TcK2 also caused a decrease in the total peroxidase activity (Fig. 8C), and an increase in the Fe-superoxide dismutase (SOD) activity when compared to wild type parasites (Fig. 8D). Consequently, the total levels of hydrogen peroxide were largely increased (Fig. 8E), independently of the presence of ascorbate. However, there was an increase in the amount of reactive oxygen species (ROS), as measured using CellRox (Fig. 8F), which was reduced by ascorbate addition. An increase from 600 fluorescence units in the case of control and 1000 fluorescence units corresponding to ROS levels was also detected using 5CM-H2DCFDA. However, this reagent provided a much higher background, due to the presence of medium. Anyway, the reduced growth of the TcK2 null cells correlated with the increased levels of ROS, probably produced by the augmented levels of H2O2, which might react with free iron or copper to generate hydroxyl radicals, causing parasite damage.
Only the active kinase restores the wild type phenotypes in TcK2 null parasites
To determine whether the changes observed in the null parasites were due to an indirect effect related to the absence of TcK2 as a protein, or to the requirement of an enzymatically active kinase, we generated parasites expressing the wild type and an enzymatically inactive TcK2 mutant. For this, we used null parasites obtained by replacement of the two copies of the endogenous kinase with Neo and Blast genes. This was done because these parasites were transfected with pROCK, which is a plasmid that integrates in the tubulin locus, being selected by hygromycin resistance [34]. The resulting constructs were expected to express constitutively the full length TcK2 gene. In one set of transfections the full-length wild type kinase was inserted, while in the other, a kinase with the lysine 695 replaced by alanine was used. The position of this mutation is indicated by an asterisk in S2 Fig., which corresponds to the lysine 622 of human PERK. This residue is located in the ATP binding site and inactivates the enzyme [35]. As shown in Fig. 9A, both forms of TcK2 were localized in similar vesicular structures in the posterior end of the parasite at the same position shown by cruzipain. The reinsertion of the wild type gene restored the growth defect, while the slow growth was maintained in the case of the mutated and probably inactive kinase. In addition, the differentiation capacity and ROS levels were restored by the wild type but not by the mutant kinase (Fig. 9C and D). Western blotting showed the presence of similar levels of the TcK2 protein in both cases, which were similar also to TcK2 expression levels in untransfected parasites, whereas the Neo/Blast null line that was used for these constructions has no TcK2 signal (Fig. 9E). The upper band was also seen in the kinase dead add-back. Nevertheless, upon incubation in PBS without hemin, the intensity of the upper phosphorylated band increased only in the original cells and in the wild type add-back of TcK2, not in the parasite line containing the kinase dead construct. These results indicate that the absence of the TcK2 activity is responsible for the growth defect and alterations in the oxidative metabolism observed in the null cells.

Importantly, TcK2 immunoprecipitated from epimastigotes previously incubated for 6 h in PBS had more eIF2α phosphorylation activity, as compared to the immunoprecipitates obtained from epimastigotes maintained in LIT medium (Fig. 9F). Control immunoprecipitations with null epimastigotes did not show any activity, confirming that the ability to phosphorylate the eIF2α in vitro was due to TcK2. Altogether, these data support the notion that the enzyme, migrating mainly as the top band is active in phosphorylating eIF2α.
Discussion
We have demonstrated that T. cruzi expresses a membrane bound protein kinase in the endosomes that is able to phosphorylate eIF2α in response to heme deprivation. In the absence of heme, the parasite undergoes differentiation in a process that is dependent on the active form of the kinase. In parasites lacking the kinase, heme accumulates in the cell, most likely in the cytosol, causing an increase in hydrogen peroxide levels, which leads to the generation of reactive oxygen species and parasite damage, as proposed in the model depicted in the Fig. 10. Therefore, this unusual eIF2α kinase is involved in the control of both cell differentiation and heme homeostasis in T. cruzi, a parasite that deals with environments presenting variable amounts of heme and other oxidant products [36].

The topology of TcK2 is very similar to PERK. Indeed, we have demonstrated that TcK2 is linked to the membrane, as shown for its orthologues in T. brucei [19] and L. infantum [21]. However, it is not predominantly in the ER, which agrees with our previous observation that T. brucei K2 is located in the flagellar pocket, a compartment related to the endocytic/exocytic pathway, and early endocytic vesicles [19]. In contrast to T. brucei, in T. cruzi the K2 is found in organelles more spread throughout the cytoplasm, which might reflect differences in protein trafficking in these organisms. Our immunofluorescence data strongly suggest that TcK2 is located in the endosomal membranes. Detection of the enzyme in purified reservosomes or other organelle, as well as their immunolocalization by electron microscopy could strengthen this conclusion. These findings do not exclude the possibility that some TcK2 is localized in ER mainly in the case of trypomastigotes, as there is some degree of co-localization with BiP. Further studies on endosome formation, its relationship to the ER and its remodeling during differentiation are required for a more detailed understanding of TcK2 trafficking in the cells.
Our data also suggest that TcK2 may be involved in the control of protein synthesis in the different parasite stages. We have previously shown that metacyclogenesis requires the inhibition of translation by phosphorylation of eIF2α [23]. In contrast to a cell line overexpressing a wild type form of eIF2α, a cell line overexpressing non-phosphorylatable form of eIF2α (Thr169Ala mutation) does not undergo metacyclogenesis under conditions that lack heme and other nutrients. Those previous data give strong support for the findings described here on TcK2 role in phosphorylating eIF2α in vivo as a requirement for differentiation.
The relatively smaller amounts of the upper band, which partially corresponds to the phosphorylated enzyme form in epimastigotes and amastigotes implies that TcK2 could be in a less active state in proliferating cells, which in fact present high rates of protein synthesis [37]. In contrast, protein synthesis is decreased in metacyclic-trypomastigotes and trypomastigotes, which is compatible with the predominant presence of relatively larger amounts of the upper phosphorylated band in the Western blotting. While epimastigotes proliferate and synthesize most of the housekeeping genes, trypomastigotes predominantly produce proteins involved in cell invasion and virulence, belonging to the trans-sialidase large family of genes and surface glycoproteins as mucins and mucin associated proteins [38]. Thus, it is possible that when hemin levels are low, the kinase is activated resulting in protein synthesis inhibition through TceIF2α phosphorylation, a requirement for T. cruzi differentiation, perhaps by allowing preferential translation of trypomastigote specific mRNAs, in agreement with recent findings that a subpopulation of mRNAs is translated in trypomastigotes [39].
Several other factors have been shown to promote T. cruzi differentiation. Examples are nutrient starvation [24], hemoglobin derived peptides [40], proteasome inhibition [41]. These factors could directly act on the differentiation, or could be part of a stress response generated by critical changes in environment, perhaps affecting heme levels or the activation of other eIF2α kinase. For example, amino acid starvation or proteasome inhibition may activate TcK1, the homolog of GCN2.
We observed that heme, but not other porphyrins, prevents parasite differentiation to metacyclic-trypomastigotes forms. In a similar way, it specifically binds and inhibits TcK2 activity in vitro. Specific binding was inferred by the saturable interaction as observed for HRI, which interacts more strongly to hemin (Fe+3-protoporphyrin) when compared to other porphyrins [42]. Further work would be necessary to precisely identify the heme binding site in TcK2. The presence of heme, but not other analogs, also promotes parasite growth by activating CaM kinase II [30]. This activation was shown to occur because of a non-deleterious increase in ROS [43].
As TcK2 is probably in the membrane of organelles, which are consumed when epimastigotes move across the insect gut, we presume that the kinase activation by heme depletion could be one key factor regulating differentiation. In fact, when parasites migrate to the midgut the amount of nutrients decrease and the electron dense structure gradually disappear [24]. As differentiation is accomplished, the content of these organelles that contain heme are consumed and cruzipain is secreted, participating in cell invasion [28, 44]. These findings are in agreement with our observations that electron lucent structures predominated in the parasites, as seen by EM in medium lacking hemin. Thus, heme levels seems to decrease initially in these organelles, then in the cytosol, most likely causing TcK2 activation on the later stages of differentiation. In fact, an increased phosphorylation of TcK2 was detected during metacyclogenesis by phosphoproteome analysis [45], supporting the notion that the enzyme is activated in trypomastigotes. Likewise, more active TcK2 was found in stressed cells compared to non-stressed epimastigotes.
The mechanism by which the organelles that accumulate heme are consumed and by which TcK2 and cruzipain disperse is unknown. It might involve events such as proteolysis, or other post-translational modifications. Interestingly, there is evidence that eIF2α phosphorylation and translation arrest are linked to membrane trafficking in mammalian cells [46], which could explain the differences in the localization and distribution of the kinase and the different endosomes.
T. cruzi has lost the capacity to synthesize heme, which is an essential growth factor for the parasite [47]. Heme appears to be internalized directly into the cytosol by ABC-transporters [27]. Therefore, heme accumulation in the endosomal compartment could occur through an unknown heme transporter in their membranes as a mechanism to store and prevent excessive levels in the cytosol. Alternatively, heme could be scavenged from the medium and stored in specific organelles through the parasite cytostome, as shown for other macromolecules [32]. T. brucei acquires heme through the haptoglobin-transferrin receptor localized in the flagellar pocket [48], where TbK2 is also localized [19]. However, the haptoglobin-transferrin receptor is found only in African Trypanosomes, which proliferate in the vertebrate blood and utilizes heme from blood. In Leishmania amazonensis, a parasite that develops in the insect gut and inside intracellular vacuoles, heme is taken up by the Leishmania heme response 1 (LHR1) protein, located in the parasite membrane and on acidic intracellular organelles [49]. A paralog of LHR1 is present in T. cruzi (TcCLB.511071.190, http://tritrypdb.org) and we are investigating its possible association and role in heme transport.
The generation of TcK2 null parasites confirms the kinase role in cellular differentiation, probably through the phosphorylation of eIF2α, as shown earlier [23]. We also inferred that the decrease in metacyclic-trypomastigote formation was not due to a less efficient cell growth, because the single knockout of K2 partially inhibited differentiation without affecting cellular proliferation. In addition, ascorbate that fully restored the growth rate did not fully restore differentiation. This was not the case for the cell invasion by metacyclic-trypomastigotes, which was largely affected by the absence of TcK2, and was even increased by adding ascorbate during the differentiation period. Most likely, it was due to the presence of high levels of ROS, causing parasite damage, or other modifications in the parasites that affected invasion.
Our results also showed that the lack of active TcK2 is directly related to the observed phenotypes as only the reintroduction of the wild type kinase, not of the protein mutated in the ATP binding site, abrogated the effects of its gene depletion. We also found that the incapacity to restore the normal growth phenotype and ROS was not due to protein mislocalization or poor expression. Moreover, when submitted to stress, only the endogenous and the wild type add-back TcK2 showed an increase in the upper band, which supports the notion that this band corresponds to an active and autophosphorylated kinase. Also, the presence of low levels of the upper band in the kinase dead mutant suggest that it could be phosphorylated by other kinases, in agreement with the presence of several phosphor-sites in the C-terminus of the protein [50], causing eIF2α phosphorylation. This modulation is also observed in mammalian PERK. Alternatively, it could be due to other modifications such as glycosylation, as shown in the T. brucei enzyme [19].
Our data also indicated that in the TcK2 null cells, the SOD dismutase activity increased and the peroxidase activity decreased, which would generate high levels of intracellular H2O2. This might occur as direct effect of the increased heme levels in the cytosol due to the failure to transport heme into endosomes. Otherwise, it could be a consequence of the differential translation mediated by increased eIF2α phosphorylation, or by phosphorylation of another substrates, as in the case of phosphorylation of the transcription factor Nrf2 in mammalian cells [51]. A possibility remains that heme is also acting as a source of iron, which can directly bind and activate superoxide dismutase, a strictly iron-dependent enzyme in trypanosomatids [52]. In addition, the reason why in the absence of TcK2 there was an increase in the heme detected in the soluble fraction after a gentle lysis is unclear at this moment. One possibility is that the kinase affects directly the heme intake into endosomes by interacting, or phosphorylating an unknown carrier. In fact, there is a link between the levels of heme and several stress elements [53], but this mechanism remains obscure. In mammalian cells, for example, PERK mediates apoptosis in response to ROS by facilitating the interaction of the ER with the mitochondria, independently of the kinase activity [54]. PERK knockouts were shown to affect largely the unfolded protein responses causing high levels of ER stress and its fragmentation [55].
The observation that ascorbate was effective at low concentrations suggests that it could protect the parasite as an antioxidant towards reactive species, donating one electron to oxidizing radicals such as hydroxyl radical [56]. This agrees with our observation that ROS levels increased in the TcK2 null parasites being restored to normal levels in cells growing in the presence of ascorbate. Therefore, it is possible that hydroxyl radicals would be generated by the Fenton reaction, which would occur with the increased levels of H2O2 produced by the parasite and minimal amounts of free Fe2+ present in the medium. This is in agreement with the results showing that inhibition of growth occurs a few days after ascorbate removal. At concentrations higher than 1 mM, ascorbate would act as a pro-oxidant agent, reducing Fe3+ to Fe2+, which would increase the production of more hydroxyl radicals in the presence of H2O2 by the Fenton reaction.
Heme as regulatory molecule controlling TcK2 activity also appears to occur in the mammalian stages of the parasite, as the kinase is active in trypomastigotes released from infected cells and it is mainly inactive in proliferating amastigotes. In addition, our observations indicate that intracellular amastigotes growth is largely diminished in the TcK2 null cells. The fact that growth is not restored by ascorbate could be due to its inability to decrease intracellular H2O2. These observations agree with recent findings indicating that iron and heme availability influences T. cruzi infection and disease progression [57]. Moreover, the intake of heme and/or iron has been found critical for Leishmania survival and infection [58] and several other hemoparasites [59].
In conclusion, our results indicate that heme affects the eIF2α kinase 2 of T. cruzi regulating development in insect and most likely in mammalian hosts. Future understanding of how this kinase regulates heme levels across the different parasite compartments and the development of specific inhibitors for this kinase could be useful in the control and in the understanding of the Chagas’ disease.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Brazilian National Council of Animal Experimentation (http://www.cobea.org.br/). The protocol was approved by the Committee on the Ethics of Animal Experiments of the Institutional Animal Care and Use at the Federal University of São Paulo (Id # CEP 1982/11).
Parasite cultures and differentiation
T. cruzi epimastigotes (Y strain) were cultivated in liver infusion tryptose (LIT) medium supplemented with 10% fetal bovine serum at 28°C [60]. For the differentiation into metacyclic-trypomastigotes, exponentially growing epimastigotes (1 × 107 cells per ml) were collected by centrifugation (2000 g for 10 min) and resuspended at a density of 5 × 108 cells per ml in Triatoma artificial urine (TAU) medium [190 mM NaCl, 17 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 8 mM phosphate buffer (pH 6.0)]. After 2 h culture at 28°C the cells were diluted to 5 × 106 in TAU supplemented with 2.5% sodium bicarbonate, 500 U/ml penicillin, 10 mM L-proline, 50 mM L-glutamic acid, 2 mM L-aspartic acid, 10 mM glucose and incubated at 28°C for 96 h [37].
Intracellular amastigotes were obtained from LLCMK2 (ATCC CCL-7) monkey kidney cells through lysis 72 h after infection. Trypomastigotes were collected from the cell medium (from 120 to 144 h after infection) as described previously [61]. Cell invasion assays were carried out by seeding the parasites onto each well of 24-well plates containing 13-mm diameter round glass coverslips coated with 2 × 104 cells. After 2 h incubation with parasites (2 × 105 parasites), the medium was removed, and the wells were washed with PBS and incubated with 1 ml of 4% paraformaldehyde in PBS at room temperature for 20 min. The coverslips were then washed with PBS and mounted in Prolong Gold Antifade Reagent (Invitrogen) in the presence of 10 μg/ml of 4-6-diamidino-2-phenylindole (DAPI). Images were analyzed in an Olympus (BX-61) fluorescence microscope.
Immunoblotting
Extracts were prepared from parasites collected by centrifugation at 2000 g for 5 min at 4°C and washed once with PBS before lysis in SDS-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. Alternatively, extracts were prepared by lysis of 1 × 107 cells in 50 μl of ice-cold 50 mM NaCl, 20 mM Tris-HCl (pH 7.4) containing 1% Triton X100, 1 mM phenylmethanesulfonyl fluoride (PMSF), and the cOmplete protease inhibitor cocktail, EDTA-free (Roche). The lysates were centrifuged at 6000 g for 10 min at 4°C and the supernatants were incubated with 5 U of calf intestinal alkaline phosphatase (Promega) for 10 min and the reaction was stopped by adding sample buffer and further incubation at 95°C for 5 min.
Immunoblots were performed with extracts of cells submitted to electrophoresis on SDS-PAGE (7.5%) and transferred to nitrocellulose membranes. The membranes were stained with 0.3% Ponceau S in 10% acetic acid and then treated with 5% nonfat milk in PBS containing 0.05% Tween-20 for 60 min. The membranes were incubated 1 h with the primary antibodies in PBS containing 0.1% Tween-20. Anti-TcK2 rabbit polyclonal antiserum, which was obtained by immunization with a recombinant protein corresponding to the kinase domain (KD) of the T. brucei K2 expressed in Escherichia coli BL21 [19]. It was used at 1 : 1500 dilution. A monoclonal antibody (mAb) 5D10, obtained as described in [62] by immunization with a recombinant protein corresponding to the residue 346 to 511 of T. cruzi K2, and expressed from pET28a (Novagen) in E. coli BL21. It was used at 1 : 500 dilution. Anti-Hsp70 [63] was used at 1 : 20,000, anti-T. cruzi β-tubulin [64] at 1 : 20,000 and anti-BiP [65] at 1 : 20,000 dilutions. The membranes were washed three times with PBS containing 0.5% Tween-20 (10 min each) followed by 1 h incubation with peroxidase labeled goat anti-mouse or rabbit IgG antibody (Invitrogen) at 1 : 10,000 dilutions. Bound antibodies were detected by ECL (Millipore) by using a LI-COR Odyssey imaging apparatus.
Immunofluorescence
Parasites were washed with PBS and attached to glass slides (Tekdon Incorporated) pretreated with 0.01% poly-L-lysine for 5 min. The excess of parasites was removed and the slides incubated with 4% paraformaldehyde in PBS at room temperature for 20 min, washed in PBS and permeabilized 5 min in PBS containing 0.1% Triton X100. Fixed and permeabilized cells were washed with PBS, incubated for 20 min with PBS containing 2% BSA and for 1 h at room temperature with primary antibodies diluted in the same buffer. Anti-human influenza hemagglutinin (HA) (Covance) was used at 1 : 1,000, the mAb of anti-5D10 at 1 : 500, anti-Binding immunoglobulin protein (BiP) at 1 : 5,000, and anti-T. cruzi cruzipain mAb (a gift of Prof. Dr. Julio Scharfstein [66], at 1 : 2,000 dilutions. The slides were washed with PBS, incubated 1 h at room temperature with Alexa Fluor 594, or 488, goat anti-rabbit or anti-mouse IgG (Invitrogen) diluted 1 : 1000, washed once more, and mounted in Prolong Gold Antifade Reagent (Invitrogen) in the presence of 10 μg of DAPI per ml. Images were acquired by using a Hamamatsu Orca R2 CCD camera coupled to an Olympus (BX-61) microscope equipped with a ×100 plan Apo-oil objective (NA 1.4). Acquisitions were at every 0.2 μm for each set of excitation/emission filters. Blind deconvolution was performed by employing the AutoQuant X2.2 software (Media Cybernetics).
Immunoprecipitation
Parasites at exponential growth phase were collected by centrifugation at 2000 g for 5 min at 4°C and resuspended at 4°C in 137 mM NaCl, 2 mM EDTA and 20 mM Tris-HCl (pH 8.0) containing protease inhibitor cocktail 1% Nonidet P40, for lysis. The lysates were centrifuged at 12000 g for 20 min and the supernatants transferred to new tubes, which were incubated at 4°C with the anti-TcK2 rabbit polyclonal antiserum for 16 h. Immune complexes were collected after 2 h incubations with Protein G Sepharose (GE). The immunoprecipitates were centrifuged at 500 g for 2 min, washed once in 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM Tris-HCl (pH 8.0) containing 1% Triton-X100.
Electron microscopy
Parasites were fixed at room temperature with 2% formaldehyde/2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2). The fixed parasites were then decanted overnight on ACLAR (Electron Microscopy Sciences) film precoated with 0.01% poly-L-lysine. The ACLAR films were washed three times with 0.1 M sodium cacodylate (pH 7.2) and then fixed with 1% osmium tetroxide in the same buffer at room temperature. The preparation was then washed with cacodylate buffer followed by three washes with water. Subsequently, the ACLARfilms were treated with 0.4% uranyl acetate, gradually dehydrated in a series of ethanol solutions, and embedded in Epon resin (Electron Microscopy Sciences). After sectioning to produce consecutive sections, the material was stained with uranyl acetate and lead citrate, mounted on Formvar grids, and observed in a JEOL 1200 EX II transmission electron microscope at 80 kV.
Generation of recombinant parasites
TcK2 knockouts were generated by gene replacement using antibiotic resistance markers flanked by untranslated regions (UTR) of the K2 gene (http://tritrypdb.org TcCLB.506559.129). The 5′-UTR region was amplified from T. cruzi (Y-strain) genomic DNA [67] using oligonucleotides 5k2fowApaI and 5revXbaI. The 3′-UTR was amplified using oligonucleotides 3′K2fowSal and 3K2RevSac. Primer sequences used in this study are presented in S1 Table. The amplified fragments were cloned into plasmids containing the neomycin phosphotransferase (Neo), hygromycin phosphotransferase (Hygro), or blasticidin (Blast) genes inserted into the XbaI and SalI sites of pGEM-T (a gift from Dr. Carlos Renato Machado from the Universidade Federal de Minas Gerais, Brazil). The blasticidin gene was amplified from pNAT6Myc [68] with oligonucleotides BlastFowXbaI and BlastRevSalI. Restriction mapping and DNA sequencing confirmed all constructs.
HA-epitope tagged TcK2 was prepared in a modified pTEX plasmid [69] containing one HA encoding sequence 5-TACCCTTACGACGTTCCTGATTACGCTAGCTGA between the BamH1 and XhoI sites. Two segments corresponding to the C-terminus sequence of TcK2 were amplified from T. cruzi genomic DNA (Y strain) with oligonucleotides K2TcXbaFor / K2TcNotRev and K2TcNotFor / K2TcBam and cloned sequentially into pTEX-HA to introduce a Not I site in the sequence and create a 900 base sequence followed by the HA tag.
Thirty micrograms of each plasmid linearized with NotI for the HA-tag, and SacI for the gene replacements, were incubated with T. cruzi epimastigotes previously washed and resuspended to 5 × 106 parasites in 100 μl of 137 mM NaCl, 5 mM KCl, 5.5 mM Na2HPO4, 0.77 mM glucose and 21 mM Hepes (pH 7.0) and pulsed with the Amaxa Nucleofactor (Lonza), program U-033. Cells were diluted and selected by growing in 0.2 to 0.5 mg/ml G418, 0.5 mg/ml Hygro, or 0.05 mg/ml Blast depending on the plasmid used for transfection. Limiting dilution was further used to clone the parasites.
The presence of resistance marker insertions was detected by PCR amplification of DNA extracted from parasite clones using oligonucleotides 5K2fow at the 5′UTR of K2 and G418Rev for G418, HigroR for Hygro, and BlastRev for Blast. The removal of K2 was confirmed by PCR amplification using oligonucleotides TcK2-KD-pGEX5X-1(F) and TcK2-KD-pGEX5X-1(R).
T. cruzi containing the original and the kinase death were obtaining by cloning both constructs in the XbaI and XhoI sites of the pROCK-GFP-HYG plasmid [34]. The full-length gene was amplified from the genomic DNA of T. cruzi Y strain by PCR using the primers Tck2Xba-fow and TcK2XhoI-rev. The mutated protein was generated by amplification of two fragments encompassing the full-length gene with two sets of primers. One of the fragments was generated with Pfx50 polymerase (Life Technologies) by using the primers Tck2XhoIrev and Tck2mut-fow and the other with the primers TceiF2-K2mut-rev and the TcK2Xbafow to replace the lysine 695 by an alanine. The two fragments were gel purified and assembled by a second PCR using the primers Tck2Xba-fow and TcK2XhoI-rev. The final fragment was inserted into pCR2.1 Topo (Life Technologies) and subcloned into the XbaI and XhoI sites of pROCK-GFP-HYG. The obtained plasmids were confirmed by restriction analysis and sequencing.
Recombinant proteins, kinase and binding assays
Soluble recombinant T. cruzi eIF2α containing a histidine-tag was expressed from pDEST17 [23] in E. coli BL21. Expression was induced for 16 h at 30°C with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG); the cells collected and resuspended in 0.25 M NaCl, 50 mM Tris-HCl (pH 8.0) and the cOmplete protease inhibitor cocktail and lysed by using a French press. The recombinant protein was purified by affinity chromatography on a nickel agarose column (Qiagen) by elution with the lysis solution containing 0.5 M imidazole. The kinase domain of TcK2 was amplified from total T. cruzi DNA with the oligonucleotides TcK2-KD-pGEX5X-1 and TcK2-KD-pGEX5X-1 and cloned into the EcoRI and XhoI sites of the plasmid pGEX5X-1 (GE) to generate a protein fused with GST. The TcK2 recombinant was expressed in E. coli BL21 after induction with 0.5 mM IPTG for 16 h at 30°C. The protein was purified from bacteria lysed by the French press by using affinity chromatography on GST-Sepharose (GE) equilibrated in PBS containing cOmplete cocktail, by elution in the same buffer containing 0.1 M glutathione. Both proteins were dialyzed in the kinase buffer (50 mM NaCl, 20 mM Tris-HCl (pH 7.5) at 4°C for 18 h. The kinase assays were performed in the presence of 5 μg/ml eIF2α, 1 μg/ml TcK2, 10 mM MgCl2, 10 μCi of [32P] γATP (3000 Ci/mmol, Perkin Elmer) and 0.5 mM ATP at 37°C for 1 h. When indicated the heme derivatives (Frontiers Scientific) were added 15 min before addition of ATP. Reactions were stopped by the addition of SDS-PAGE sample buffer. The labeling was quantified by using 9200 Typhon imaging apparatus (GE) after electrophoresis and Coomassie R-250 staining. For the binding assays hemin at indicated concentrations were incubated 30 min with the indicated with 5 μg/ml of recombinant proteins in 0.1 M Tris-HCl (pH 7.4) and 50 mM NaCl and the absorbance read in a SpectraMax M3 reader (Molecular Devices), at 25°C. Dissociation constant determination was estimated by using the Prisma 6 software (Graphpad).
Reactive species of oxygen, H2O2 and peroxidases
Parasites at 2 × 107 cells per ml were pre-loaded with 5 μM of CellRox (Invitrogen) in PBS for 30 min, or with 5 μM CM-H2DCFDA (Invitrogen). In the case of this later reagent, cells were further incubated for 30 min in LIT medium. Reactive species of oxygen were then detected by flow cytometry using a FACS Accuri C6 (BD Biosciences) as recommended by the manufacturer. Hydrogen peroxide levels were measured by incubating 2 × 107 parasites per ml of PBS with 1 U/ml horseradish peroxidase in the presence of 2 μM Amplex Red (Invitrogen) for 30 min. The parasites were then centrifuged and the resulting fluorescence measured in the supernatants (excitation 530 nm and emission 590 nm) by using a SpectraMax M3 Reader. Total peroxidase activity was measured in the supernatant of 2 × 107 parasites lysed in 50 μl of ice-cold 50 mM NaCl, 20 mM Tris-HCl (pH 7.4) containing 1% Triton X100. The activity was quantified after 30 min incubation at 25°C by adding 2 μM Amplex Red (Invitrogen) and 2 mM H2O2. SOD activity was measured in the supernatant of 1 × 107 parasites previously lysed by three sonication cycles (20 sec each) in 200 μl of 50 mM potassium phosphate buffer (pH 7.4) and 0.1 mM EDTA (SOD buffer). Reactions were done at 25°C by the addition of 25 μl of the lysate into 200 μl of SOD buffer containing 10 mg/ml β-nicotinamide adenine dinucleotide and 28 mg/ml nitrotetrazolium blue chloride (both from Sigma) followed by 25 μl of 3.3 mM phenazine methosulfate (Sigma). The SOD activity was determined by the kinetics of nitrotetrazolium blue chloride reduction followed by absorption at 560 nm [70].
Intracellular heme levels and incorporation of heme analogues
We employed the method described in [71]. Briefly, 2 × 107 cells were centrifuged, washed with PBS, and resuspended in 1 ml of 10 mM Tris-HCl (pH 7.4) containing the cOmplete cocktail. The cells were lysed by freezing and thawing (three cycles), the insoluble material removed by centrifugation at 10,000 g for 10 min and 840 μl transferred to a new tube containing 100 μl of 1 M NaOH and 200 μl of pyridine. Heme levels were then measured in a 1 ml cuvette after addition of 2 mg of solid sodium dithionite at 450 nm by using the Spectra Max M3 Reader.
The incorporation of the fluorescent Zn(II) Mesoporphyrin IX (Frontier Scientific) was performed in 2 × 107 parasites resuspended in 100 μl of PBS. Ten μM of the porphyrin was added and the mixture incubated for 5 min followed by the addition of the indicated concentrations of hemin as a competitor. After more 30 min, the cells were chilled by adding 800 μl of cold PBS, centrifuged and washed with cold PBS and lysed in PBS containing 0.5% Triton X-100. The fluorescent porphyrin was measured in the supernatant (excitation 405 nm and emission at 578 nm).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Sonenberg N, Dever TE (2003) Eukaryotic translation initiation factors and regulators. Curr Opin Struct Biol 13 : 56–63. 12581660
2. Sonenberg N, Hinnebusch A (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136 : 731–745. doi: 10.1016/j.cell.2009.01.042 19239892
3. Donnelly N, Gorman AM, Gupta S, Samali A (2013) The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci 70 : 3493–3511. doi: 10.1007/s00018-012-1252-6 23354059
4. Castilho BA, Shanmugam R, Silva RC, Ramesh R, Himme BM, et al. (2014) Keeping the eIF2 alpha kinase Gcn2 in check. Biochim Biophys Acta.1843 : 1848–1968. doi: 10.1016/j.bbamcr.2014.04.006 24732012
5. Rothenburg S, Deigendesch N, Dey M, Dever TE, Tazi L (2008) Double-stranded RNA-activated protein kinase PKR of fishes and amphibians: varying the number of double-stranded RNA binding domains and lineage-specific duplications. BMC Biology 6 : 12. doi: 10.1186/1741-7007-6-12 18312693
6. Garcia MA, Meurs EF, Esteban M (2007) The dsRNA protein kinase PKR: virus and cell control. Biochimie 89 : 799–811. 17451862
7. Dey M, Cao C, Dar AC, Tamura T, Ozato K, et al. (2005) Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 122 : 901–913. 16179259
8. Ron D, Harding HP (2012) Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harb Perspect Biol 4 : 1–13. 23209157
9. Chen JJ, London IM (1995) Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Bioch Sci 20 : 105–108. 7709427
10. Wek RC, Jiang HY, Anthony TG (2006) Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 34 : 7–11. 16246168
11. Zhang M, Joyce BR, Sullivan WJ Jr., Nussenzweig V (2013) Translational control in Plasmodium and Toxoplasma parasites. Eukariot Cell 12 : 161–167. doi: 10.1128/EC.00296-12 23243065
12. Vonlaufen N, Kanzok SM, Wek RC, Sullivan WJ Jr. (2008) Stress response pathways in protozoan parasites. Cell Microbiol 10 : 2387–2399. doi: 10.1111/j.1462-5822.2008.01210.x 18647172
13. Zhang M, Fennell C, Ranford-Cartwright L, Sakthivel R, Gueirard P, et al. (2010) The Plasmodium eukaryotic initiation factor-2alpha kinase IK2 controls the latency of sporozoites in the mosquito salivary glands. J Exp Med 207 : 1465–1474. doi: 10.1084/jem.20091975 20584882
14. Fennell C, Babbitt S, Russo I, Wilkes J, Ranford-Cartwright L, et al. (2009) PfeIK1, a eukaryotic initiation factor 2alpha kinase of the human malaria parasite Plasmodium falciparum, regulates stress-response to amino-acid starvation. Malar J 8 : 99. doi: 10.1186/1475-2875-8-99 19435497
15. Zhang M, Mishra S, Sakthivel R, Rojas M, Ranjan R, et al. (2012) PK4, a eukaryotic initiation factor 2alpha(eIF2alpha) kinase, is essential for the development of the erythrocytic cycle of Plasmodium. Proc Natl Acad Sci USA 109 : 3956–3961. doi: 10.1073/pnas.1121567109 22355110
16. Konrad C, Wek RC, Sullivan WJ Jr. (2011) A GCN2-like eukaryotic initiation factor 2 kinase increases the viability of extracellular Toxoplasma gondii parasites. Eukariot Cell 10 : 1403–1412. doi: 10.1128/EC.05117-11 21908594
17. Joyce BR, Queener SF, Wek RC, Sullivan WJ Jr. (2010) Phosphorylation of eukaryotic initiation factor-2{alpha} promotes the extracellular survival of obligate intracellular parasite Toxoplasma gondii. Proc Natl Acad Sci USA 107 : 17200–17205. doi: 10.1073/pnas.1007610107 20855600
18. Narasimhan J, Joyce BR, Naguleswaran A, Smith AT, Livingston MR, et al. (2008) Translation regulation by eukaryotic initiation factor-2 kinases in the development of latent cysts in Toxoplasma gondii. J Biol Chem 283 : 16591–16601. doi: 10.1074/jbc.M800681200 18420584
19. Moraes MCS, Jesus TCL, Hashimoto NN, Dey M, Schwartz KJ, et al. (2007) Novel membrane-bound eIF2a kinase in the flagellar pocket of Trypanosoma brucei. Eukariot Cell 6 : 1979–1991. 17873083
20. Cloutier S, Laverdiere M, Chou M-N, Boilard N, Chow C, et al. (2012) Translational control through eIF2alpha phosphorylation during the Leishmania differentiation process. PLoS One 7: e35085. doi: 10.1371/journal.pone.0035085 22693545
21. Chow C, Cloutier S, Dumas C, Chou MN, Papadopoulou B (2011) Promastigote to amastigote differentiation of Leishmania is markedly delayed in the absence of PERK eIF2alpha kinase-dependent eIF2alpha phosphorylation. Cell Microbiol 13 : 1059–1077. doi: 10.1111/j.1462-5822.2011.01602.x 21624030
22. Hope R, Ben-Mayor E, Friedman N, Voloshin K, Biswas D, et al. (2014) Phosphorylation of the TATA-binding protein activates the spliced leader silencing pathway in Trypanosoma brucei. Sci Signal 7: ra85. doi: 10.1126/scisignal.2005234 25185157
23. Tonelli RR, Augusto LdS, Castilho BA, Schenkman S (2011) Protein Synthesis Attenuation by Phosphorylation of eIF2alpha Is Required for the Differentiation of Trypanosoma cruzi into Infective Forms. PLoS One 6: e27094. doi: 10.1371/journal.pone.0027904 22114724
24. Figueiredo RC, Rosa DS, Soares MJ (2000) Differentiation of Trypanosoma cruzi epimastigotes: metacyclogenesis and adhesion to substrate are triggered by nutritional stress. J Parasitol 86 : 1213–1218. 11191893
25. Zeledon R, Bolanos R, Rojas M (1984) Scanning electron microscopy of the final phase of the life cycle of Trypanosoma cruzi in the insect vector. Acta Trop 41 : 39–43. 6143481
26. Figueiredo RC, Rosa DS, Gomes YM, Nakasawa M, Soares MJ (2004) Reservosome: an endocytic compartment in epimastigote forms of the protozoan Trypanosoma cruzi (Kinetoplastida: Trypanosomatidae). Correlation between endocytosis of nutrients and cell differentiation. Parasitol 129 : 431–438. 15521631
27. Cupello MP, Souza CFD, Buchensky C, Soares JBRC, Laranja GAT, et al. (2011) The heme uptake process in Trypanosoma cruzi epimastigotes is inhibited by heme analogues and by inhibitors of ABC transporters. Acta Trop 120 : 211–218. doi: 10.1016/j.actatropica.2011.08.011 21903090
28. Pereira MG, Nakayasu ES, Sant’Anna C, De Cicco NN, Atella GC, et al. (2011) Trypanosoma cruzi epimastigotes are able to store and mobilize high amounts of cholesterol in reservosome lipid inclusions. PLoS One 6: e22359. doi: 10.1371/journal.pone.0022359 21818313
29. Porto-Carreiro I, Attias M, Miranda K, de Souza W, Cunha e S (2000) Trypanosoma cruzi epimastigote endocytic pathway: cargo enters the cytostome and passes through an early endosomal network before storage in reservosomes. Eur J Cell Biol 79 : 858–869. 11139150
30. Souza CF, Carneiro AB, Silveira AB, Laranja GA, Silva-Neto MA, et al. (2009) Heme-induced Trypanosoma cruzi proliferation is mediated by CaM kinase II. Biochem Biophys Res Commun 390 : 541–546. doi: 10.1016/j.bbrc.2009.09.135 19818332
31. Lara Fa, Sant’anna C, Lemos D, Laranja GaT, Coelho MGP, et al. (2007) Heme requirement and intracellular trafficking in Trypanosoma cruzi epimastigotes. Biochem Biophys Res Commun 355 : 16–22. 17292866
32. Sant’anna C, de Souza W, Cunha e S (2004) Biogenesis of the reservosomes of Trypanosoma cruzi. Micros Microanal 10 : 637–646. 15525436
33. Liu R, Hu J (2011) HemeBIND: a novel method for heme binding residue prediction by combining structural and sequence information. BMC bioinformatics 12 : 207. doi: 10.1186/1471-2105-12-207 21612668
34. DaRocha WD, Silva RA, Bartholomeu DC, Pires SF, Freitas JM, et al. (2004) Expression of exogenous genes in Trypanosoma cruzi: improving vectors and electroporation protocols. Parasitol Res 92 : 113–120. 14634799
35. Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397 : 271–274. 9930704
36. Piacenza L, Alvarez MN, Peluffo G, Radi R (2009) Fighting the oxidative assault: the Trypanosoma cruzi journey to infection. Curr Opin Microbiol 12 : 415–421. doi: 10.1016/j.mib.2009.06.011 19616990
37. Contreras VT, Salles JM, Thomas N, Morel CM, Goldenberg S (1985) In vitro differentiation of Trypanosoma cruzi under chemically defined conditions. Mol Biochem Parasitol 16 : 315–327. 3903496
38. Goldenberg S, Avila AR (2011) Aspects of Trypanosoma cruzi stage differentiation. Adv Parasit 75 : 285–305. doi: 10.1016/B978-0-12-385863-4.00013-7 21820561
39. De Gaudenzi JG, Carmona SJ, Aguero F, Frasch AC (2013) Genome-wide analysis of 3′-untranslated regions supports the existence of post-transcriptional regulons controlling gene expression in trypanosomes. Peer Journal 1: e118. doi: 10.7717/peerj.118 23904995
40. Garcia ES, Gonzalez MS, De Azambuja P, Baralle FE, Fraidenraich D, et al. (1995) Induction of Trypanosoma cruzi metacyclogenesis in the gut of the hematophagous insect vector, Rhodnius prolixus, by hemoglobin and peptides carrying alpha D-globin sequences. Exp Parasitol 81 : 255–261. 7498422
41. Cardoso J, Soares MJ, Menna-Barreto RF, Le Bloas R, Sotomaior V, et al. (2008) Inhibition of proteasome activity blocks Trypanosoma cruzi growth and metacyclogenesis. Parasitol Res 103 : 941–951. doi: 10.1007/s00436-008-1081-6 18581141
42. Igarashi J, Murase M, Iizuka A, Pichierri F, Martinkova M, et al. (2008) Elucidation of the heme binding site of heme-regulated eukaryotic initiation factor 2alpha kinase and the role of the regulatory motif in heme sensing by spectroscopic and catalytic studies of mutant proteins. J Biol Chem 283 : 18782–18791. doi: 10.1074/jbc.M801400200 18450746
43. Nogueira NP, de Souza CF, Saraiva FM, Sultano PE, Dalmau SR, et al. (2011) Heme-induced ROS in Trypanosoma cruzi activates CaMKII-like that triggers epimastigote proliferation. One helpful effect of ROS. PLoS One 6: e25935. doi: 10.1371/journal.pone.0025935 22022475
44. Sant’Anna C, Pereira MG, Lemgruber L, de Souza W, Cunha e Silva NL (2008) New insights into the morphology of Trypanosoma cruzi reservosome. Microsc Res Techniq 71 : 599–605. doi: 10.1002/jemt.20592 18452191
45. Marchini FK, de Godoy LM, Rampazzo RC, Pavoni DP, Probst CM, et al. (2011) Profiling the Trypanosoma cruzi phosphoproteome. PLoS One 6: e25381. doi: 10.1371/journal.pone.0025381 21966514
46. Kloft N, Neukirch C, von Hoven G, Bobkiewicz W, Weis S, et al. (2012) A subunit of eukaryotic translation initiation factor 2alpha-phosphatase (CreP/PPP1R15B) regulates membrane traffic. J Biol Chem 287 : 35299–35317. doi: 10.1074/jbc.M112.379883 22915583
47. Salzman TA, Stella AM, Wider de Xifra EA, Batlle AM, Docampo R, et al. (1982) Porphyrin biosynthesis in parasitic hemoflagellates: functional and defective enzymes in Trypanosoma cruzi. Comparative Biochmestry and Physiology B 72 : 663–667. 6751683
48. Vanhollebeke B, De Muylder G, Nielsen MJ, Pays A, Tebabi P, et al. (2008) A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans. Science 320 : 677–681. doi: 10.1126/science.1156296 18451305
49. Huynh C, Yuan X, Miguel DC, Renberg RL, Protchenko O, et al. (2012) Heme uptake by Leishmania amazonensis is mediated by the transmembrane protein LHR1. PloS Pathog 8: e1002795. doi: 10.1371/journal.ppat.1002795 22807677
50. de Godoy LM, Marchini FK, Pavoni DP, Rampazzo Rde C, Probst CM, et al. (2012) Quantitative proteomics of Trypanosoma cruzi during metacyclogenesis. Proteomics 12 : 2694–2703. doi: 10.1002/pmic.201200078 22761176
51. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, et al. (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23 : 7198–7209. 14517290
52. Dufernez F, Yernaux C, Gerbod D, Noel C, Chauvenet M, et al. (2006) The presence of four iron-containing superoxide dismutase isozymes in trypanosomatidae: characterization, subcellular localization, and phylogenetic origin in Trypanosoma brucei. Free Radic Biol Med 40 : 210–225. 16413404
53. Koskenkorva-Frank TS, Weiss G, Koppenol WH, Burckhardt S (2013) The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic Biol Med 65 : 1174–1194. doi: 10.1016/j.freeradbiomed.2013.09.001 24036104
54. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, et al. (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19 : 1880–1891. doi: 10.1038/cdd.2012.74 22705852
55. Huang G, Yao J, Zeng W, Mizuno Y, Kamm KE, et al. (2006) ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J Cell Sci 119 : 153–161. 16352659
56. Du J, Cullen JJ, Buettner GR (2012) Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta 1826 : 443–457. doi: 10.1016/j.bbcan.2012.06.003 22728050
57. Paiva CN, Feijo DF, Dutra FF, Carneiro VC, Freitas GB, et al. (2012) Oxidative stress fuels Trypanosoma cruzi infection in mice. J Clin Invest 122 : 2531–2542. doi: 10.1172/JCI58525 22728935
58. Andrews NW (2012) Oxidative stress and intracellular infections: more iron to the fire. J Clin Invest 122 : 2352–2354. doi: 10.1172/JCI64239 22728929
59. Toh SQ, Glanfield A, Gobert GN, Jones MK (2010) Heme and blood-feeding parasites: friends or foes? Parasit Vectors 3 : 108–108. doi: 10.1186/1756-3305-3-108 21087517
60. Camargo EP (1964) Growth and differentiation in Trypanosoma cruzi: Origin of metacyclic trypomastigotes in liquid media. Rev Inst Med Tropical S Paulo 6 : 93–100.
61. Abuin G, Freitas-Junior LHG, Colli W, Alves MJ, Schenkman S (1999) Expression of trans-sialidase and 85 kDa glycoprotein genes in Trypanosoma cruzi is differentially regulated at the post-transcriptional level by labile protein factors. J Biol Chem 274 : 13041–13047. 10224055
62. Cornejo A, Oliveira CR, Wurtele M, Chung J, Hilpert K, et al. (2012) A novel monoclonal antibody against the C-terminus of beta-tubulin recognizes endocytic organelles in Trypanosoma cruzi. Protein Pept Lett 19 : 636–643. 22519535
63. McDowell MA, Ransom DM, Bangs JD (1998) Glycosylphosphatidylinositol-dependent secretory transport in Trypanosoma brucei. Biochem J 335 : 681–689. 9794811
64. Chung J, Rocha AA, Tonelli RR, Castilho BA, Schenkman S (2013) Eukaryotic initiation factor 5A dephosphorylation is required for translational arrest in stationary phase cells. Biochem J 451 : 257–267. doi: 10.1042/BJ20121553 23368777
65. Bangs JD, Uyetake L, Brickman MJ, Balber AE, Boothroyd JC (1993) Molecular cloning and cellular localization of a BiP homologue in Trypanosoma brucei. Divergent ER retention signals in a lower eukaryote. J Cell Sci 105 : 1101–1113. 8227199
66. Monteiro AC, Abrahamson M, Lima AP, Vannier-Santos MA, Scharfstein J (2001) Identification, characterization and localization of chagasin, a tight-binding cysteine protease inhibitor in Trypanosoma cruzi. J Cell Sci 114 : 3933–3942. 11719560
67. Medina-Acosta E, Cross GAM (1993) Rapid isolation of DNA from trypanosomatid protozoa using a simple ‘mini-prep’ procedure. Mol Biochem Parasitol 59 : 327–330. 8341329
68. Alsford S, Horn D (2008) Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol Biochem Parasitol 161 : 76–79. doi: 10.1016/j.molbiopara.2008.05.006 18588918
69. Kelly JM, Ward HM, Miles MA, Kendall G (1992) A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Res 20 : 3963–3969. 1324472
70. Ewing JF, Janero DR (1995) Microplate superoxide dismutase assay employing a nonenzymatic superoxide generator. Anal Biochem 232 : 243–248. 8747482
71. Miguel DC, Flannery AR, Mittra B, Andrews NW (2013) Heme uptake mediated by LHR1 is essential for Leishmania amazonensis virulence. Infect Immun 81 : 3620–3626. doi: 10.1128/IAI.00687-13 23876801
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Control of Murine Cytomegalovirus Infection by γδ T Cells
- Dimorphism in Fungal Pathogens of Mammals, Plants, and Insects
- ATPaseTb2, a Unique Membrane-bound FoF1-ATPase Component, Is Essential in Bloodstream and Dyskinetoplastic Trypanosomes
- Rational Development of an Attenuated Recombinant Cyprinid Herpesvirus 3 Vaccine Using Prokaryotic Mutagenesis and In Vivo Bioluminescent Imaging
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy