
Ontogeny of Recognition Specificity and Functionality for the Broadly Neutralizing Anti-HIV Antibody 4E10
4E10 is an antibody that neutralizes a broad variety of HIV strains. However, 4E10 is uncommon in infected patients and has not been successfully elicited by any vaccine approach attempted. Hurdles to re-eliciting 4E10 include the accumulation of many mutations during development, demonstrated reactivity against host proteins and significant structural flexibility. Lacking a confirmed sequence for precursors of 4E10, we studied the recognition and biophysical properties of an ensemble of eight of the likeliest candidates. Surprisingly, 4E10 gained host reactivity and structural flexibility, but lost stability during development when compared to candidate precursors. However, recognition of HIV was remarkably conserved, despite a considerable improvement in binding. Since these results run counter to those expected from conventional vaccination protocols, 4E10 is unlikely to serve as the basis of a useful HIV vaccine.
Published in the journal:
. PLoS Pathog 10(9): e32767. doi:10.1371/journal.ppat.1004403
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004403
Summary
4E10 is an antibody that neutralizes a broad variety of HIV strains. However, 4E10 is uncommon in infected patients and has not been successfully elicited by any vaccine approach attempted. Hurdles to re-eliciting 4E10 include the accumulation of many mutations during development, demonstrated reactivity against host proteins and significant structural flexibility. Lacking a confirmed sequence for precursors of 4E10, we studied the recognition and biophysical properties of an ensemble of eight of the likeliest candidates. Surprisingly, 4E10 gained host reactivity and structural flexibility, but lost stability during development when compared to candidate precursors. However, recognition of HIV was remarkably conserved, despite a considerable improvement in binding. Since these results run counter to those expected from conventional vaccination protocols, 4E10 is unlikely to serve as the basis of a useful HIV vaccine.
Introduction
An effective HIV vaccine will likely need to elicit broadly-neutralizing antibodies (bnAbs) that target the viral envelope protein (Env) as part of a protective immune response [1]–[6]. Env-derived and reverse-engineered immunogen-based vaccines, however, have consistently failed to elicit bnAbs. Possible explanations include that: (1) immunogens may be unable to bind germline-encoded precursors (GEPs) of bnAbs with sufficient affinity to initiate B cell activation and affinity maturation, which has a ∼micromolar threshold [7]–[9]; (2) rearranged VH and VL genes compatible with the development of bnAbs may not be common in human or animal model vaccinee GEP repertoires; (3) some bnAbs are autoreactive, which hinders their elicitation through self-tolerance mechanisms; (4) the unusual characteristics inherent to bnAbs, such as long complementarity determining regions (CDRs), functionally-required polyspecificity, and a high degree of somatic mutation (typically observed in Abs elicited in response to chronic infections, including bnAbs), may not be easily achieved through conventional vaccination strategies; (5) imperfect immunogens may elicit off-target (non-neutralizing or non-Env) or humoral responses with limited breadth; and, finally, (6) neutralization mechanisms may involve complexities beyond simply binding a particular epitope on Env (e.g., inducing specific conformational changes), which may be difficult to recapitulate, since selective expansion of particular B cell clones is based solely on BCR binding properties, not higher-order functionalities [10], [11].
The bnAb 4E10, the focus of our studies, has a conserved, linear epitope (core epitope: 671NWFD/NIT676) immediately adjacent to the viral membrane in the Env gp41 subunit membrane proximal external region (MPER) [12], [13]. While 4E10 displays admirable breadth [14], has been the target of a successful design effort to reverse-engineer tight-binding immunogens [15], has recognizable GEPs present at finite frequencies in human naïve repertoires [16], and arguably does not require a high degree of polyspecificity to neutralize HIV [17], [18], its viability as a vaccine target is hampered by limited potency, demonstrated autoreactivity and exceptional combining site flexibility [17], [19], [20]. The neutralization mechanism of 4E10 also has not been clearly defined and may involve higher order effects [17], [21], [22]. The ontogeny of 4E10, therefore, must be elucidated in order to understand how these properties were acquired and to what degree they impose constraints that might hinder re-elicitation by vaccination.
Mutations acquired during Ab maturation occur preferentially in the CDRs, which make up the six loops (CDRs 1, 2, and 3 on the heavy (HCDRs) and light (LCDRs) chains) comprising the combining site [23], [24]. CDRs are responsible for the majority of direct contacts to an antigen, as opposed to the intervening framework regions (FWRs), which form the immunoglobulin β-sheet structure stabilizing the combining site, helping define CDR loop conformations. While CDR mutations are typically thought to more directly affect antigen binding and neutralization, bnAbs consistently depend on FWR substitutions to a surprising degree, though 4E10 is an exception to this exception [4]. bnAbs are notorious for their high degree of somatic hypermutation, the product of a long process of affinity maturation against a rapidly mutating virus during a persistent, chronic infection [25]. While typical affinity-matured Abs have acquired 15 to 20 VH mutations, bnAbs accumulate up to 100 VH mutations [4]. These mutations are crucial because reversion to germline sequences drastically reduces epitope affinity and neutralization potency and breadth. In many cases, bnAb GEPs are unable to bind Env, though the actual eliciting isolate may not be known [26]–[31]. In addition, bnAbs can contain extraordinarily long HCDR3s, up to 33 residues long versus an average of 13 for non-HIV bnAbs [32], [33]. Using phylogenetically-inferred GEP sequences [16], [34] (Fig. 1), 4E10 has acquired between 33 and 35 mutations during maturation, 20 to 22 in VH, depending on gene segment selection, and 13 in VL, and has an HCDR3 22 residues long, values at the less exceptional end of the bnAb spectrum and not unheard of for conventional Abs.

Affinity-matured Abs display univalent equilibrium binding constants (KD) for their cognate antigens, typically ranging from 10−6 to 10−10 M, that are orders-of-magnitude stronger than their GEPs [35]. Multiple approaches, including computational analyses and biophysical comparisons of affinity-matured Abs and their associated GEPs, have generated a consensus model for the molecular mechanism of affinity maturation [36]–[47], perhaps better understood as binding optimization, that traces its roots back to Pauling [48]. In the consensus model, the antigen specificities of the naïve, germline-encoded repertoire, while diverse and extensive, are further extended by encoding a high degree of polyspecificity into GEPs. This is accomplished partly through increased combining site plasticity in GEPs, more formally stated as the ability of GEP CDRs to dynamically sample a broader ensemble of structural conformers. In response to immunogen stimulation, Ab binding properties are iteratively optimized through cycles of somatic hypermutation and selection, resulting in improved binding affinities, kinetics and thermodynamics. While mutations have been observed to improve or add direct contacts to antigen, typically improving enthalpies of interaction and off-rates (kd), a majority of measurably favorable mutations do not directly contact antigen. These mutations indirectly optimize binding by: (1) increasing shape complementarity between paratopes and epitopes through more global effects on structure; (2) increasing antibody stability, typically measured as solution thermostabilities (Tm), thus compensating for deleterious effects of other mutations that improve affinity; and (3) structurally rigidifying the combining site conformer optimal for binding antigen from the accessible ensemble. Rigidifying the combining site can affect measured interaction parameters in different ways depending on the mechanism of binding. The two mechanistic extremes are known as “conformer-selection” and “instructive-encounter”, or “induced-fit”, binding. In conformer-selection mode, binding does not occur until the compatible conformer is adopted. Rigidification of the binding site through mutation then typically improves entropies of interaction and on-rates (ka). In instructive-encounter mode, initial binding occurs to sub-optimal conformers which affects the rate of interchange with the optimal conformer. Rigidification of the binding site through mutation then typically improves affinity through changes distributed over kd and ka. However, the consensus of studies of binding proteins and enzymes suggests that conformer selection is the preeminent recognition mechanism [49].
Surprisingly, comparisons of the bound and unbound structures of 4E10 revealed that this affinity-matured bnAb incorporates considerable HCDR conformational flexibility [17], in excess of what has been observed in most other antibodies, mature or GEP, suggesting that the ontogeny of 4E10 may be an exception to the consensus model and may pose unique challenges as a vaccine target. In order to fully understand the ontogeny of this unique bnAb and consequences for vaccine development, we characterized the unbound and complex crystal structures, and the functional and binding properties, of 4E10 and an ensemble of its most likely GEPs. GEPs showed detectable, but extremely weak, binding to soluble Env gp140s and extremely limited neutralization potency, though some reverse engineered epitope-scaffolds (ESs) showed robust GEP affinities, well above the B cell activation threshold. 4E10 and GEP paratopes displayed a remarkable degree of structural conservation in the antigen-bound state, with little improvement in overall shape complementarity. Multi-log improvements in affinity for ESs were the result of improved off-rates or combined improvements in on - and off-rates, with a small number of enhanced contacts to antigen observed in the crystal structures. However, minimal mutations of GEP sequences to include these enhanced direct contacts only marginally increases affinity. FWR mutations had little discernable effect on global or local structure. Controverting the consensus model of ontogeny, 4E10 thermostability was appreciably worse than its GEPs; while 4E10 and GEPs displayed similarly constrained VH/VL interdomain movements upon binding, 4E10 maturation involved negligible combining site rigidification, with both 4E10 and GEP HCDRs sampling extensive conformer ensembles. The narrowing of polyspecificity assumed to concur with maturation was not observed with 4E10, as both 4E10 and its GEPs showed similar patterns of limited polyspecificity to a phage-displayed human peptidome (Phage Immunoprecipitation Sequencing; PhIP-Seq) [50]. While 4E10 is demonstrably autoreactive, GEPs exhibited a distinct profile of autoantigen recognition by PhIP-Seq. When combined, these results inform efforts to re-elicit 4E10 by vaccination and its mechanism of neutralization.
Results
Prediction of an ensemble of likeliest GEP sequences
Lacking access to the original donor, identification of a single GEP sequence with high confidence for many bnAbs, including 4E10, is complicated by extensive editing and TdT N-nucleotide insertion during rearrangement [26], leading to our decision to study an ensemble of the 12 likeliest candidates (Fig. 1). Due to the prediction that the sequence differences introduced by alternate heavy chain J gene segments may not affect any discernable GEP property, the initial ensemble was limited to eight GEPs (IGHJ4*02 paired with all six D segments plus IGHJ1*01 paired with IGHD1-1*01 and IGHD6-19*01), with the intention of generating additional GEPs if the IGHJ4*02/IGHJ1*01 substitution exhibited any differences in structure or binding properties on the IGHD1-1*01 or IGHD6-19*01 backgrounds. These eight GEPs also recapitulated some of the variability seen in potential 4E10 GEPs identified by deep sequencing of uninfected individuals (Fig. 1) [16], providing an additional justification for studying the ensemble. However, GEPs in our ensemble differed from one another by no more than four mutations, though the mutations were quite non-conservative.
GEP protein production and validation
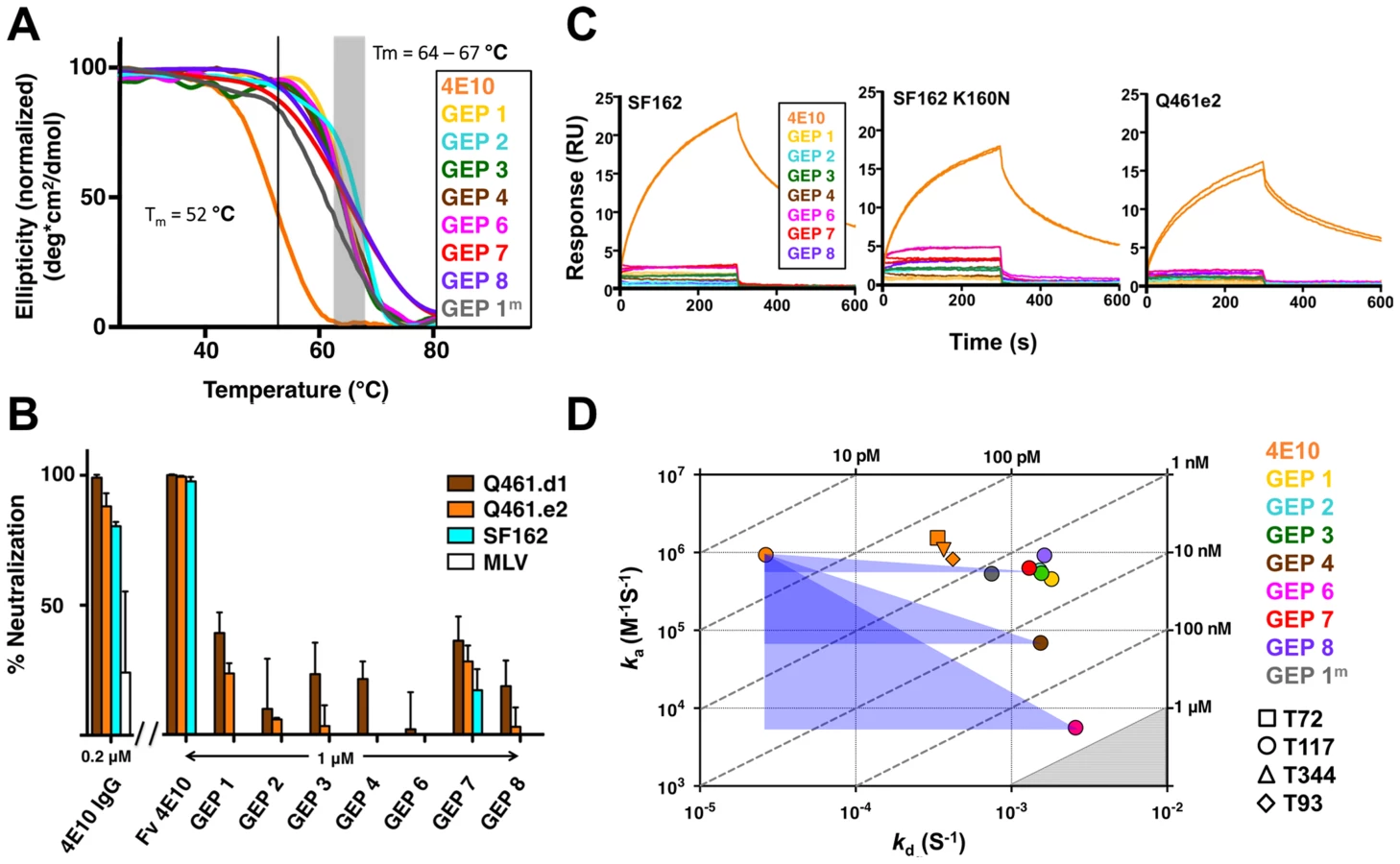
GEPs were engineered as cleavable, single-chain, variable domain cassette (Fv; VH+VL) constructs to ease expression, analysis, and crystallization and to prevent monobody-diabody interchange, following protocols developed for 4E10 [18]. The prior study confirmed that these Fv constructs retained the structural and binding properties of Fab fragments of intact 4E10. All eight GEP Fvs expressed at high levels as bacterial inclusion bodies and, in all but one case (GEP 5), were refolded in vitro with what in our experience were exceptionally high efficiencies of 20 to 40%. GEP 5 was not included in subsequent experiments because its extremely poor in vitro refolding efficiency suggested that this was not a viable pairing. GEP constructs were stable and monodisperse in solution, running exclusively as monomers by size exclusion chromatography (SEC). Reduced/non-reduced PAGE analysis of GEPs confirmed purity and proper disulfide bond formation. GEP Tm values (Fig. 2A), determined by circular dichroism (CD) spectroscopy as previously described [18], narrowly ranged from 64.2°C to 67.0°C, showing that GEP Fvs were well folded, but had even higher Tm values than 4E10 Fv (52.8°C).
GEP neutralization potencies were dramatically reduced
The neutralization potency of GEPs was tested against clade A (Q461.d1, Q461.e2 [51]) and B (SF162 [52]) HIV-1 isolates using standard TZM-bl assays (Fig. 2B) [53]. Overall, GEP Fv potencies were markedly reduced in comparison with 4E10 Fv. GEPs displayed only very weak and likely insignificant neutralization potencies, though with a trend of greater effect against the clade A isolates, particularly the neutralization-sensitive strain Q461.d1, and with GEP 1 and GEP 7 showing marginally better potencies across tested isolates.
GEPs bound Env proteins and engineered antigens, but with reduced affinities
In order to characterize the change in binding properties during 4E10 ontogeny, the binding of 4E10 and GEP Fvs to three (SF162, SF162K160N and Q461.e2) soluble Env gp140 trimers (gp1403) [54] and four engineered 4E10 ES immunogens (T72, T93, T117 and T344) [15], [55] was evaluated by surface plasmon resonance (SPR) interaction analysis (Figs. 2C, 2D, Figs. 3–7, Table 1). Isolates and ESs were selected to span a range of binding properties, where previous studies had shown 4E10 bound a free peptide spanning its linear epitope with a KD of 12 nM, SF162 gp1403 with a KD of 98 nM, and ESs with KDs of either ≤10 picomolar (T117) or ∼100 picomolar (T72, T93 and T344) [15],[18],[55]. All seven GEPs showed unquantifiably weak, but detectable binding to chip-coupled clade A (Q461.e2) and clade B (SF162, SF162K160N) gp1403 and T72, T93, and T344 in qualitative SPR analyses, with KDs all estimated to be well above the ∼micromolar B cell activation threshold. Quantitative SPR analyses of GEPs binding to T117 showed KDs ranging from the low nanomolar to low micromolar range. 4E10 interactions with ESs ranged from 100 - to 10,000-fold stronger than GEPs, which was qualitatively consistent with the observed difference in 4E10 versus GEP interactions with gp1403. Since the GEP/gp1403 interactions were too weak to quantitate, peptide binding studies were not performed on the expectation that they would also be too weak to measure accurately. Five GEPs (1, 2, 3, 7 and 8) bind T117 with nearly identical behavior, including GEP 1 and GEP 7, which differ only by alternate J segment utilization, showing that the two incorporated mutations did not affect binding, so no further IGHJ1*01 GEPs were produced for study. GEP 4 and GEP 6, which both incorporate differences from G96H in 4E10 (A or V), showed approximately 10-fold (GEP 4) or 100-fold (GEP 6) reductions in affinity relative to the cluster of other GEPs. Kinetically, the five clustering GEPs (1, 2, 3, 7 and 8) showed affinity reductions for T117 relative to 4E10 overwhelmingly through faster off-rates (kds). GEPs 4 and 6, in addition to comparable increases in kd, also showed progressive reductions in on-rates (kas).






4E10 - and GEP-ES complex structures show binding site conservation
In order to shed light on potential structural differences accounting for reduced GEP binding affinities, crystal structures of GEP 1, 2 and 7 in complex with T117 were determined at resolution values of 2.9 Å, 1.8 Å, and 3.1 Å respectively, rebuilt and refined with good statistics (Table 2), and compared to two reference 4E10/antigen complex structures: 4E10 bound to an epitope peptide (2FX7.pdb [12]) or a related ES, T88 (3LH2.pdb [15]). Superpositions showed that almost all direct contacts to the core epitope (NWFDIT) and epitope conformation are conserved between 4E10 and GEP complexes to a remarkable degree (Fig. 8, Tables 3 and 4). Only six of the 35 predicted somatic mutations affect sequence positions making direct contacts to the core epitope: Y/K32L, S/Q93L, T/S31H, I/V51H, F/L54H and T/I56H. However, residues at two of these positions (93L, 51H) contacted the epitope solely through main-chain interactions, which were not affected by the mutated side-chains. Replacements at three other positions (31H, 54H, 56H) nearly perfectly recapitulated contacts, also conserving the interface. The 4E10 and GEP paratope/epitope interface is largely hydrophobic, thus binding is mainly mediated by Van der Waals contacts and desolvation entropy, which is conserved through equivalent positioning of nonpolar groups. This was reflected in the close concordance between the surface areas (SA) buried in the complexes by core epitope and the corresponding shape complementarity (Sc) [56] between Ab and core epitope. Only two somatic mutations were predicted to contribute to a stronger binding interaction. The Y/K32L mutation replaces a hydrogen bond involving the tyrosine hydroxyl with a salt bridge. The P/L95H mutation, involving non-contacting residues, restructured LCDR3 to reposition the side-chain of the conserved serine at 94L from a non-contacting position in GEPs to one contributing a hydrogen bond in 4E10 complex structures. However, the Y/K32L-P/L95H double mutation made on a GEP 1 background (GEP 1m) did not affect Tm, only marginally increased affinity by about three-fold, and did not improve neutralization potency (Figs. 2A, 2D, 6 and Table 1), arguing that affinity and neutralization are largely influenced by somatic mutations through indirect effects beyond the ability to adopt the optimal binding conformer.




GEP structures revealed that structural plasticity was retained during affinity maturation
In order to determine whether the 4E10 combining site rigidified during affinity maturation, undergoing binding site preconfiguration, the crystal structures of GEP 7 and GEP 1 were determined in the unbound state at resolution values of 1.9 Å and 1.7 Å respectively, rebuilt and refined with good statistics (Table 2), and compared to the unbound structure of 4E10 (4LLV.pdb [17]). Superpositions of 4E10 and GEP bound and unbound structures showed that interdomain movements, while present, were limited compared to CDR rearrangements (Fig. 9), and that 4E10 retained at least as much CDR flexibility as was present in GEPs, particularly in HCDR2 and HCDR3, while LCDRs were relatively constrained (Figs. 10A and 10B). Calculated root mean square deviations (RMSDs; Fig. 10C) of VH and VL superpositions, with and without CDRs, confirmed that while the FWR regions of GEPs and 4E10 were nearly identical, the bulk of rearrangements observed during binding occurred in HCDRs, with 4E10 movements as large, or larger, than observed in GEPs. LCDR movements, while much smaller overall, contributed less to rearrangements in 4E10 than GEPs, suggesting some minimal degree of rigidification during maturation. Comparison of the surfaces directly contacting epitopes (Movies S1 and S2) showed that GEPs retained higher degrees of structural conservation than 4E10 between the bound and unbound states. These analyses need to be interpreted cognizant of the constraints imposed by crystallization, which involved varying non-physiological solution conditions and variable crystal contacts between structures. However, these caveats were minimized by comparing multiple structures with multiple copies per asymmetric unit.


Engineered ESs can make extensive contacts outside of the targeted epitope
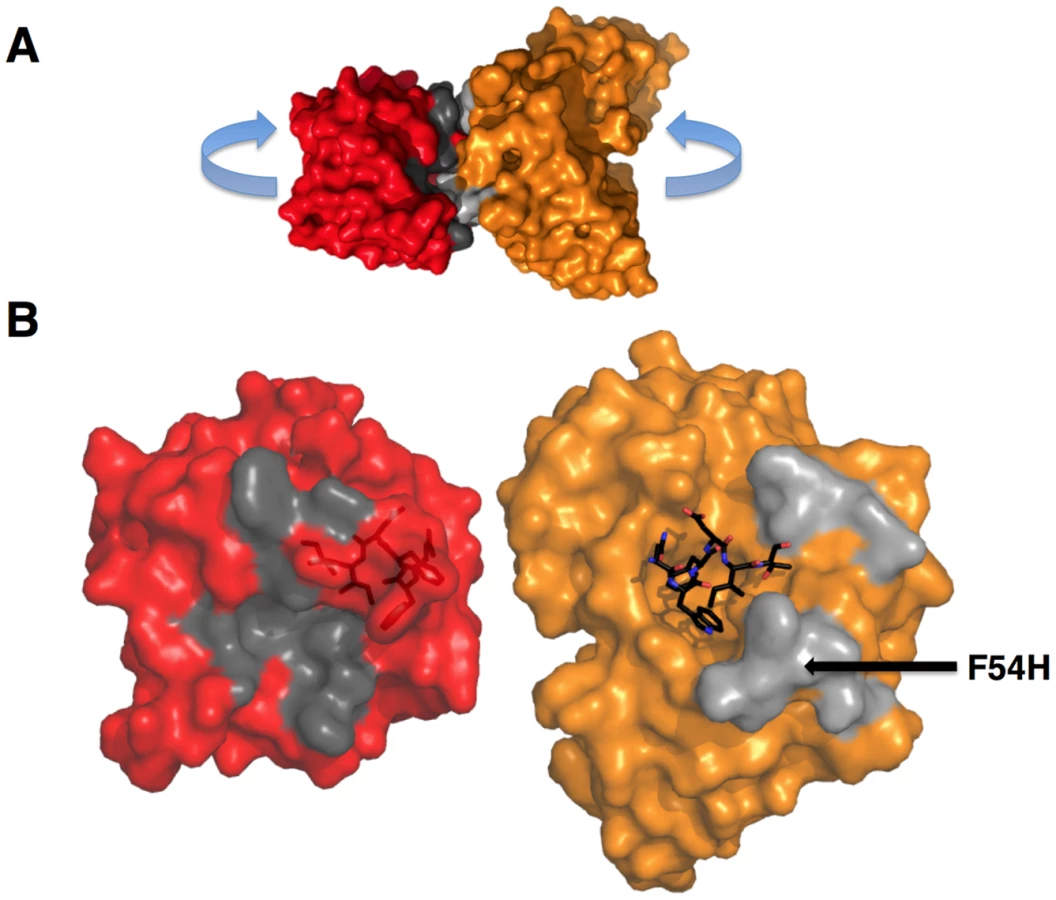
The goal of reverse engineering an Ab is to scaffold the desired epitope to re-elicit Abs that solely recognize the epitope [6], [57]. However, the 4E10 linear epitope, as currently defined, is smaller than typical Ab/antigen interfaces, which poses the design challenge of isolating humoral responses to the epitope and not contiguous scaffold surfaces. Previous crystal structures of 4E10/ES complexes showed that many ESs achieved this goal well, including T93 [15], [55]. However, GEP/T117 complex structures reported here showed extensive GEP/scaffold contacts (SA for GEP2 contacts to scaffold minus epitope = 308 Å2) (Fig. 11). A dominant feature of these interactions was the binding of the side-chain of the GEP-specific residue F54H in a deep hydrophobic cleft of the T117 scaffold protein, a putative phosphotransferase from S. typhimurium. These additional contacts raise the concern that Abs elicited by T117 immunizations may have off-target (non-Env) specificities. Nevertheless, the T117 scaffold is highly complementary to 4E10 (Sc for GEP2/T117 = 0.69), which may help explain the increased affinity of T117 for 4E10 and GEPs, and may be ideal for preferentially targeting GEPs through the F54H interaction (F54H is present in the heavy chain gene used by all GEPs). The additional T117 contacts appear to have had the effect of increasing the affinity for T117 over other ESs by two orders of magnitude for 4E10 and by one to three orders of magnitude for the GEPs. However, structural superpositions show that these extra Ab/scaffold interactions do not affect Ab/epitope interactions, or the structure of the epitope in the complexes, which are highly conserved (Fig. 8B).
GEPs displayed a similar degree of limited polyspecificity to 4E10, but different autoreactivity
A validated phage-displayed library consisting of 413,611 overlapping 36-mer peptides spanning the entire human proteome combined with phage immunoprecipitation sequencing (PhIP-Seq) was used to assess the polyspecificity and autoantigen recognition profiles of GEPs in comparison to 4E10 [17], [50] (Fig. 12A, Tables 5, 6, and 7). GEP 2 and GEP 4 were selected to represent both the clustering (GEP 2) and 96H mutant (GEP 4) GEPs, and an affinity-matured, murine anti-canine CD28 Ab, 1C6 [58], was included for comparison (Fig. 12A, Table 8). The top hit in the 4E10 Fv PhIP-Seq analysis reported here, a peptide derived from the type 2 inositol 1,4,5-trisphosphate receptor (IP3R), matches the top hit from the previous PhIP-Seq analysis of IgG 4E10 [17]. However, overall scores were considerably reduced in the PhIP-Seq analysis of 4E10 as an Fv construct versus intact IgG (replicate average −Log10 P-values for the top ten scoring peptides of 35.3 to 255 for IgG versus 4.35 to 12.3 for Fv), likely due to decreased accessibility of coupled Fv relative to IgG, an increased chance of inactivating Fv versus IgG during chemical coupling, and the inherent increase in local avidity of bivalent IgG versus univalent Fv on potentially sparsely-coupled beads. Given these caveats, overall scoring behavior was very similar across the Fvs tested, with 4E10 and 1C6, the affinity-matured Abs, showing the highest average scores. GEP 4 and 1C6 showed the largest number of high-scoring hits, with 61 and 194 peptides with replicate-averaged −Log10 P-values of ≥4.0 respectively; 4E10 and GEP2 had 12 and 20 peptides, respectively, scoring ≥4.0. Qualitatively, the results were not dramatically different, but with GEP 4 and 1C6 showing nominally greater spreads of top-scoring peptides rising above the bulk responses. None of the top-scoring three-dozen 4E10 peptides appeared in the top three-dozen hits from either GEP; however two of the top-scoring dozen GEP peptides (derived from zinc finger Ran-binding domain-containing protein 3 or hyaluronidase-3 isoform 1 precursor) were in common between GEP 2 and GEP 4, scoring 1 and 12 (GEP 2) and 4 and 2 (GEP 4), respectively. However, no common peptide motifs could be identified within or between Fv results. None of the IP3R peptides scored in the top 65,000 GEP hits. While 1C6 showed the highest scoring spread of top hits (replicate average −Log10 P-values for the top ten scoring peptides of 12.14 to 30.9), the top ten scoring peptides displayed a considerably higher average hydrophobic character (Φ), with average Φ values of: 4E10 = 0.37; GEP 2 = 0.42; GEP 4 = 0.45; 1C6 = 0.71 (higher values are more hydrophobic). Using relative hydrophobicity of the top-scoring PhIP-Seq peptides as a surrogate measure of the overall hydrophobicity of the combining site was consistent with the structures of the Abs (Fig. 12B), where the 1C6 combining site is structured as a large, broad, very hydrophobic concavity and 4E10 and its GEPs sporting smaller, convex hydrophobic surfaces.





Pre-binding of 4E10 at the MPER affects the binding of bnAb b12 at the CD4 binding site
In order to test the hypothesis that 4E10 may induce global conformational changes in gp1403 as part of a higher-order neutralization mechanism [17], [21], consistent with downstream effects such as gp120 release, binding of Abs to epitopes distant from the MPER (the V3 loop, by 447-52D [59], and the CD4 binding site, by b12 [60]) was assayed by SPR in the presence or absence of saturating 4E10 (Fig. 13). The qualitative results show that 4E10 pre-binding does not affect 447-52D binding to the flexible, extended V3 loop, but does alter the association kinetics of b12 at the CD4 binding site, suggesting that 4E10 binding induced global conformational changes in gp1403 registering at distant sites. Since b12 dissociation kinetics were unaffected, 4E10 did not preclude achieving a similar b12 bound-state conformation.

Discussion
Reverse engineered ESs are ideal reagents for studying the binding properties of GEPs. Where peptides and Env do not display sufficient affinities, the best ESs display strong affinities across the GEP ensemble, allowing for the biophysical characterization of 4E10 ontogeny by using ESs to compare 4E10 and GEP binding kinetics and bound-state crystal structures. Consistent with current theory and previous results comparing GEPs with matured bnAbs or other Abs, 4E10 displays orders-of-magnitude better affinities than candidate GEPs for both Env gp1403 proteins and engineered ESs. This gain in affinity is potentially sufficient to account for the concurrent gain in neutralization potency over GEPs. Previous results showing minimal contributions from FWR mutations [61] and comparisons of complex structures of 4E10 with GEP/T117 complex structures, which showed near-complete conservation of the recognition interface (Fig. 8), argue that affinity maturation was likely due, at least in part, to the small number of somatic mutations in the CDR regions, e.g. 32L and 95H, that add or improve direct contacts to epitope. This limited number of mutations is presumably readily achievable during conventional vaccination. Based on PhIP-Seq peptidome binding results (Fig. 12A), 4E10 and GEPs have distinct autoantigen profiles, suggesting that 4E10 autoreactivity was acquired during ontogeny and not inherent in GEPs, consistent with recognizable GEPs populating naïve repertoires of mature B cells at relatively high frequencies [16]. Among ESs, T117 in particular interacts with GEPs sufficiently strongly to drive B cell activation and maturation (Fig. 2D) and selectively interacts with GEP-specific structural features, e.g. F54H (Fig. 11). These aspects of 4E10 combine to seemingly argue that 4E10 ontogeny follows a relatively short path and that directed re-elicitation of 4E10, and perhaps other bnAbs, may be an achievable goal.
In many aspects, however, 4E10 ontogeny appears more convoluted. Contrasting with the consensus model of Ab ontogeny and results from other systems [38], 4E10 is considerably less thermostable than GEPs, which was difficult to account for only from static views of structures. This lowers the headroom available to tolerate mutations that increase affinity or potency that otherwise might degrade stability, potentially adding a significant hurdle to re-elicitation of 4E10, but clearly does not limit the ability of 4E10 to bind to or neutralize HIV. The conformer ensemble sampled by GEP combining sites echoes the startling plasticity of 4E10, with structural comparisons of bound versus unbound states (Figs. 9 and 10) showing perhaps a small degree of 4E10 LCDR rigidification, but an increase in the conformer ensemble sampled by the HCDRs. This directly contradicts the consensus model, and specific examples [40], of Ab maturation, showing alternatively that preconfiguration of 4E10 does not occur during, or contribute to, affinity maturation. HCDR flexibility is highlighted by F29H, which is able to flip out into solvent in the unbound 4E10 and GEP 1 structures, allowing HCDR1 to dynamically sample multiple conformers. A high number of conserved glycine residues may contribute to 4E10 and GEP HCDR mobility. The reduced affinities due to decreased kas (Fig. 2D), relative to the clustering GEPs, of GEP 4 and 6 with non-glycine residues at position 96H, predicted to restrict HCDR3 conformer sampling, suggests that HCDR3 mobility was needed to achieve the bound-state conformation with optimal kinetics by destabilizing non-optimal conformers. Retention of significant combining site plasticity also strongly argues for a functional role other than polyspecificity, which is unremarkable in comparison to other Abs, based on PhIP-Seq peptidome binding results (Fig. 12A). Minimal focusing of apparent polyspecificity also contradicts the consensus model, and specific examples [62], of maturation. Also unexpectedly, the ∼100-fold increase in affinity of 4E10 over GEPs to T117 is through a decreased kd (Fig. 2D), suggesting that improvements in binding affinity arose from a more favorable bound state and not a decrease in entropic barriers imposed by more plastic GEPs. Thermodynamic studies of 4E10 and GEPs were not possible because no single antigen, needed for valid comparisons, bound to both 4E10 and GEPs with parameters accessible to measurement. However, while crystal structures showed a highly-conserved bound state in both 4E10 and GEPs, the small number of improved contacts could not account fully for the observed improvement in affinity. Complicating this analysis, and its use as a vaccine immunogen, are the extensive contacts between GEPs, and presumably also 4E10, and the scaffold of T117 outside of the stabilized MPER epitope (Fig. 11). The interaction with T117 also highlights another possibility. The 4E10 combining site extends beyond the minimal, linear epitope in T117, making contacts to the scaffold. Unless the MPER epitope forms an isolated structure extended away from the rest of Env, 4E10 may make contacts to elements of Env outside of the linear 4E10 epitope, as it does with T117. HCDR conformer dynamics may therefore be understood as enabling such interactions, which may foster conformational changes in Env leading to neutralization by any of several mechanisms. Prebinding of 4E10 to gp1403 affected b12 binding at the CD4 site distant from the MPER (Fig. 13), indirectly demonstrating a global conformational change consistent with this supposition. Additional interactions between GEPs and the scaffold moiety of T117 would also be predicted to drive rigidification of an even greater portion of the combining site during conventional immunization, increasing the distinction between 4E10 and a matured Ab derived from a 4E10 GEP stimulated with T117.
T117 was the product of a cutting-edge design effort to generate a structurally-stabilized MPER epitope with optimized binding properties for use as a vaccine immunogen to drive 4E10 ontogeny based on the current paradigm of affinity maturation. However, 4E10 appears to take the affinity maturation pathway less traveled, one that contrasts, in many fundamental ways, the current paradigm. This raises the concern that conventional immunization protocols, based on the current paradigm of structural stabilization of optimal binding conformers, will not successfully re-elicit 4E10 or other unconventional Abs. Understanding unconventional maturation pathways then becomes paramount for the future of molecular vaccinology, allowing efforts to focus on re-eliciting Abs identified as the products of conventional ontogeny in the near term while developing unconventional vaccination strategies to target exceptional Abs in the long term.
Materials and Methods
Protein prediction, expression, purification and characterization
The 4E10 heavy and light chain nucleotide sequences were analyzed using JoinSolver [63], IMGT/V-QUEST [64], [65], Ab-Origin [66], SoDA [67] and iHMMune [68] to compositely identify segments with the fewest nucleotide mismatches, generating a combinatorial ensemble of 12 GEPs (Fig. 1). Each derived GEP candidate consisted of a single VL domain sequence assigned with high confidence (IGKV3-20*01 plus IGKJ1*01), one VH gene segment also confidently assigned (IGHV1-69*06), plus one of six likely D segments (IGHD1-1*01, IGHD6-19*01, IGHD7-27*01, IGHD6-25*01, IGHD1-26*01, IGHD4-17*01) and either of two likely J segments (IGHJ4*02, IGHJ1*01). The CDR3 junctions and presumed N-nucleotide insertions in mature 4E10 were retained in the candidate GEPs. Sequences are numbered following Kabat [24]. 4E10, eight of the 12 GEPs, and 1C6 Fvs were engineered, expressed as inclusion bodies in E. coli BL21(DE3) RIL cells (Invitrogen), refolded, purified, and validated by SEC and PAGE as previously described [18]. Circular dichroism (CD) spectra of 4E10 Fv and GEPs were measured on a J-815 spectrometer (Jasco) at a concentration of 10 µM in 10 mM Na2HPO4/KH2PO4 (pH = 7.4). Temperature melts were performed at 210 nM with a temperature ramp from 25°C to 95°C at a slope of 2°C/minute, data pitch of 2°C, and delay time of 10 s (Fig. 2). Tms were determined by nonlinear least-squares analysis using a linear extrapolation model with Spectra Analysis software (Jasco). Fabs of 447-52D and 4E10 were prepared by digestion of IgG with papain (Pierce), affinity chromatography with protein A (Pierce) and preparative SEC (Superdex 200 16/10 column; GE Healthcare); IgG 4E10 was purchased from Polymun Scientific, and IgG 447-52D and Fab b12 were kindly provided by Pamela Bjorkman (Caltech).
Neutralization assays
Neutralization assays (Fig. 2B) were performed using single-round entry-competent viruses and TZM-bl cells as previously described [53]. Percent neutralization at concentrations of 0.96 µM (4E10 and GEP Fvs) or 0.17 µM (4E10 IgG) was calculated as previously described [18], [53].
SPR interaction analyses
All SPR experiments were performed at 25°C on a Biacore T100 instrument with the T200 sensitivity enhancement (GE Healthcare). For analyses of the binding of 4E10 and GEP Fv analytes to chip-captured ES ligands (Fig. 2D, Table 1), ESs at ∼1 µg/mL were captured on a SA sensor chip (GE Healthcare) following either carboxy (T117) or amine (T72, T93, T344) biotinylation, following the manufacturer's recommended protocol (EZ-Link, Thermo Scientific), in a running buffer of HBS-EP+ (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% P-20; GE Healthcare) plus 0.1 mg/mL bovine serum albumin. A reference flow cell was left blank. Duplicate 4E10 and GEP Fv analyte injections were randomized and run at a flow rate of 50 µL/minute. Regeneration, if needed, was achieved by injection of 10 mM glycine at 50 µL/minute followed by buffer stabilization. Sensorgrams obtained from SPR measurements were double-reference subtracted [69] with BIAevaluation 2.0.3 software (GE Healthcare) employing previously described methodology [18]; data were fit with either 1∶1 or steady-state binding models. For analyses of the binding of 4E10 and GEP Fv analytes to gp1403 (Fig. 2C), gp1403 at 30 µg/mL in 10 mM sodium acetate (pH = 5.0) were direct amine coupled at a density of ∼2200 RUs on a CM5 sensor chip (GE Healthcare); a reference surface was prepared by activating and deactivating a flow cell without the addition of protein; gp1403 were prepared as previously described [18]. Duplicate 5 minute, 300 nM injections of 4E10 and GEP Fv were made in HBS-EP+ at 50 µL/minute followed by 5 minutes of dissociation, regeneration with 10 mM glycine (pH = 1.5) for 5 seconds and then 6 minutes of stabilization. Assays of Fab b12 or Fab 447-52D binding to SF162 gp1403 in the presence or absence of 4E10 Fv (Fig. 13) were conducted in HBS-EP+ buffer. Using standard amine coupling chemistry, 1273 RUs of SF162 gp1403 were coupled to a CM5 sensor chip. A reference surface was prepared by activating and deactivating a flow cell without the addition of protein. Two samples were injected at a flow rate of 50 µL/minute using the dual injection command in the T100 control software (v2.0.3, GE Healthcare) with injection 1 at 5 minutes, injection 2 at 2 minutes and a final dissociation time of 5 minutes. Fab-alone curves were generated by injecting HBS-EP+ followed by an injection of 10 nM Fab b12 or 447-52D and double referenced by subtracting a dual injection of HBS-EP+ followed by HBS-EP+. Fab with 4E10 Fv curves were generated by injecting 3 µM 4E10 Fv (∼90 RUs bound) followed by an injection of 10 nM Fab b12 or 447-52D in the presence of 3 µM 4E10 Fv and double referenced by subtracting a dual injection of 3 µM 4E10 Fv followed by 3 µM 4E10 Fv. Optimal regeneration was achieved by injection of 10 mM glycine (pH = 1.5) at a flow rate of 50 µL/minute for 5 seconds followed by a 6 minute buffer stabilization phase. Sensorgrams were corrected by the double-subtraction method [69] in Scrubber 2.0b software (BioLogic Software). Fig. 13 shows the second sample of each set of injections with baselines zeroed just prior to the second injection.
Crystallization and crystallography
Crystals of GEPs and 4E10 Fv were grown by the hanging-drop vapor diffusion method at 25°C with the following well solutions:
-
GEP 1 : 25% w/v PEG 3350, 0.2 M NaCl, 0.1 M Tris (pH = 8.5)
-
GEP 1/T117: unbuffered 12% w/v PEG 8000, 0.1 M KCl, 5% v/v glycerol
-
GEP 2/T117 : 1.6 M Li2SO4, 0.1 M sodium acetate (pH = 5.0)
-
GEP 7 : 20% w/v PEG 2000, 0.2 M Trimethylamine N-oxide, 0.1 M Tris (pH = 9.0)
-
GEP 7/T117 : 10% w/v PEG 10000, 0.1 M NH4OAc, 0.1 M Bis-Tris (pH = 5.5)
-
1C6 : 3.5 M sodium formate, 0.1 M Bis-Tris Propane (pH = 7.0)
Crystals were cryopreserved in mother liquor containing 15% v/v glycerol (GEP 1, GEP 1/T117, GEP 7/T117), mother liquor containing 10% (1C6) or 20% (GEP 7) v/v glycerol, or 20% w/w sucrose (GEP 2/T117). Diffraction data for GEP 1, 4E10, and 1C6 were collected at the Advanced Light Source beamline 5.0.2 (Lawrence Berkeley National Laboratory, Berkeley, CA) and reduced using HKL-2000 [70] or d*TREK (Rigaku) [71]. Diffraction data for GEP 7, GEP 1/T117, GEP 7/T117 and GEP 2/T117 were collected in house with CuKα radiation on a R-AXIS IV++ image plate detector with HR optics (Rigaku) at −170°C. Initial phase information for all data sets was determined by molecular replacement, as implemented in the CCP4i program suite [72]–[74], using 3LF6.pdb (T117), 3LH2.pdb (GEPs) and 1JP5.pbd (1C6) as initial search models. Phases were improved by subsequent rounds of model building and refinement using COOT [75] and REFMAC [76]. Structure validation was carried out with PROCHECK [77], the MolProbity server [78], and the RCSB ADIT validation server. Data collection and structure refinement statistics are shown in Table 2. Crystals of GEP 2 alone could not be grown, despite considerable effort.
PhIP-Seq analysis
4E10, GEP and 1C6 Fvs were coupled to beads and analyzed in duplicate. For each Fv analyzed, 3 mg of magnetic beads (Invitrogen M-270 Epoxy Dynabeads) were resuspended in 60 µL 0.1 M NaPO4 (pH = 7.4). Beads were rocked at ambient temperature for 24 hrs with 60 µg of each Fv in 1 M (NH4)2SO4 and then washed with 10 mM glycine in PBS to cap unreacted epoxy groups. Activity of Fv coupled beads was confirmed by epitope-scaffold binding prior to PhIP-Seq analyses. PhIP-Seq analyses were performed as previously described [17], [50]. Results were plotted as replicate #1 versus replicate #2 −Log10 P-values; highly discordant, and therefore spurious, hits falling near the axes were excluded from analysis. 241 peptides were also discarded because they displayed nonspecific binding to all Abs tested [17]. Peptide hydrophobicity was determined with the Sigma-Aldrich PEPscreen Library Design Tool and overall charge at neutral pH was determined with the Innovagen Peptide Property Calculator.
Accession numbers
Coordinates and structure factor amplitudes have been deposited in the Protein Data Bank [79] under accession codes: 4M8Q.pdb (ligand-bound GEP 1), 4LRN.pdb (unbound GEP 1), 4M62.pdb (ligand-bound GEP 2), 4ODX.pdb (ligand-bound GEP 7), 4OB5.pdb (unbound GEP 7), and 4LCI.pdb (1C6).
Supporting Information
Zdroje
1. StephensonKE, BarouchDH (2013) A global approach to HIV-1 vaccine development. Immunol Rev 254 : 295–304.
2. HaynesBF, McElrathMJ (2013) Progress in HIV-1 vaccine development. Curr Opin HIV AIDS 8 : 326–332.
3. BonsignoriM, AlamSM, LiaoHX, VerkoczyL, TomarasGD, et al. (2012) HIV-1 antibodies from infection and vaccination: insights for guiding vaccine design. Trends Microbiol 20 : 532–539.
4. KleinF, MouquetH, DosenovicP, ScheidJF, ScharfL, et al. (2013) Antibodies in HIV-1 vaccine development and therapy. Science 341 : 1199–1204.
5. StamatatosL (2012) HIV vaccine design: the neutralizing antibody conundrum. Curr Opin Immunol 24 : 316–323.
6. BurtonDR (2002) Antibodies, viruses and vaccines. Nat Rev Immunol 2 : 706–713.
7. LangJ, JacksonM, TeytonL, BrunmarkA, KaneK, et al. (1996) B cells are exquisitely sensitive to central tolerance and receptor editing induced by ultralow affinity, membrane-bound antigen. J Exp Med 184 : 1685–1697.
8. QiH, EgenJG, HuangAY, GermainRN (2006) Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science 312 : 1672–1676.
9. BatistaFD, NeubergerMS (1998) Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity 8 : 751–759.
10. KimM, SunZY, RandKD, ShiX, SongL, et al. (2011) Antibody mechanics on a membrane-bound HIV segment essential for GP41-targeted viral neutralization. Nat Struct Mol Biol 18 : 1235–1243.
11. JulienJP, HuarteN, MaesoR, TanevaSG, CunninghamA, et al. (2010) Ablation of the complementarity-determining region H3 apex of the anti-HIV-1 broadly neutralizing antibody 2F5 abrogates neutralizing capacity without affecting core epitope binding. J Virol 84 : 4136–4147.
12. CardosoRM, ZwickMB, StanfieldRL, KunertR, BinleyJM, et al. (2005) Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22 : 163–173.
13. ZwickMB, LabrijnAF, WangM, SpenlehauerC, SaphireEO, et al. (2001) Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol 75 : 10892–10905.
14. BinleyJM, WrinT, KorberB, ZwickMB, WangM, et al. (2004) Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J Virol 78 : 13232–13252.
15. CorreiaBE, BanYE, HolmesMA, XuH, EllingsonK, et al. (2010) Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure 18 : 1116–1126.
16. LarimoreK, McCormickMW, RobinsHS, GreenbergPD (2012) Shaping of Human Germline IgH Repertoires Revealed by Deep Sequencing. J Immunol 189 (6) 3221–30..
17. FintonKA, LarimoreK, LarmanHB, FriendD, CorrentiC, et al. (2013) Autoreactivity and exceptional CDR plasticity (but not unusual polyspecificity) hinder elicitation of the anti-HIV antibody 4E10. PLoS Pathog 9: e1003639.
18. XuH, SongL, KimM, HolmesMA, KraftZ, et al. (2010) Interactions between lipids and human anti-HIV antibody 4E10 can be reduced without ablating neutralizing activity. J Virol 84 : 1076–1088.
19. Doyle-CooperC, HudsonKE, CooperAB, OtaT, SkogP, et al. (2013) Immune tolerance negatively regulates B cells in knock-in mice expressing broadly neutralizing HIV antibody 4E10. J Immunol 191 : 3186–3191.
20. ChenY, ZhangJ, HwangKK, Bouton-VervilleH, XiaSM, et al. (2013) Common tolerance mechanisms, but distinct cross-reactivities associated with gp41 and lipids, limit production of HIV-1 broad neutralizing antibodies 2F5 and 4E10. J Immunol 191 : 1260–1275.
21. RuprechtCR, KrarupA, ReynellL, MannAM, BrandenbergOF, et al. (2011) MPER-specific antibodies induce gp120 shedding and irreversibly neutralize HIV-1. J Exp Med 208 : 439–454.
22. AlamSM, MorelliM, DennisonSM, LiaoHX, ZhangR, et al. (2009) Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc Natl Acad Sci U S A 106 : 20234–20239.
23. AmzelLM, PoljakRJ (1979) Three-dimensional structure of immunoglobulins. Ann Rev Biochem 48 : 961–997.
24. WuTT, KabatEA (1970) An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med 132 : 211–250.
25. BredenF, LepikC, LongoNS, MonteroM, LipskyPE, et al. (2011) Comparison of antibody repertoires produced by HIV-1 infection, other chronic and acute infections, and systemic autoimmune disease. PLoS One 6: e16857.
26. XiaoX, ChenW, FengY, ZhuZ, PrabakaranP, et al. (2009) Germline-like predecessors of broadly neutralizing antibodies lack measurable binding to HIV-1 envelope glycoproteins: implications for evasion of immune responses and design of vaccine immunogens. Biochem Biophys Res Commun 390 : 404–409.
27. ScheidJF, MouquetH, UeberheideB, DiskinR, KleinF, et al. (2011) Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333 : 1633–1637.
28. ZhouT, GeorgievI, WuX, YangZY, DaiK, et al. (2010) Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329 : 811–817.
29. McGuireAT, HootS, DreyerAM, LippyA, StuartA, et al. (2013) Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. J Exp Med 210 : 655–663.
30. BatesJT, KeeferCJ, UtleyTJ, CorreiaBE, SchiefWR, et al. (2013) Reversion of somatic mutations of the respiratory syncytial virus-specific human monoclonal antibody Fab19 reveal a direct relationship between association rate and neutralizing potency. J Immunol 190 : 3732–3739.
31. WuX, ZhouT, ZhuJ, ZhangB, GeorgievI, et al. (2011) Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333 : 1593–1602.
32. JohnsonG, WuTT (2000) Kabat database and its applications: 30 years after the first variability plot. Nucleic Acids Res 28 : 214–218.
33. JohnsonG, WuTT (1998) Preferred CDRH3 lengths for antibodies with defined specificities. Int Immunol 10 : 1801–1805.
34. MaBJ, AlamSM, GoEP, LuX, DesaireH, et al. (2011) Envelope deglycosylation enhances antigenicity of HIV-1 gp41 epitopes for both broad neutralizing antibodies and their unmutated ancestor antibodies. PLoS Pathog 7: e1002200.
35. FooteJ, EisenHN (1995) Kinetic and affinity limits on antibodies produced during immune responses. Proc Natl Acad Sci U S A 92 : 1254–1256.
36. ThomsonCA, BrysonS, McLeanGR, CreaghAL, PaiEF, et al. (2008) Germline V-genes sculpt the binding site of a family of antibodies neutralizing human cytomegalovirus. EMBO J 27 : 2592–2602.
37. ScharfL, WestAPJr, GaoH, LeeT, ScheidJF, et al. (2013) Structural basis for HIV-1 gp120 recognition by a germ-line version of a broadly neutralizing antibody. Proc Natl Acad Sci U S A 110 : 6049–6054.
38. WangF, SenS, ZhangY, AhmadI, ZhuX, et al. (2013) Somatic hypermutation maintains antibody thermodynamic stability during affinity maturation. Proc Natl Acad Sci U S A 110 : 4261–4266.
39. LingwoodD, McTamneyPM, YassineHM, WhittleJR, GuoX, et al. (2012) Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 489 : 566–570.
40. SchmidtAG, XuH, KhanAR, O'DonnellT, KhuranaS, et al. (2013) Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc Natl Acad Sci U S A 110 : 264–269.
41. WedemayerGJ, PattenPA, WangLH, SchultzPG, StevensRC (1997) Structural insights into the evolution of an antibody combining site. Science 276 : 1665–1669.
42. ManivelV, SahooNC, SalunkeDM, RaoKV (2000) Maturation of an antibody response is governed by modulations in flexibility of the antigen-combining site. Immunity 13 : 611–620.
43. BaborM, KortemmeT (2009) Multi-constraint computational design suggests that native sequences of germline antibody H3 loops are nearly optimal for conformational flexibility. Proteins 75 : 846–858.
44. SethiDK, AgarwalA, ManivelV, RaoKV, SalunkeDM (2006) Differential epitope positioning within the germline antibody paratope enhances promiscuity in the primary immune response. Immunity 24 : 429–438.
45. YinJ, BeuscherAEt, AndryskiSE, StevensRC, SchultzPG (2003) Structural plasticity and the evolution of antibody affinity and specificity. J Mol Biol 330 : 651–656.
46. YinJ, MundorffEC, YangPL, WendtKU, HanwayD, et al. (2001) A comparative analysis of the immunological evolution of antibody 28B4. Biochemistry 40 : 10764–10773.
47. NguyenHP, SetoNO, MacKenzieCR, BradeL, KosmaP, et al. (2003) Germline antibody recognition of distinct carbohydrate epitopes. Nat Struct Biol 10 : 1019–1025.
48. PaulingL (1940) Theory of the Structure and Process of Formation of Antibodies. J Am Chem Soc 62 : 2643–2657.
49. ChangeuxJP, EdelsteinS (2011) Conformational selection or induced fit? 50 years of debate resolved. F1000 Biol Rep 3 : 19.
50. LarmanHB, ZhaoZ, LasersonU, LiMZ, CicciaA, et al. (2011) Autoantigen discovery with a synthetic human peptidome. Nat Biotechnol 29 : 535–541.
51. BlishCA, NedellecR, MandaliyaK, MosierDE, OverbaughJ (2007) HIV-1 subtype A envelope variants from early in infection have variable sensitivity to neutralization and to inhibitors of viral entry. AIDS 21 : 693–702.
52. Cheng-MayerC, LevyJA (1988) Distinct biological and serological properties of human immunodeficiency viruses from the brain. Ann Neurol 23 Suppl: S58–61.
53. DerbyNR, KraftZ, KanE, CrooksET, BarnettSW, et al. (2006) Antibody responses elicited in macaques immunized with human immunodeficiency virus type 1 (HIV-1) SF162-derived gp140 envelope immunogens: comparison with those elicited during homologous simian/human immunodeficiency virus SHIVSF162P4 and heterologous HIV-1 infection. J Virol 80 : 8745–8762.
54. DavenportTM, FriendD, EllingsonK, XuH, CaldwellZ, et al. (2011) Binding interactions between soluble HIV envelope glycoproteins and quaternary-structure-specific MAbs PG9 and PG16. J Virol 85 : 7095–7107.
55. CorreiaBE, BanYE, FriendDJ, EllingsonK, XuH, et al. (2011) Computational protein design using flexible backbone remodeling and resurfacing: case studies in structure-based antigen design. J Mol Biol 405 : 284–297.
56. LawrenceM, ColmanPM (1993) Shape complementarity at protein/protein interfaces. J Mol Biol 234 : 946–950.
57. BurtonDR (2010) Scaffolding to build a rational vaccine design strategy. Proc Natl Acad Sci U S A 107 : 17859–17860.
58. GravesSS, StoneDM, LoretzC, PetersonLJ, LesnikovaM, et al. (2011) Antagonistic and agonistic anti-canine CD28 monoclonal antibodies: tools for allogeneic transplantation. Transplantation 91 : 833–840.
59. GornyMK, ConleyAJ, KarwowskaS, BuchbinderA, XuJY, et al. (1992) Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J Virol 66 : 7538–7542.
60. RobenP, MooreJP, ThaliM, SodroskiJ, BarbasCF3rd, et al. (1994) Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J Virol 68 : 4821–4828.
61. KleinF, DiskinR, ScheidJF, GaeblerC, MouquetH, et al. (2013) Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153 : 126–138.
62. LavoieTB, MohanS, LipschultzCA, GrivelJC, LiY, et al. (1999) Structural differences among monoclonal antibodies with distinct fine specificities and kinetic properties. Mol Immunol 36 : 1189–1205.
63. Souto-CarneiroMM, LongoNS, RussDE, SunHW, LipskyPE (2004) Characterization of the human Ig heavy chain antigen binding complementarity determining region 3 using a newly developed software algorithm, JOINSOLVER. J Immunol 172 : 6790–6802.
64. BrochetX, LefrancMP, GiudicelliV (2008) IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36: W503–508.
65. GiudicelliV, ChaumeD, LefrancMP (2004) IMGT/V-QUEST, an integrated software program for immunoglobulin and T cell receptor V-J and V-D-J rearrangement analysis. Nucleic Acids Res 32: W435–440.
66. WangX, WuD, ZhengS, SunJ, TaoL, et al. (2008) Ab-origin: an enhanced tool to identify the sourcing gene segments in germline for rearranged antibodies. BMC Bioinformatics 9 Suppl 12: S20.
67. VolpeJM, CowellLG, KeplerTB (2006) SoDA: implementation of a 3D alignment algorithm for inference of antigen receptor recombinations. Bioinformatics 22 : 438–444.
68. GaetaBA, MalmingHR, JacksonKJ, BainME, WilsonP, et al. (2007) iHMMune-align: hidden Markov model-based alignment and identification of germline genes in rearranged immunoglobulin gene sequences. Bioinformatics 23 : 1580–1587.
69. MyszkaDG (1999) Improving biosensor analysis. J Mol Recognit 12 : 279–284.
70. Otwinowski Z, Minor W (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Jr CWC, Sweet RM, editors. Meth Enzymol. NY: Academic Press. pp. 307–326.
71. PflugrathJW (1999) The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr 55 : 1718–1725.
72. McCoyAJ, Grosse-KunstleveRW, AdamsPD, WinnMD, StoroniLC, et al. (2007) Phaser crystallographic software. Journal of Applied Crystallography 40 : 658–674.
73. Collaborative Computational Project N (1994) The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst D50 : 760–763.
74. PottertonE, BriggsP, TurkenburgM, DodsonE (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr 59 : 1131–1137.
75. EmsleyP, CowtanK (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 : 2126–2132.
76. MurshudovGN, VaginAA, DodsonEJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53 : 240–255.
77. LaskowskiRA, MacArthurMW, MossDS, ThorntonJM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography 26 : 283–291.
78. DavisIW, Leaver-FayA, ChenVB, BlockJN, KapralGJ, et al. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35: W375–383.
79. BermanHM, WestbrookJ, FengZ, GillilandG, BhatTN, et al. (2000) The Protein Data Bank. Nucleic Acids Research 28 : 235–242.
80. PettitFK, BareE, TsaiA, BowieJU (2007) HotPatch: a statistical approach to finding biologically relevant features on protein surfaces. J Mol Biol 369 : 863–879.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- The Secreted Peptide PIP1 Amplifies Immunity through Receptor-Like Kinase 7
- Symbionts Commonly Provide Broad Spectrum Resistance to Viruses in Insects: A Comparative Analysis of Strains
- MIF Contributes to Associated Immunopathogenicity Development
- The Ins and Outs of Rust Haustoria
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy