
Evidence for Induction of Integron-Based Antibiotic Resistance by the SOS Response in a Clinical Setting
Bacterial resistance to β-lactams may rely on acquired β-lactamases encoded by class 1 integron-borne genes. Rearrangement of integron cassette arrays is mediated by the integrase IntI1. It has been previously established that integrase expression can be activated by the SOS response in vitro, leading to speculation that this is an important clinical mechanism of acquiring resistance. Here we report the first in vivo evidence of the impact of SOS response activated by the antibiotic treatment given to a patient and its output in terms of resistance development. We identified a new mechanism of modulation of antibiotic resistance in integrons, based on the insertion of a genetic element, the gcuF1 cassette, upstream of the integron-borne cassette blaOXA-28 encoding an extended spectrum β-lactamase. This insertion creates the fused protein GCUF1-OXA-28 and modulates the transcription, the translation, and the secretion of the β-lactamase in a Pseudomonas aeruginosa isolate (S-Pae) susceptible to the third generation cephalosporin ceftazidime. We found that the metronidazole, not an anti-pseudomonal antibiotic given to the first patient infected with S-Pae, triggered the SOS response that subsequently activated the integrase IntI1 expression. This resulted in the rearrangement of the integron gene cassette array, through excision of the gcuF1 cassette, and the full expression the β-lactamase in an isolate (R-Pae) highly resistant to ceftazidime, which further spread to other patients within our hospital. Our results demonstrate that in human hosts, the antibiotic-induced SOS response in pathogens could play a pivotal role in adaptation process of the bacteria.
Published in the journal:
. PLoS Pathog 8(6): e32767. doi:10.1371/journal.ppat.1002778
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002778
Summary
Bacterial resistance to β-lactams may rely on acquired β-lactamases encoded by class 1 integron-borne genes. Rearrangement of integron cassette arrays is mediated by the integrase IntI1. It has been previously established that integrase expression can be activated by the SOS response in vitro, leading to speculation that this is an important clinical mechanism of acquiring resistance. Here we report the first in vivo evidence of the impact of SOS response activated by the antibiotic treatment given to a patient and its output in terms of resistance development. We identified a new mechanism of modulation of antibiotic resistance in integrons, based on the insertion of a genetic element, the gcuF1 cassette, upstream of the integron-borne cassette blaOXA-28 encoding an extended spectrum β-lactamase. This insertion creates the fused protein GCUF1-OXA-28 and modulates the transcription, the translation, and the secretion of the β-lactamase in a Pseudomonas aeruginosa isolate (S-Pae) susceptible to the third generation cephalosporin ceftazidime. We found that the metronidazole, not an anti-pseudomonal antibiotic given to the first patient infected with S-Pae, triggered the SOS response that subsequently activated the integrase IntI1 expression. This resulted in the rearrangement of the integron gene cassette array, through excision of the gcuF1 cassette, and the full expression the β-lactamase in an isolate (R-Pae) highly resistant to ceftazidime, which further spread to other patients within our hospital. Our results demonstrate that in human hosts, the antibiotic-induced SOS response in pathogens could play a pivotal role in adaptation process of the bacteria.
Introduction
Transferable genes encoding antibiotic resistance to major antibiotics (e.g. β-lactams, aminoglycosides) are often carried by class 1 integrons in Gram negative pathogens [1]. In these genetic elements the antibiotic resistance genes are carried inside mobile structures called gene cassettes, which generally correspond to a promoterless gene associated to a recombination site called attC, formerly called 59-be [2], [3]. Gene cassette expression is driven by a promoter located in the integron platform upstream of the attI site, the primary site of cassette integration, and in the case of the class 1 integron, inside the intI1 gene which encodes the cassette recombinase [4]. This organization allows a positional regulation of the cassette's expression: the closer a gene cassette is located to attI, the higher is its expression [1]. In addition to this transcriptional attenuation along the cassette array, the decrease in expression can be due to problems of translational coupling [5], [6]. Thus, gene expression in these elements can be modulated by the site-specific recombination events mediated by the integrase IntI1 [1], [3].
The SOS response is a conserved regulatory network that is induced in response to DNA damage [7]. It also promotes integron rearrangements by controlling the expression of integrases with promoters that contain a LexA-binding motif [8], [9]. During the SOS response, the RecA protein, bound to single stranded DNA, stimulates the cleavage of the repressor LexA, thus releasing the transcription of the LexA-controlled genes. The adaptations resulting from integron activity are thought to influence bacterial evolution, especially in Proteobacteria, where integrons are extremely common [3]. We have recently shown that common horizontal gene transfer processes, such as conjugation [10], trigger the SOS response and ultimately the integron integrase expression. However, the most medically relevant SOS induction is certainly the one directed by antibiotic treatments. Hence, a number of antibiotics, including the β-lactams, aminoglycosides and fluoroquinolones, have been found to directly or indirectly provoke this stress response [11]–[13]. Despite this evidence from in vitro studies, the clinical significance of the SOS response on integron rearrangement and the dynamics of integron-based bacterial adaptation during human infections are unknown. So far, there are no examples of SOS-mediated antibiotic resistance occurring during therapeutic use of antibiotics, even though, as mentioned above, many of them stimulate the SOS response.
Here, we witnessed the emergence in a hospitalized patient of an isolate of Pseudomonas aeruginosa highly resistant to the third generation cephalosporin ceftazidime, associated with the production of an extended-spectrum β-lactamase encoded by a class 1 integron-borne gene. This strain, highly resistant to ceftazidime, further became epidemic within the hospital. We discovered the mechanism, based on the excision of a gene cassette originally located upstream of the β-lactamase-encoding gene cassette, which modulated the expression of the transferable resistance gene. This patient had been previously treated with ceftazidime (to treat the infection by P. aeruginosa) and metronidazole (to treat an infection by anaerobes). This led us to suspect the involvement of this treatment in the integron cassette array remodeling, through SOS response induction inside the patient. We demonstrated that the metronidazole, not an anti-pseudomonal antibiotic, is able to trigger the SOS response in P. aeruginosa, and subsequently activates the integrase IntI1 and cassette rearrangement. Deletion of the gcuF1 cassette and full expression the β-lactamase were obtained at high rates in vitro, supporting this scenario to explain the genesis, in the patient, of the R-Pae isolate from S-Pae after metronidazole treatment.
Results
Enzymatic resistance mechanisms to β-lactams in an epidemic multidrug resistant P. aeruginosa
We detected the same clone of a multi-drug resistant clone of P. aeruginosa (R-Pae) in 13 adult patients by pulsed-field gel electrophoresis (Figure S1). R-Pae was resistant to potent anti-pseudomonal agents, including ceftazidime, cefepime, aztreonam, aminoglycosides, and fluoroquinolones (Table S1). A double-disk synergy test revealed a weak synergy between β-lactamase substrates (ceftazidime or cefepime) and β-lactamase inhibitors (imipenem or clavulanate) in these bacteria, suggesting production of an extended-spectrum β-lactamase (oxacillinase) of Ambler class D (Figure S2) [14]. We searched for the most common genes encoding extended-spectrum oxacillinases in P. aeruginosa [15] by PCR for the blaOXA-10, blaOXA-2 and blaOXA-1 groups [16]. Only the PCR specific to blaOXA-10 using primers OXA-10A and B (see Table S2) was positive. As blaOXA-10 and variants are found as gene cassettes often borne by class 1 integrons [17], we performed PCR experiments using primers specific to these integron platforms, directed to the conserved sequences flanking the variable cassette array, usually called 5′-CS and 3′-CS (see Table S2). A single 1673-bp amplicon was thus obtained, which was subsequently sequenced to reveal two resistance gene cassettes, namely aacA4 that determines a 6′-N-aminoglycoside acetyltransferase conferring high resistance to gentamicin and tobramycin [18], and blaOXA-28 that encodes the extended-spectrum oxacillinase OXA-28 [19] (Figure 1A). Additionally, quantification of the specific mRNA transcripts by RT-qPCR showed that the R-Pae1 isolate overexpressed the ampC gene encoding the intrinsic chromosomal cephalosporinase AmpC, when compared to the wild type reference strain of P. aeruginosa PAO1 (Figure 2A).
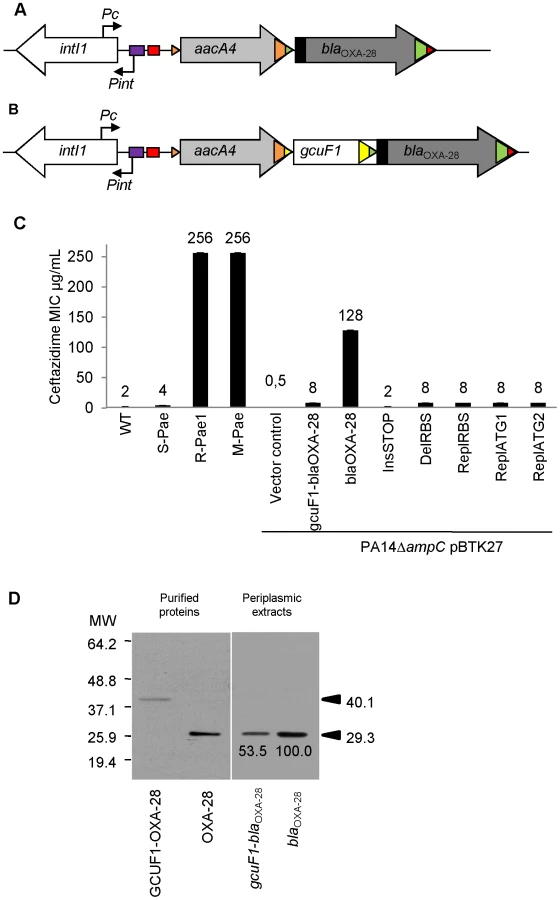

To determine whether bacterial resistance had emerged during the course of treatment, we retroactively analyzed the P. aeruginosa isolates archived from the first colonized patient (patient 1) within 2 months before the isolation of R-Pae. The clonal strain S-Pae was isolated from the sputum of patient 1, 28 days before the isolation of R-Pae in the patient's lung. R-Pae1 was isolated after a treatment with ceftazidime (to treat the infection by P. aeruginosa) and metronidazole (to treat an infection by anaerobes). To our surprise, S-Pae was susceptible to ceftazidime (MIC, 4 µg/ml) despite the presence of an intact blaOXA-28 gene in its genome (Figure 1B, C). S-Pae and R-Pae1 demonstrated an equivalent expression level of the cephalosporinase-encoding ampC (Figure 2A). The other resistance mechanisms found in S-Pae and R-Pae1 (efflux pump overproduction and porin loss) do not alter the susceptibility to ceftazidime (see Text S1). Because of the lack of evidence for classical resistance mechanisms accounting for the difference in β-lactam susceptibilities in these isolates, we investigated the possible mechanism of resistance modulation.
OXA-28 oxacillinase production in R-Pae1 and S-Pae
S-Pae differed from R-Pae1 by a 10-fold lower amount of blaOXA-28 transcripts (Figure 2B) and by the presence of a 319-bp cassette, gcuF1, inserted immediately upstream of blaOXA-28 (Figure 1B). Using nested-PCR, we demonstrated the presence of free circular cassettes of gcuF1 in S-Pae (Figure 3A, B), demonstrating that the recombination between its own attC site and the aacA4 attC site was occurring, though at extremely low level [2]. Computational analysis of the nucleotide sequence of the S-Pae integron predicted the translation of a new ORF, a fused protein consisting of gcuF1 and blaOXA-28 (GCUF1-OXA-28). We were able to show that these two cassettes (gcuF1 and blaOXA-28) could be transcribed in a single transcript. Hence, we could retrieve a specific amplicon after PCR amplification using cDNA prepared from S-Pae RNA as the matrix and with primers overlapping the junction between gcuF1 and blaOXA-28 (Figure S3). The GCUF1-OXA-28 peptide (368 residues) was predicted to have a molecular weight of 40.1 kDa, compared with the 29.3-kDa native OXA-28 (266 residues). To confirm these data, both ORFs were expressed in Escherichia coli BL21 from plasmid pET-28a which adds an N-terminal polyHis tag. After purification, we found that their molecular weights estimated by SDS-PAGE were in full agreement with our predictions (Figure 1D). This protein contained the original 19-residue long signal peptide now misplaced between the GCUF1 and the OXA-28 domains at position 103–121 of the GCUF1-OXA-28 protein (Figure 1B and 4A). Since β-lactamases are periplasmic proteins and are produced as preproteins with an N-terminal peptide signal [20], one would expect that the misplacement of the signal peptide in the GCUF1-OXA-28 protein will abolish the periplasmic process of the β-lactamase. However, cellular production of this altered protein conferred a residual resistance to ceftazidime (MIC of ceftazidime, 8 µg/ml; gcuF1-blaOXA-28 in Figure 1C), suggesting the presence of an active and processed OXA-28 in the periplasm of the GCUF1-OXA28-producing isolate. To clarify this point, we cloned the blaOXA-28 and gcuF1-blaOXA-28 sequences into the broad host range vector pBTK27 to encode C-terminal His-tagged polypeptides that were expressed in the reference strain P. aeruginosa PA14ΔampC. Western-blot analysis of periplasmic extracts of GCUF1-OXA28-producing bacteria revealed the presence of a reduced amount of processed periplasmic OXA-28 (Figure 1D), consistent with the lower resistance to ceftazidime when the gcuF1 cassette is inserted upstream of the blaOXA-28 (Figure 1C). We used a directed mutagenesis approach to clarify the origin of periplasmic OXA-28 in S-Pae and determine whether it is due to the export processing of the fusion protein or to an internal translational initiation at the original OXA-28 start codon (Figure 4A). In P. aeruginosa PA14ΔampC carrying a plasmid-borne gcuF1-blaOXA-28, the in-frame insertion of a stop codon just upstream of blaOXA-28 reduced the resistance level to ceftazidime down to 2 µg/ml. We also tested the effect of the in frame deletion of the blaOXA-28 ribosome binding site (GAAGGT), or its substitution by a sequence with no ribosome binding properties (CTCTCT). Finally, we tested the substitution of the ATG start codon with either a GTC or a GTG valine codons, which have no or weak translation initiation power [21]. None of these mutations led to a change in resistance level (Figure 1C). Hence, we confirmed that detected OXA-28 came entirely from the processing of the ORF2-OXA28 fusion protein, and that the inefficiency of its maturation was in part responsible for the low resistance level to ceftazidime. Additionally, the putative ribosome binding site of gcuF1 (TTAGG) is predicted to have a poor translation initiation efficiency [22], [23] (Figure 4A), likely leading to reduced translation of gcuF1-blaOXA-28.


SOS response induction by metronidazole treatment
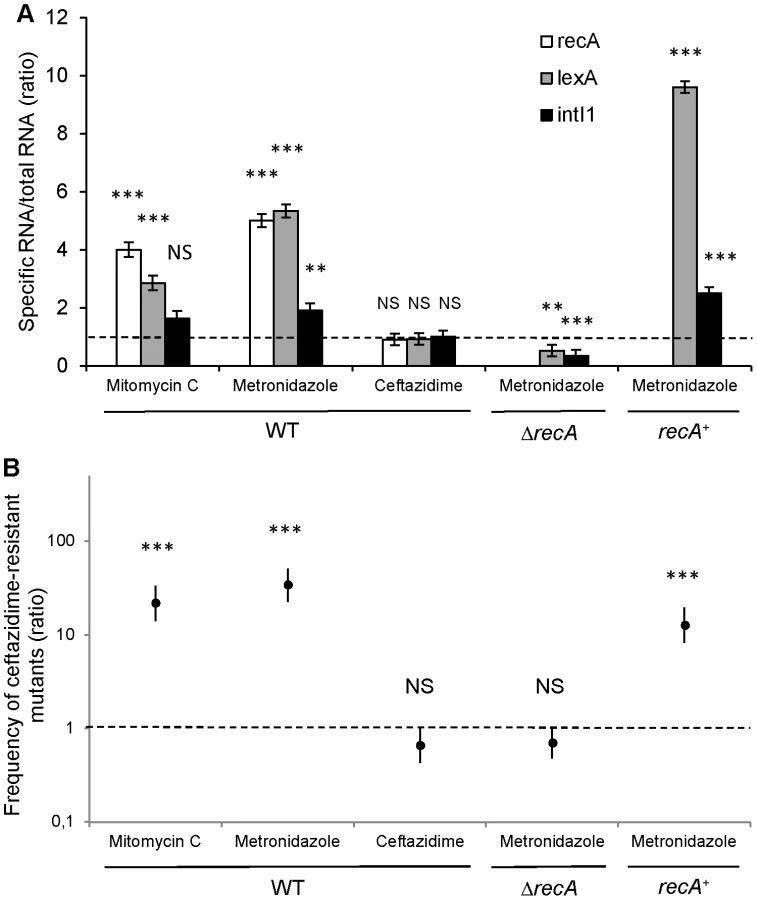
In patient 1, the transition from S-Pae to R-Pae1 was observed after treatment with two antibiotics, ceftazidime and metronidazole, both known to activate the bacterial SOS response [12], [24]. Cassette expression in class 1 integrons can be controlled by two promoters, Pc and P2, which can exist under various forms [4]. P2 is created by the insertion of three guanines between the potential −35 and −10 regions, but this insertion also disrupts the LexA binding box (also called the SOS box) of the intI1 promoter and abrogates the SOS control of intI1 expression [4]. Analysis of the S-Pae class 1 integron revealed a functional LexA-binding box overlapping the −10 box of the intI1 promoter [25], thus the P2 promoter is absent and the cassettes' expression only relies on the strong Pc promoter variant PcS (Figure 4B). It has been previously found that the encoded integrase IntI1 (IntI1R32_N39) displayed the second highest excision activity of the four known existing variants [4]. In agreement with this, using nested-PCR, we confirmed the presence of free circular gcuF1 cassettes in S-Pae (Figure 3A, B), occurring through recombination between its own attCgcuF1 site and the aacA4 attCaacA4 site, as observed previously for a few other cassettes [2]. LexA is the transcriptional repressor that binds the SOS box sequences to silence transcription. RecA, once activated by the presence of abnormal single strand DNA produced by a variety of stimuli that includes antibiotic exposure, induces the LexA autoproteolysis and releases the transcriptional silencing driven by LexA binding to SOS boxes. We hypothesized that the excision of the gcuF1 cassette by the integrase IntI1 and subsequent emergence of full resistance to ceftazidime in R-Pae1 was a result of the SOS response induced by antibiotic therapy in the patient.
We quantified the expression of SOS pathway genes recA and lexA, as well as the integrase encoding gene intI1 by RT-qPCR after in vitro induction with metronidazole and ceftazidime. Our results indicated that in vitro exposure of S-PaeΔampC to the minimal inhibitory concentration of ceftazidime (2 µg/ml) neither triggered the SOS response (recA and lexA were not induced), nor enhanced the expression of the intI1 gene (Figure 5A). We measured the frequency of ceftazidime-resistant mutant emergence by gcuF1 cassette excision in the same experimental conditions. Consistent with the integrase expression data, we found that the gcuF1 cassette excision frequency remained basal after exposure to ceftazidime (Figure 5B). On the contrary, in vitro exposure of S-PaeΔampC to therapeutic concentrations of metronidazole (50 µg/ml for recA wild-type strain and 25 µg/ml for recA mutants) triggered the SOS response, as indicated by the increased recA and lexA expression, and the increase in intI1 expression, gcuF1 cassette excision and a subsequent 34-fold enhancement of the frequency of emergence of ceftazidime-resistant mutants (Figure 5A, B). As shown with the results obtained in recA-deleted and recA-complemented strains, the effect of metronidazole on the integron rearrangement fully depended on the presence of recA, confirming the role of the SOS induction for the cassette rearrangement (Figure 5A, B). In mutants isolated both in patient 1 (R-Pae1) and in vitro (M-Pae), the gcuF1 cassette excision provoked full blaOXA-28 expression and a massive increase in resistance to ceftazidime (Figure 1C).
Discussion
In this study, we identified a new mechanism of modulation of antibiotic resistance in integrons. The positional regulation of gene cassette expression was already documented, but this was considered so far as only relying on the transcription attenuation process and on the lack of transcriptional coupling between genes carried in consecutive cassettes [6]. What we describe here is the presence of a genetic element, the gcuF1 cassette, upstream of the integron-borne β-lactamase cassette blaOXA-28 which modulates the transcription, translation, and secretion of this enzyme, all at once. The poor ribosome-binding site found upstream of gcuF1 (Figure 4A) [23] is likely responsible for the low production of the fusion protein detected, but is also likely responsible for the low level of blaOXA-28 mRNA. Indeed, it has been shown that a reduced ribosome binding to RBS can destabilize mRNA, which then becomes more vulnerable to endonucleolytic attack [26]. GcuF1 shares 78% identity with integron-borne orfD gene cassettes of unknown function that are frequently found in clinical strains of Pseudomonas sp. and Enterobacteriaceae [1]. The insertion of gcuF1 generates a fusion protein GCUF1-OXA-28 with a misplaced signal peptide between the GCUF1 and the OXA-28 domains. However, the GCUF1-OXA-28-producing bacteria still demonstrated residual resistance to ceftazidime, consistent with the presence of small amount of the processed OXA-28 in its periplasm. Using various mutants constructed in this aim, we established that the OXA-28 produced from gcuF1-blaOXA-28 was exclusively derived from cleavage at position 121 of the fusion protein GCUF1-OXA-28 (Figure 4A). These data confirm that a protein with a misplaced cleavable leader sequence (i.e. outside the N-terminus) can be exported, although less efficiently, into the periplasm [27].
We showed that the gcuF1 cassette can be excised by the IntI1 integrase, leading to the production of a circularized cassette. The gcuF1 cassette carries an attC site with an unusual R″ box, with a T instead of a C in last position, as in the large majority of integron cassettes (Figure 4A). The gcuF1 closest relative is a cassette found in the Acidovorax sp. JS42 genome (GenBank accession number CP000539), which is 87% identical over the whole cassette sequence, but shows a CC at this precise position. Thus the substitution of this dinucleotide by a single T explains why the spacer between the R″ and L″ boxes is reduced to 4 nucleotides, instead of the usual 5 nucleotides in gcuF1 (Figure 4A). The last base of R″ is normally pairing with the first base of the R′ box in the single strand recombinogenic form of the attC site [28]. Frumerie and colleagues tested all possible base pairs (C/G, A/T, G/C and T/A) at this position in the predicted annealed R″/R′, and found that all deeply decreased the recombination rate, by more than a hundred fold factor [29]. However the effect of single base substitutions at these positions is so far unknown, the observations made in our study suggest that substitution of the conserved C in R″ by a T does not abolish the attC recognition and recombination, but the effect of this mutation, as well as the one brought by the R″/L″ spacer reduction, on the rate of recombination needs to be established.
We found that excision of the gcuF1 cassette from the original cassette array leads to increased resistance to ceftazidime (Figure 1C). As the expression of IntI1 is controlled by the SOS response, we surmised the antibiotic treatment given in first instance to this patient (ceftazidime and metronidazole) to be responsible for the SOS induction episode that ultimately led to the IntI1-mediated gcuF1 deletion. We found that, in contrast to ceftazidime, in vitro exposure to therapeutic concentrations of metronidazole, an antimicrobial against which P. aeruginosa is naturally resistant, greatly enhanced the frequency of emergence of ceftazidime-resistant mutants. This phenotype is dependent on the excision of the gcuF1 cassette that is fully dependent on the SOS response, as attested by the lack of excision in a recA mutant (Figure 5B). We speculate that in patient 1, metronidazole likely promoted the SOS-dependent transition from S-Pae to R-Pae1, which was further selected by ceftazidime therapy.
Interestingly, Cipriano Souza et al. showed that previous consumption of metronidazole was an independent risk factor for acquisition of multi-drug resistant P. aeruginosa by hospitalized patients [30]. Metronidazole and related 5-nitroimidazoles are redox-active prodrugs. Metronidazole is widely used to treat anaerobic bacteria infections, (e.g. Clostridium difficile), protozoa, and the microaerophilic Helicobacter pylori [31]. Bacterial nitroreductases, such as RdxA in H. pylori, catalyze the conversion of metronidazole to mutagenic products that directly interact with DNA bases [32], [33]. This causes DNA helix destabilization and single - and double-strand DNA breakage [34] that activate the SOS response [7], [24]. The effect of metronidazole in P. aeruginosa, in terms of DNA damage has still to be established, but one can speculate that the RdxA homolog in P. aeruginosa (PA5190 in the PAO1 genome, http://www.pseudomonas.com) could play a similar role in the metabolism of the metronidazole, and explain how this antibiotic triggers the SOS induction.
Our data suggest that SOS induction by antibiotics can result in the development of integron-based resistance in vivo. SOS also enhances the rate of mutations [7]. This is of particular concern in P. aeruginosa in which multidrug resistance mainly arise from chromosomal mutations [35]. More generally, it may lead to undesired changes in the behavior of bacteria and their faster adaptation to hostile environments. This is alarming because apart from metronidazole, other major classes of antibiotics (e.g. β-lactams, aminoglycosides, trimethoprim and fluoroquinolones) can trigger the bacterial SOS response [11]–[13].
The expression of horizontally acquired antibiotic resistance mechanisms is tightly regulated; this may reduce the biological cost associated with resistance expression and account for the dissemination of susceptible strains carrying hidden resistance determinants [36], [37]. Here, in S-Pae, expression of antibiotic resistance is silenced until antibiotic exposure triggers expression. This could represent an efficient evolutionary pathway for resistance determinants to be “switchable” and render bacteria fitness-neutral in the absence of antibiotic selection pressure [37]. Current policies for controlling the spread antibiotic resistance often rely on the detection of resistant bacteria, and on the assumption that resistance has a functional cost [38]. Future antibiotic restriction guidelines should consider the fact that resistance genes can spread latently in susceptible isolates with low biological cost.
In summary, we describe a reversible mechanism modulating an acquired antibiotic resistance in bacteria. The metronidazole-induced SOS response favored the emergence in a patient of bacteria highly resistant to ceftazidime that could then spread to twelve other patients which were under antibiotic pressure.
The suppression of the SOS response activation has been reported to enhance killing by antibiotics of E. coli and to increase survival of infected mice [39], [40]. Efforts have been made to identify small molecules and short peptides that inhibit RecA activity, although the absence of potential adverse effects on Rad51 (the human RecA homologue) needs to be demonstrated [41]–[43]. Our results suggest an adaptive role for the antibiotic-induced SOS response in bacterial genome rearrangement in vivo within humans. Altogether, this supports the hypothesis that inhibition of RecA is a plausible therapeutic adjuvant in combined therapy to reduce the capacity to generate antibiotic-resistant mutants.
Materials and Methods
Bacterial isolates
We identified a multidrug-resistant P. aeruginosa strain (R-Pae) in 13 patients hospitalized in the hematological ward of the University Hospital of Besançon (France) from March 2004 (Patient 1) to December 2009 (Patient 13). The genetic similarity of P. aeruginosa clinical isolates was investigated by pulsed field gel electrophoresis (PFGE; CHEF-DR III; Bio-Rad, Hercules, California) with the use of DraI enzyme, as described elsewhere [44]. We retroactively analyzed the bacterial isolates of patient 1's early specimens. Twenty-eight days before pulmonary infection with R-Pae1, this patient was colonized with S-Pae, a clonally-related isolate that was more susceptible to β-lactams than R-Pae. Early and late sputums only contented S-Pae and R-Pae, respectively. In review of the patient record, patient 1 was treated with ceftazidime (4 g/day for 8 days) for P. aeruginosa and also with metronidazole (500 mg/day for 7 days) for infections by anaerobes prior to the isolation of R-Pae1. Oligonucleotides, bacterial strains and plasmids used for this study are detailed in the Tables S2, S3 and S4, respectively.
Determination of the resistance level to antibiotics
The minimal inhibitory concentrations (MICs) of selected antibiotics were determined by the conventional Mueller-Hinton agar (MHA) dilution method, and interpreted according to CLSI (Clinical and Laboratory Standards Institute) guidelines [45]. The wild-type reference strain of P. aeruginosa PA14 was used as a control in susceptibility testing. MHA was supplemented with 1 mM of IPTG for strains carrying pBTK27 derivatives (Table S4).
Real time quantitative RT-PCR (RT-qPCR)
Total RNA was isolated from cultures at an absorbance at 600 nm of 1.0 (or otherwise stated) using the Qiagen RNeasy protocol (Qiagen, Valencia, California). The RNA samples were treated with DNase (Turbo DNAse; Ambion, Austin, Texas) and further cleaned according to the manufacturer's protocol. Total RNA was quantified using the RiboGreen RNA Quantitation Kit (Molecular Probes, Carlsbad, California). Total RNA was reverse transcribed with Superscript III reverse transcriptase (Invitrogen, Carlsbad, California) as specified by the supplier. Quantitative PCR was performed on an Mx4000 Multiplex QPCR System (Stratagene, Santa Clara, California) using samples in triplicate with 25 ng of total RNA in a 20 µl reaction using SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, California) and specific primers for housekeeping gene rpsL, blaOXA-28, ampC, recA, lexA, and intI1 (Table S2). PCR cycling conditions consisted of 95°C for 10 min, and 40 cycles of 95°C for 15 s, 60°C for 1 min. After each assay, a dissociation curve was run to confirm specificity of all PCR amplicons. The mRNA levels of ampC and blaOXA-28 were normalized to that of reference gene rpsL [46] and expressed as a ratio to the levels in the isolate PA14 (for ampC) or R-Pae1 (for blaOXA-28) in which the values were set at 1.00. For recA, lexA, and intI1 genes, resulting Ct values were converted to nanograms, normalized to total RNA and expressed as the average of triplicate samples.
Evidence of circular forms of gcuF1 cassette
We assessed the presence in S-Pae isolate of free circular forms of the gcuF1 cassette. Total DNA from isolates R-Pae1 (without gcuF1, taken as a control) and S-Pae (with gcuF1) were PCR amplified with primers circ1 and circ2 (Figure 3A, Table S2). The purified PCR products were used as templates for a second nested PCR with primers circ3 and circ4. PCR products were visualized on an agarose gel. The PCR fragment obtained from S-Pae DNA was further sequenced to verify its specificity.
Evidence of gcuF1-blaOXA-28 transcripts
To determine whether the DNA element gcuF1-blaOXA-28 could transcribe a functional transcript, we carried out RT-PCR reactions by using PCR primers overlapping the gcuF1-blaOXA-28 junction (overlap1 and overlap2, Table S1), and cDNA prepared from S-Pae RNA as the matrix (see above). The nucleotide sequence of the amplicon was determined to check for the specificity of the reaction.
Deletion mutant construction
For the deletion of ampC from PA14 and S-Pae, approximately 1000-bp specific fragments upstream (with primers AmpCdel1F/AmpCdel1R) and downstream (with primers AmpCdel2F/AmpCdel2R) of ampC were PCR amplified from PA14 and S-Pae total DNAs, and used in an overlap extension reaction to create a single 2,000-bp product (Table S1). These products were cloned into Gateway-compatible gene replacement vector pEX18AmpGW [47], yielding the plasmids pDelAmpC-PA14 and pDelAmpC-SPae (Table S3), which were then transformed into E. coli DH5α.
For recA inactivation in S-PaeΔampC, 5′ (ca. 450-bp) and 3′ (ca. 530-bp) portions of recA were amplified separately with primer pairs RecAdel1F/RecAdel1R and RecAdel2F/RecAdel2R, respectively. The tetA gene was amplified from plasmid mini-CTX1 with primers TetF and TetR (Table S1). These three fragments were cloned simultaneously in a 4 way ligation in the EcoRI/HindIII sites of pEX18ap to yield plasmid pDelRecA (Table S3).
The plasmids for ampC or recA deletion were transferred into the recipient strains (PA14 or S-PaeΔampC) by triparental mating that included the donor strain E. coli DH5α with strain E. coli HB101 (containing helper plasmid pRK2013), followed by selection with irgasan (25 µg/ml) and carbenicillin (150 µg/ml for PA14, 500 µg/ml for S-PaeΔampC) and screening for P aeruginosa transconjugants with the deletion as previously described [48]. Deletion of the ampC and inactivation of recA were verified by PCR and sequencing.
Construction of plasmids expressing blaOXA-28 and derivatives
The resistance level to ceftazidime conferred by the production of OXA-28, GCUF1-OXA-28, and their derivatives was assessed by cloning gcuF1-blaOXA-28 (PCRed from S-Pae with primers 1 and 3) and blaOXA-28 (PCRed from R-Pae1 with primers 2 and 3) sequences into the broad host range vector pBTK27. This yielded plasmids pBTK/gcuF1-oxa28 and pBTK/oxa28, respectively, encoding C-terminal polypeptides that were expressed in the reference strain P. aeruginosa PA14ΔampC (Table S3). We used the plasmid pBTK/gcuF1-oxa28 as template for various mutageneses with a QuikChange kit (Stratagene). We inserted a TGA stop codon downstream gcuF1 with mutagenic primers stop-F and stop-R, yielding plasmid pInsSTOP. We deleted in frame the GAAGGT sequence including the natural blaOXA-28 ribosome binding site (GAAGG) with mutagenic primers delRBS-F and delRBS-R, yielding plasmid pDelRBS, which encodes this GCUF1-OXA-28 variant missing amino acids E100 and G101. We also substituted the sequence harboring the blaOXA-28 ribosome binding site with a sequence with no translation initiation power (GAAGGT by CTCTCT) using mutagenic primers replRBS-F and replRBS-R, yielding plasmid pReplRBS. The ATG start codon from blaOXA-28 was substituted by GTC or by GTG with mutagenic primers (RepATG1-F/RepATG1-R and RepATG2-F/RepATG2-R, respectively) yielding plasmid pReplATG1 and pReplATG2, respectively, which encodes the M103V GCUF1-OXA-28 variant (Tables S2 and S4). All pBTK27-derivated plasmids were introduced into the reference strain P. aeruginosa PA14ΔampC by triparental mating (see above) to assess the resistance to ceftazidime.
OXA-28 and GCUF1-OXA-28 purification
To determine the size of the encoded proteins, blaOXA-28 (PCRed from R-Pae1 with primers 5 and 6) and gcuF1-blaOXA-28 sequences (PCRed from S-Pae with primers 4 and 6) were cloned into the pET-28a vector (Kmr; Novagen-Merck, Darmstadt, Germany) at NheI/XhoI, yielding plasmids pET/oxa28 and pET/gcuF1-oxa28, respectively, encoding N-terminal His-tagged polypeptides. The cloned gene products were expressed in E. coli BL21(DE3) by IPTG induction (0.2 mM) to the exponentially growing cells (A600 of 0.8) and left overnight at 20°C with shaking. Bacteria were harvested and lysed using standard protocols. The lysates were applied on a 5 ml Ni-NTA column (Qiagen). His-tagged peptides were eluted with PBS supplemented with 250 mM imidazole. Eluted fractions were separated by 12% SDS-PAGE and transferred to nitrocellulose filters. Filters were hybridized with the His-detector Ni-HRP reagent (KPL) and the immune complexes were detected by the ECL-Plus chemiluminescent system (GE Healthcare, Buckinghamshire, United Kingdom).
OXA-28 and GCUF1-OXA-28 subcellular localization
To assess the presence of His-tagged OXA-28 or GCUF1-OXA-28 in the periplasm, the plasmids pBTK/oxa28 or pBTK/gcuF1-oxa28 in P. aeruginosa PA14ΔampC was induced by 1 mM IPTG for 4 h at 37°C. Periplasmic fractions were prepared by using Peripreps Periplasting kit (Epicentre Biotechnologies, Madison, Wisconsin) and analyzed by SDS-PAGE, transfer, and hybridization (see below). The raw integrated density of the blots was assessed using the ImageJ 1.44p software (National Institute of Health).
Construction of plasmid expressing recA
We cloned recA (PCRed from S-Pae with primers RecA1 and RecA2) sequence into the broad host range vector pBTK27, yielding plasmid pBTK/recA (Tables S2 and S4). Plasmid pBTK/recA was introduced into the strain S-PaeΔampCΔrecA by triparental mating (see above).
Frequency of emergence of ceftazidime-resistant mutants by gcuF1 cassette excision
S-PaeΔampC, S-PaeΔampCΔrecA, and S-PaeΔampCΔrecA carrying pBTK/recA or pBTK27 plasmids were used to determine the frequency of emergence of ceftazidime-resistant mutants by gcuF1 cassette excision. The gene ampC was deleted to avoid the emergence of resistant mutants overproducing this intrinsic β-lactamase. Bacteria were grown in LB broth (Luria-Bertani) overnight, then diluted 1∶250 and grown until OD600 = 0.3. Half of the cultures were then exposed to antibiotics (mitomycin C, 1 MIC for 1.5 h; ceftazidime, 1 MIC for 1 h; metronidazole, 1/40 MIC for 15 h). MICs of mitomycin C were 1.0/0.3 µg/ml, those of ceftazidime were 2/2 µg/ml and those of metronidazole were 2000/1000 µg/ml for S-Pae derivatives with or without the recA gene, respectively. Exposure to antibiotics in these conditions did not alter the growth rates of the bacteria. Metronidazole concentrations used for SOS response induction experiments (25 and 50 µg/ml) were in the range of those found in the plasma of treated patients [49]. Cultures were collected by centrifugation and washed twice with 0.9% NaCl. Appropriate dilutions in 0.9% NaCl were plated on MH plates with or without ceftazidime at 50 µg/ml. We checked the gcuF1 excision by PCR in 133 clones growing on ceftazidime-containing media, obtained after exposure to mitomycin C, metronidazole and ceftazidime. Only one of them (0.8%) still displayed the gcuF1 cassette. The gcuF1 excision rate under antibiotic stress was then estimated as the number of ceftazidime-resistant colonies divided by the number of plated cells. The result was expressed as the ratio of the excision rates with and without antibiotic (for wild-type and ΔrecA strains) and with or without recA (for recA-complemented strain). All assays were independently performed at least 3 times. All the samples were subjected to microscopic observation to ascertain that no filamented cells were present. We confirmed that the isolate S-Pae was not a hypermutator (see Text S1).
Ethics statement
Approval and written informed consent from all subjects or their legally authorized representatives were obtained before study initiation. The study was approved by the ethical committee ‘Comité d'Etude Clinique’ of the Besançon hospital, Besançon, France.
Statistical analysis
Student's t-tests were used to determine statistical significance for comparisons of gene expression (Figure 5A) and frequencies of emergence of ceftazidime-resistant mutants (by gcuF1 excision; Figure 5B) with and without antibiotic (for wild-type and ΔrecA strains) and with or without recA (for recA-complemented strain). Data were log transformed and variance estimates were pooled over similar experiments, resulting in pooled estimates of standard error of 0.24 with 65 degrees of freedom for the t-tests of Figure 2A and of 0.54 with 27 degrees of freedom for the t-tests of Figure 5B. Graphical examination supports the assumption of normality and homogeneous variation across experiments for the gene expression data Figure 5A and frequency the emergence of ceftazidime-resistant mutant data Figure 5B expressed on a log scale. The chosen significance threshold was 0.05 for all tests.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. PartridgeSRTsafnatGCoieraEIredellJR 2009 Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol Rev 33 757 784
2. CollisCMHallRM 1992 Gene cassettes from the insert region of integrons are excised as covalently closed circles. Mol Microbiol 6 2875 2885
3. CambrayGGueroutAMMazelD 2010 Integrons. Annu Rev Genet 44 141 166
4. JovéTDa RéSDenisFMazelDPloyMC 2010 Inverse correlation between promoter strength and excision activity in class 1 integrons. PLoS Genet 6 e1000793
5. CoyneSGuigonGCourvalinPPérichonB 2010 Screening and quantification of the expression of antibiotic resistance genes in Acinetobacter baumannii with a microarray. Antimicrob Agents Chemother 54 333 340
6. JacquierHZaouiCSanson-le PorsM-JMazelDBerçotB 2009 Translation regulation of integrons gene cassette expression by the attC sites. Mol Microbiol 72 1475 1486
7. ErillICampoySBarbeJ 2007 Aeons of distress: an evolutionary perspective on the bacterial SOS response. FEMS Microbiol Rev 31 637 656
8. GuerinECambrayGSanchez-AlberolaNCampoySErillI 2009 The SOS response controls integron recombination. Science 324 1034
9. CambrayGSanchez-AlberolaNCampoySGuerinEDa ReS 2011 Prevalence of SOS-mediated control of integron integrase expression as an adaptive trait of chromosomal and mobile integrons. Mobile DNA 2 6
10. BaharogluZBikardDMazelD 2010 Conjugative DNA transfer induces the bacterial SOS response and promotes antibiotic resistance development through integron activation. PLoS Genet 6 e1001165
11. BaharogluZMazelD 2011 Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: a route towards multiresistance. Antimicrob Agents Chemother 55 2438 2441
12. BlazquezJGomez-GomezJMOliverAJuanCKapurV 2006 PBP3 inhibition elicits adaptive responses in Pseudomonas aeruginosa. Mol Microbiol 62 84 99
13. MillerCThomsenLEGaggeroCMosseriRIngmerH 2004 SOS response induction by β-lactams and bacterial defense against antibiotic lethality. Science 305 1629 1631
14. HocquetDDehecqBBertrandXPlésiatP 2011 A strain-tailored double-disc synergy test detects extended-spectrum oxacillinases in Pseudomonas aeruginosa. J Clin Microbiol 49 2262 2265
15. HocquetDPlésiatPDehecqBMariottePTalonD 2010 Nationwide investigation of extended-spectrum β-lactamases, metallo-β-lactamases, and extended-spectrum oxacillinases produced by ceftazidime-resistant Pseudomonas aeruginosa strains in France. Antimicrob Agents Chemother 54 3512 3515
16. BertFBrangerCLambert-ZechovskyN 2002 Identification of PSE and OXA β-lactamase genes in Pseudomonas aeruginosa using PCR-restriction fragment length polymorphism. J Antimicrob Chemother 50 11 18
17. FluitACSchmitzFJ 2004 Resistance integrons and super-integrons. Clin Microbiol Infect 10 272 288
18. GalimandMLambertTGerbaudGCourvalinP 1993 Characterization of the aac(6′)-Ib gene encoding an aminoglycoside 6′-N-acetyltransferase in Pseudomonas aeruginosa BM2656. Antimicrob Agents Chemother 37 1456 1462
19. PoirelLGirlichDNaasTNordmannP 2001 OXA-28, an extended-spectrum variant of OXA-10 β-lactamase from Pseudomonas aeruginosa and its plasmid - and integron-located gene. Antimicrob Agents Chemother 45 447 453
20. KoshlandDBotsteinD 1980 Secretion of β-lactamase requires the carboxy end of the protein. Cell 20 749 760
21. O'DonnellSMJanssenGR 2001 The initiation codon affects ribosome binding and translational efficiency in Escherichia coli of cI mRNA with or without the 5′ untranslated leader. J Bacteriol 183 1277 1283
22. ShineJDalgarnoL 1974 The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci U S A 71 1342 1346
23. SalisHMMirskyEAVoigtCA 2009 Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 27 946 950
24. QuillardetPHofnungM 1993 The SOS chromotest: a review. Mutat Res 297 235 279
25. GuérinEJovéTTabesseAMazelDPloyM-C 2011 High-level Gene cassette transcription prevents integrase expression in class 1 integrons. J Bacteriol 193 5675 5682
26. DeanaABelascoJG 2005 Lost in translation: the influence of ribosomes on bacterial mRNA decay. Gene Dev 19 2526 2533
27. KuhnA 1987 Bacteriophage M13 procoat protein inserts into the plasma membrane as a loop structure. Science 238 1413 1415
28. BouvierMDucos-GalandMLootCBikardDMazelD 2009 Structural features of single-stranded integron cassette attC sites and their role in strand selection. PLoS Genet 5 e1000632
29. FrumerieCDucos-GalandMGopaulDNMazelD 2010 The relaxed requirements of the integron cleavage site allow predictable changes in integron target specificity. Nucleic Acids Res 38 559 569
30. Cipriano SouzaRVicenteACVieiraVVMarquesSGSoaresMdGA 2008 Clindamycin and metronidazole as independent risk factors for nosocomial acquisition of multidrug-resistant Pseudomonas aeruginosa. J Hosp Infect 69 402 403
31. LöfmarkSEdlundCNordCE 2010 Metronidazole is still the drug of choice for treatment of anaerobic infections. Clin Infect Dis 50 S16 S23
32. SissonGJeongJ-YGoodwinABrydenLRosslerN 2000 Metronidazole activation is mutagenic and causes DNA fragmentation in Helicobacter pylori and in Escherichia coli containing a cloned H. pylori rdxA+ (nitroreductase) gene. J Bacteriol 182 5091 5096
33. TocherJHEdwardsDI 1994 Evidence for the direct interaction of reduced metronidazole derivatives with DNA bases. Biochem Pharmacol 48 1089 1094
34. MenéndezDRojasEHerreraLALópezMCSordoM 2001 DNA breakage due to metronidazole treatment. Mutat Res 478 153 158
35. LivermoreDM 2002 Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis 34 634 640
36. DepardieuFPodglajenILeclercqRCollatzECourvalinP 2007 Modes and modulations of antibiotic resistance gene expression. Clin Microbiol Rev 20 79 114
37. FoucaultMLDepardieuFCourvalinPGrillot-CourvalinC 2010 Inducible expression eliminates the fitness cost of vancomycin resistance in enterococci. Proc Natl Acad Sci U S A 107 16964 16969
38. AnderssonDIHughesD 2010 Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8 260 271
39. LuTKCollinsJJ 2009 Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci U S A 106 4629 4634
40. CirzRTChinJKAndesDRde Crécy-LagardVCraigWA 2005 Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol 3 e176
41. ClineDJHoltSLSingletonSF 2007 Inhibition of Escherichia coli RecA by rationally redesigned N-terminal helix. Org Biomolec Chem 5 1525 1528
42. SextonJZWigleTJHeQHughesMASmithGR 2010 Novel inhibitors of Escherichia coli RecA ATPase activity. Curr Chem Genomics 4 34 42
43. WigleTJSextonJZGromovaAVHadimaniMBHughesMA 2009 Inhibitors of RecA activity discovered by high-throughput screening: cell-permeable small molecules attenuate the SOS response in Escherichia coli. J Biomol Screen 14 1092 1101
44. TalonDCailleauxVThouverezMMichel-BriandY 1996 Discriminatory power and usefulness of pulsed-field gel electrophoresis in epidemiological studies of Pseudomonas aeruginosa. J Hosp Infect 32 135 145
45. CLSI 2009 Method for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard — eighth edition, CLSI document M7-A7
46. DumasJ-Lvan DeldenCPerronKKöhlerT 2006 Analysis of antibiotic resistance gene expression in Pseudomonas aeruginosa by quantitative real-time-PCR. FEMS Microbiol Lett 254 217 225
47. ChoiKHSchweizerHP 2005 An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5 30
48. SchweizerHP 1992 Allelic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol Microbiol 6 1195 1204
49. DilgerKFuxRRöckDMörikeKGleiterCH 2007 Effect of high-dose metronidazole on pharmacokinetics of oral budesonide and vice versa: a double drug interaction study. J Clin Pharmacol 47 1532 1539
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Protecting against Pneumococcal Disease: Critical Interactions between Probiotics and the Airway Microbiome
- Manipulation of Costimulatory Molecules by Intracellular Pathogens: Veni, Vidi, Vici!!
- Highly Efficient Prion Transmission by Blood Transfusion
- A Highly Intensified ART Regimen Induces Long-Term Viral Suppression and Restriction of the Viral Reservoir in a Simian AIDS Model
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy