
PK-sensitive PrP Is Infectious and Shares Basic Structural Features with PK-resistant PrP
One of the main characteristics of the transmissible isoform of the prion protein (PrPSc) is its partial resistance to proteinase K (PK) digestion. Diagnosis of prion disease typically relies upon immunodetection of PK-digested PrPSc following Western blot or ELISA. More recently, researchers determined that there is a sizeable fraction of PrPSc that is sensitive to PK hydrolysis (sPrPSc). Our group has previously reported a method to isolate this fraction by centrifugation and showed that it has protein misfolding cyclic amplification (PMCA) converting activity. We compared the infectivity of the sPrPSc versus the PK-resistant (rPrPSc) fractions of PrPSc and analyzed the biochemical characteristics of these fractions under conditions of limited proteolysis. Our results show that sPrPSc and rPrPSc fractions have comparable degrees of infectivity and that although they contain different sized multimers, these multimers share similar structural properties. Furthermore, the PK-sensitive fractions of two hamster strains, 263K and Drowsy (Dy), showed strain-dependent differences in the ratios of the sPrPSc to the rPrPSc forms of PrPSc. Although the sPrPSc and rPrPSc fractions have different resistance to PK-digestion, and have previously been shown to sediment differently, and have a different distribution of multimers, they share a common structure and phenotype.
Published in the journal:
. PLoS Pathog 8(3): e32767. doi:10.1371/journal.ppat.1002547
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002547
Summary
One of the main characteristics of the transmissible isoform of the prion protein (PrPSc) is its partial resistance to proteinase K (PK) digestion. Diagnosis of prion disease typically relies upon immunodetection of PK-digested PrPSc following Western blot or ELISA. More recently, researchers determined that there is a sizeable fraction of PrPSc that is sensitive to PK hydrolysis (sPrPSc). Our group has previously reported a method to isolate this fraction by centrifugation and showed that it has protein misfolding cyclic amplification (PMCA) converting activity. We compared the infectivity of the sPrPSc versus the PK-resistant (rPrPSc) fractions of PrPSc and analyzed the biochemical characteristics of these fractions under conditions of limited proteolysis. Our results show that sPrPSc and rPrPSc fractions have comparable degrees of infectivity and that although they contain different sized multimers, these multimers share similar structural properties. Furthermore, the PK-sensitive fractions of two hamster strains, 263K and Drowsy (Dy), showed strain-dependent differences in the ratios of the sPrPSc to the rPrPSc forms of PrPSc. Although the sPrPSc and rPrPSc fractions have different resistance to PK-digestion, and have previously been shown to sediment differently, and have a different distribution of multimers, they share a common structure and phenotype.
Introduction
The prion (PrPSc) is the infectious agent responsible for a suite of different rare animal and human diseases known as transmissible spongiform encephalopathies (TSEs) [1], [2], [3], [4], [5]. PrPSc is able to convert a normal cellular prion protein (PrPC) into PrPSc when both isoforms make contact, and thereby propagate an infection. The conversion of PrPC into PrPSc involves a conformational change of the protein in which the total amount of β-sheet increases and that of α-helical secondary structure decreases or perhaps disappears [6], [7]. PrPSc is a multimer, while PrPC is monomeric [8], [9]. These conformational differences are the only demonstrated structural differences between PrPSc and PrPC [10]. Detailed mass spectrometric analysis showed they have identical amino acid sequences [11]. No post-translational differences have been found between PrPSc and PrPC: both share one disulfide bond, two or less sugar antennae and a single glycophosphatidylinositol (GPI) anchor [12]. The composition of the sugar antennae and the GPI anchor vary similarly in both PrPSc and PrPC [13]. On the other hand, the conformational change and consequent aggregation makes PrPSc insoluble in non-denaturing detergents and partially resistant to PK digestion [1]. Thus, treatment of a sample with 50 µg/ml of PK for 1 hour at 37°C completely destroys PrPC, while, typically, PrPSc is partially cleaved at the amino terminal portion, leaving a PK-resistant core termed PrP 27–30.
In 1998 Safar et al. reported the existence of a subset of PrPSc molecules that are completely degraded by PK, which hence were termed, PK-sensitive PrPSc (sPrPSc) [14]. Tzaban et al. later demonstrated for the first time that prion-infected tissues contain sPrPSc molecules that form low molecular weight aggregates [15]. These authors subjected brain homogenates from scrapie-infected animals to sucrose gradients, and found that PrPSc was distributed in a continuum of aggregation sizes. The more dense fractions, corresponding to larger multimers, were PK-resistant, whereas the intermediate fractions, corresponding to smaller multimers, were not. It has also been described that as much as 90% of total PrPSc in the brains of individuals who had died as a consequence of Creutzfeldt-Jakob disease (CJD) was estimated to be sPrPSc [16].
Different studies on other protein misfolding diseases, such as Alzheimer's disease, suggest that large amyloid fibrils may be a means of protecting the host by sequestering the smaller and more toxic multimers as larger less toxic fibrils [17]. In the case of prion diseases, infectivity studies of the different sized fractions of hamster PrPSc revealed a several-fold increase in infectivity for non-fibrillar particles with masses equivalent to 14–28 PrP molecules [18]. These PrPSc particle sizes correspond to the sPrPSc fraction according to Tzaban and our own group's characterization using sucrose gradient ultracentrifugation [15], [19]. Although the definition of sPrPSc is operational, a question arises: are sPrPSc and rPrPSc two populations with different conformations or simply different sized multimers with the same conformation?
To address this question, we investigated first the infectivity of the sPrPSc vs. the PK-resistant (rPrPSc) fraction of hamster PrPSc (263K strain). sPrPSc was further characterized by limited proteolysis and mass spectrometry. Then, PrPSc infectivity and strain characteristics were assessed by inoculation into Syrian hamsters and comparison of the resulting incubation period and lesion distribution with that obtained after inoculation with either total PrPSc or the PK-resistant fraction of PrPSc. The obtained results are reported here.
Results
We obtained sPrPSc and rPrPSc (263K ) fractions from our starting total PrPSc by our previously described ultracentrifugation-based method [19]. When these fractions were subjected to PK digestion for 1 h at 37°C, virtually complete degradation of sPrPSc occurred after treatment with 50 µg/ml of PK whereas partial resistance occurred with lower concentrations of the enzyme (Figure 1). In contrast, rPrPSc was much more resistant to PK under these conditions (Figure 1). These results fully agree with our previously published work [19]. The sPrPSc, rPrPSc, purified PrPSc, and the unpurified PrPSc material was used in our subsequent experiments.

We performed a bioassay of normalized preparations of four prion isolates, purified PrPSc, sPrPSc, rPrPSc and the unpurified PrPSc-containing 0.1% brain homogenate. The amount of PrP present in each sample was determined by a mass spectrometry-based quantitative method [20]. The amount of residual PrPC present in the purified samples represents a negligible contribution [21]. Each of the preparations was then diluted, so that each contained approximately similar amounts of PrP. In addition, three dilutions of each of the four normalized samples were prepared: 1/10, 1/100, and 1/1,000 for the 0.1% brain homogenate sample and 1/100, 1/1,000, and 1/10,000 for the other three ones. Each of these dilution series (16 in total) was inoculated into a set of eight Syrian hamsters (LVG) (128 total). The dates of the appearance of clinical signs and the date of euthanization at terminal disease were recorded [22]. These data were plotted as a Kaplan-Meier estimate graph (Figure 2) for each dilution of each preparation. For further clarity, they are also presented in table form (Table S1).

Animals inoculated with the undiluted sPrPSc preparation got sick sooner than those inoculated with any of the other three undiluted preparations (P<0.01). By the 1/1000 dilution all of the hamsters inoculated with the purified PrPSc (sPrPSc, rPrPSc, and total purified PrPSc) had a similar incubation time, which indicates a roughly similar infectivity. When the rPrPSc and purified PrPSc preparations were diluted 1/10,000 all of the animals became sick. In contrast, of the eight animals inoculated with the 1/10,000 dilution of sPrPSc, only seven became infected. Even after 240 days, the eighth animal showed no clinical signs and there was no detectable PrPSc in its brain by mass spectrometry-based analysis [20]. Paradoxically the sPrPSc shows a shorter incubation time in the undiluted preparation (Table S2). Upon dilution of the sPrPSc preparation the incubation time disproportionately increases compared to the other purified preparations (purified PrPSc and rPrPSc). At this point, it was not clear if these differences were due to the possibility that our isolation procedure facilitates the isolation of different prion strains, as has been described by Bessen and Marsh [23], or if these differences were caused by kinetic factors, due to the smaller size of the PrPSc multimers present in the sPrPSc fraction [15], [19].
These results show that sPrPSc is infectious, and that its infectivity is comparable to that of rPrPSc. We wanted to see how sPrPSc behaved after PK treatment. We took a sample of sPrPSc and divided it into two portions. One was untreated and the other was digested with PK (50 µg/ml of PK; 37°C; 1 h). These samples were inoculated (ic) into two sets of six hamsters. The disease course in both sets was observed. In the group inoculated with PK-treated sPrPSc, animals succumbed 15 days later than in the untreated group (Figure S1). This time interval corresponds to a 100 fold dilution of the sPrPSc fraction (Figure 2). Granting that only an approximate correlation between incubation times and titers can be surmised in our experimental conditions, given that we did not perform a full calibration [20], this result suggests that PK destroyed approximately 99% of the infectivity present in the sPrPSc fraction. Unlike the unpurified 0.1% brain homogenate, where the loss of infectivity upon treatment with PK is much greater (∼99%) than the loss of WB signal (∼70%) [24], the loss of signal and loss of infectivity are proportionately large (∼99%) with the sPrPSc fraction (Figure S1).
To assess whether sPrPSc is also infectious via the ip route, we inoculated intraperitoneally two groups of animals with equal amounts of sPrPSc and rPrPSc, respectively. Both groups succumbed to infection, and although not statistically significant, animals inoculated with the sPrPSc fraction showed an incubation time slightly shorter (116±9 dpi) than the ones inoculated with the rPrPSc fraction (123±14 dpi) (Figure S2). As expected, animals inoculated ip survived longer than those inoculated ic (intracerebral) [25], [26].
After determining that there were phenotypic differences (incubation times) between sPrPSc and a complete mixture of PrPSc, we explored the possibility that there were structural differences between these two PrPSc fractions. We have previously reported that, in addition to the well-known N-terminal PK cleavage sites, around position G-90, there are other additional cleavage sites [27]. In that study, we prepared a cleavage map for total PrPSc after treatment with PK and then treatment with NTCB. The N-terminal cleavage sites include: residues 86 (G-86→D-178), 90 (G-90→D-178), 92 (G-92→D-178), 98 (Q-98→D-178), and 101 (K-101→D-178). Additional cleavage sites comprise residues 117 (A-117→D-178), 119 (G-119→D-178), 135 (S-135→D-178), 139 (M-139→D-178), 142 (G-142→D-178), and 154 (M-154→D-178) ([27] and Figure S3). We used an analogous approach to prepare a cleavage map of sPrPSc. Briefly, sPrPSc was digested with 1 µg/ml of PK for 30 minutes and subsequently treated with NTCB. This reagent cyanylates free cysteines and NaOH is subsequently used to cleave amide bonds at the N-terminal side of the modified cysteine residues [27], [28]. Given the difficulty in identifying the GPI anchor or sugar-containing peptides by mass spectrometry, only those free of sugar moieties were analyzed. The NTCB reagent cleaves at the cysteine residue at position 179, thereby separating the GPI-anchor and glycosylated portion of the protein from the set of amino-terminal truncated peptides. These peptides contain no post-translational modifications and were analyzed by MALDI-TOF.
The MALDI-TOF analysis of the NTCB treated sPrPSc fraction showed cleavage sites identical to the ones previously described for total PrPSc, namely, 117, 119, 135, and 139 (Figure 3a). Curiously, the more abundant peptides from the classic N-terminal PK-cleavage sites (86, 90, and 92) are present in the total PrPSc samples [27], but not in the sPrPSc fraction (Figure 3). This suggests that the N-terminal portion of the PrP molecules present in the sPrPSc fraction might be more exposed and therefore more susceptible to PK digestion than is the N-terminal portion present in the complete mixture of PrPSc. However, it should be remembered that MALDI-TOF is not a quantitative technique and that the signal response of larger peptides, such as (G-86→D-178), or (G-90→D-178), is poorer than that of smaller ones. Indeed, cleavages at around position 90, and perhaps slightly more amino-terminal sites, are prominent in sPrPSc as shown by WB analysis (vide infra).

The similarity of the PK-cleavage pattern between sensitive and total PrPSc is also evident when comparing the digested samples by Western blot assay using a C-terminal antibody after PK-treatment and deglycosylation (Figure 3b). Besides the intense band corresponding to cleavages around position 90, 6–7 additional lower molecular weight bands are seen in both cases, as we have previously described for total PrPSc [27]. Although we do not know the identity of each band, the apparent molecular weight of some of them could correspond with cleavages seen in MALDI-TOF. While the relative intensities of some of the bands vary between sPrPSc and total PrPSc, (the main band centered at 19–20 kDa smears a bit more into the 20–21 kDa region in the sPrPSc sample, the ∼17 kDa band is more intense in total PrPSc, bands around 15, and finally, bands at ∼10 and ∼6 kDa are more intense in sPrPSc), the overall pattern is very similar.
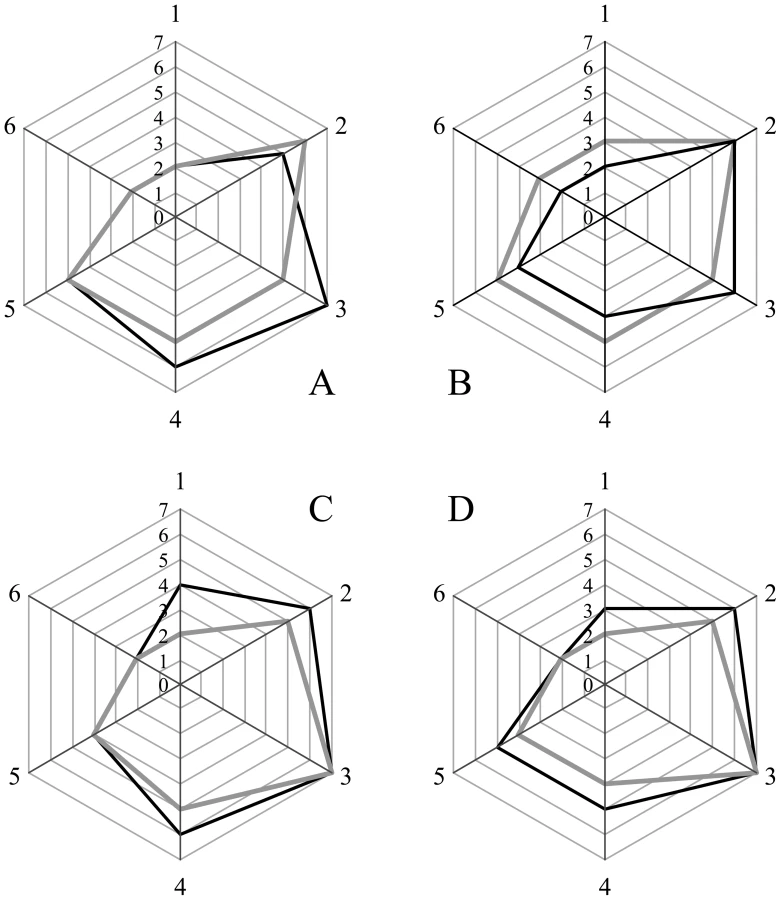
We wanted to ensure that in fractionating PrPSc, we had not inadvertently isolated a new prion strain. We performed comparative histopathological and immunohistochemical studies on the left hemisphere of brains from hamsters infected with each of the four PrPSc preparations. Spongiform change and PrP deposition were detected in all cases with no detectable differences in the PrPSc distribution or the morphology of the PrPSc aggregates among all four groups (Figure 4). The PrPSc appeared as diffuse as well as small, punctate aggregates. To assess whether there were differences in the brain regions targeted, lesion profiles of the hamsters inoculated with four different prion preparations was also performed. We compared the spongiform degeneration and PrPSc deposition in 6 brain regions. The results of this lesion profile analysis suggest that the isolation procedure did not yield different strains (Figure 5).
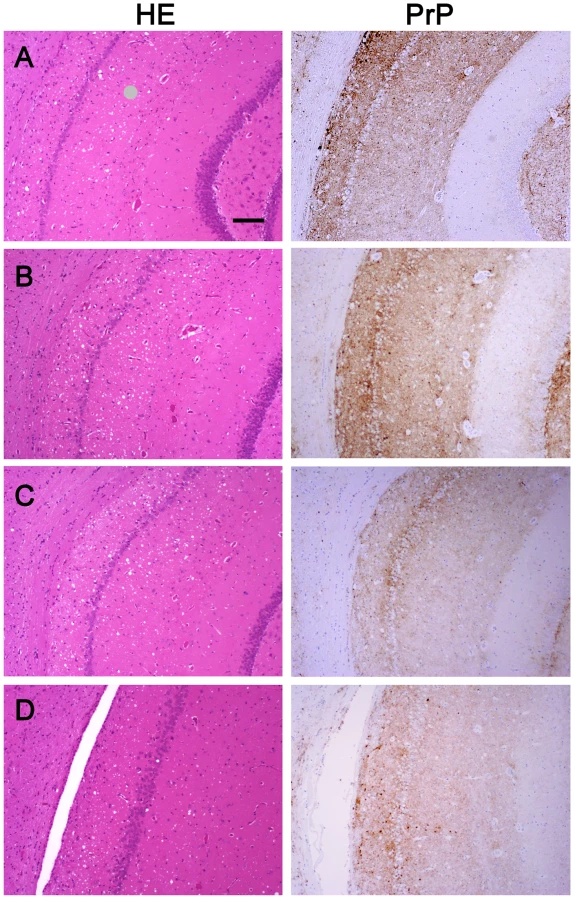
Western blot analysis of brain homogenates from the four inoculated groups showed the presence of PK resistant PrP in all groups. No differences in band patterns were detected (Figure S4). The relative amount of sPrPSc in each group was assessed by subtracting the amount PrPSc present in an aliquot digested with PK from the amount of PrPSc present in an untreated aliquot. The amount of PrPSc was determined by a previously reported mass spectrometry-based method [20]. The proportion of sPrPSc present in the brains of animals inoculated with either unpurified brain homogenate, purified prions, rPrPSc or sPrPSc was 0.3, 0.3, 0.6 or 0.5, respectively (means of duplicate analyses). In our previous work, we determined that sPrPSc accounted for between 35 and 60% of the total PrPSc present in the sample [19]. Thus the relative amount of sPrPSc in all groups is similar to that present in an unpurified brain homogenate.
We chose to use the drowsy (Dy) strain of hamster-adapted prion disease in order to compare the results with those obtained from 263K-derived prions. The phenotype of the Dy strain is very different from that of 263K [29]. It has a much longer incubation period and the typical symptoms are progressive lethargy and kyphosis [23]. The Dy strain is highly susceptible to PK digestion [30]. We wondered whether such susceptibility correlated with a higher ratio of sensitive to resistant fraction and if this would provide some insight into the nature of sPrPSc isolated from the 263K strain.
We isolated Dy sPrPSc using our method of isolating PK-sensitive prions (Figure S5) [19]. The sensitive fraction of Dy was shown to constitute approximately half of the total PrPSc, whereas in 263K, the analogous PK-sensitive fraction represents a considerably lower proportion of the total PrPSc (Figure S6). This could partially explain why Dy is more PK-susceptible than 263K. These results agree with those of Safar et al. who reported that different strains of hamster scrapie prions contain different ratios of sensitive to resistant PrPSc [14]. It should be noted that, although our PrPSc purification protocol involves sonication and therefore fragmentation of large aggregates, the sonication conditions used in 263K and Dy PrPSc isolation were the same.
The MALDI-TOF spectrum of the Dy sensitive fraction after digestion with 0.1 µg/ml of PK and NTCB cleavage showed the same internal cleavage sites as in 263K (117, 119, 135, and 139) plus others (101 (K-101→D-178) and 92 (G-92→D178)) (Figure 6) also previously described for total PrPSc (Dy strain) (Figure S3 and [27]).

Discussion
Most prions have a degree of resistance to proteinase K digestion. Digestion of prions with proteinase K yields a characteristic prion core referred to as PrP 27–30 and a loss of infectivity that is disproportionate to the loss of protein. As researchers examined hamster strains they realized that not all strains of prions were equally resistant to proteinase K digestion, as is the case with the hyper (Hy) and drowsy (Dy) strains of hamster-adapted transmissible mink encephalopathy [23], [30]. Other researchers observed a similar phenomenon with other prions [31], [32], [33], [34]. Later Safar showed that PrPSc contained both a proteinase K sensitive and proteinase K resistant fraction [14]. These results indicated that PrPSc can be both infectious and proteinase K sensitive and that these phenotypes are a general characteristic of prions.
Since Safar et al. reported the existence of a PK-sensitive fraction in PrPSc, little has been published about its structural characteristics and infectious properties [14]. Although its infectivity has been suggested [14], [35], it had not been proven. In this work we demonstrate that the sensitive fraction of PrPSc is roughly as infectious as the resistant one. Highly significant is the fact that both fractions presented similar incubation times even when the respective inocula were diluted 1000 times. It has been shown that when PrPSc was fragmented by sonication, infectious titer was reduced because the rate of PrPSc clearance from the brain increased [36]. PrPSc is rapidly cleared from the brain once inoculated and the rate of clearance is influenced by the particle size [37]. Our results indicate that both rPrPSc and sPrPSc fractions have the same degree of infectivity indicating that the aggregation state of PrPSc does not affect its infectivity. This is in contrast with a previous report where the majority of sPrPSc was found to be not infectious when measured by the scrapie cell assay [38]. Also, Weber et al. found that extended sonication of PrPSc was associated with a loss of infectivity as measured by the prolongation of the incubation time [39]. In this case some PrPSc may be degraded during sonication. In our case, total PrPSc is isolated first, when sonication occurred, and then both fractions are separated at an intermediate centrifugation speed with no further sonication. On the other hand, hamster 263K ( = Sc237) prions did not show altered incubation times when the prion rods were fragmented by sonication into spherical particles [40].
Several explanations may account for the similarity in infectivity values in sPrPSc and rPrPSc. Namely, common and highly infectious species are present in both fractions and thus are subject to the same rate of clearance. Another possibility is the presence of one or more cofactors that copurify with both fractions of PrPSc. PrP 27–30 rods that dissociated into much smaller detergent-lipid-protein complexes and liposomes led to a 100-fold increase in infectivity as measured by endpoint titration [41]. Perhaps the binding of PrPSc to the cell membrane increases the efficiency of disease transmission from cell to cell.
Limited proteolysis studies on the sPrPSc fraction of 263K and Dy indicate that, as in total PrPSc, the sensitive fraction is also composed of alternating PK-resistant stretches interspersed with PK-susceptible stretches. The relatively small variations in intensity of some of the PK-resistant bands detected by SDS-PAGE (Fig. 3b), could be interpreted as corresponding to differences in accessibility of the PK-resistant stretches involved, but the global emerging picture is one of a shared basic structural organization.
This result emphasizes the fact that the difference between sensitive and resistant PrPSc seems to be the previously described different size of the aggregates and not the conformation of their respective aggregates [15], [19]. Our data provide information on the N-terminal half of the protein only, therefore we cannot rule out differences in the C-terminal portion.
As we mentioned in the introduction, the sensitive fraction of 263K varies between 20–40% of the total PrPSc. Despite this variability, the amount of sensitive PrPSc obtained for 263K was always less than that for Dy. This result, previously reported by Safar et al. [14], would indicate that a specific ratio of sensitive to resistant exists for each strain. This author also reported that the incubation time of a given prion strain is directly proportional to the level of protease-sensitive PrPSc [14]. According to Deleault et al., variations in the sPrPSc∶rPrPSc ratio between different prion strains appear to be an incidental product rather than a strain property [42]. Nevertheless, we want to point out that for hamster-adapted prion diseases the PK-susceptibility seems to directly correlate with the proportion of sPrPSc present in the strain.
In summary, we have shown that sPrPSc is fully infectious, and that its infectivity can be reduced by approximately 99% by treatment with PK (50 µg/mL; 1 hr). sPrPSc appears to share the same basic architecture with rPrPSc, as judged by the generation of similar fragmentation patterns by limited proteolysis with PK as seen in the WB and MALDI analysis. This, together with the fact that sPrPSc induces a disease with signs and histopathological and lesion profiles that are essentially identical to those induced by total or rPrPSc, strongly suggests that sPrPSc differs from rPrPSc, as previously shown, just in its smaller size, i.e., being made up of fewer PrP units [15], [19]. On the other hand, the fact that different prion strains are characterized by different relative ratios of sPrPSc∶rPrPSc suggests that sPrPSc might play an important role in key properties of strains. Finally, sPrPSc, which is soluble in detergent-containing solutions, might prove to be useful in studies aimed at elucidating the structure of PrPSc.
Materials and Methods
Ethics statement
Animal experiments were carried out in accordance with the recommendations contained in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The procedures were governed by a protocol that was approved by the Institutional Animal Care and Use Committee of the United States Department of Agriculture, Agricultural Research Service, Albany, CA (Protocol Number: P-10-3). All surgery was performed under isoflurane anaesthesia, and all efforts were made to minimize the suffering of the animals. The small numbers of experiments carried out at the University of Santiago de Compostela were approved by the University Ethics Committee, in accordance with the European Union Council Directive 86/609/EEC.
Isolation of total PrPSc and sPrPSc
PrPSc was isolated from brains of terminally ill Syrian hamsters infected intracranially (ic) with the 263K strain of scrapie using a slightly modified version of the procedure of Diringer et al. [43]. A cocktail of protease inhibitors (Complete, Roche Diagnostics, Penzberg, Germany) at a final concentration of 1× was used in all buffers throughout the procedure up to the penultimate pellet, as defined in the mentioned study [43]. The final pellet was resuspended in 20 mM Tris (pH 8.5) containing 1% sarkosyl and no protease inhibitors, at concentrations between 1–2 µg/µl. The stock suspension thus prepared was aliquoted and frozen until further use; its purity was assessed by SDS-PAGE with Coomassie blue staining, and estimated to be approximately 80%. sPrPSc was isolated from total PrPSc by ultracentrifugation at an intermediate speed [19]. A 50–150 µl portion of the PrPSc stock was homogenized by application of 3 sonication pulses of 1 s each, and spun in a Beckman TLX ultracentrifuge (Beckman, Fullerton, CA) using a TLA-120-1 rotor at 40,000 rpm (56,806 g) for 2 hours at 20°C. The supernatant was collected and the pellet was resuspended by sonication in a volume of 20 mM Tris (pH 8.5) containing 1% sarkosyl equivalent to that of the supernatant. Fractions of supernatant and pellet were treated with 50 µg/ml of PK at 37°C during 1 hour; the reaction was terminated with 2 mM Pefabloc (Fluka, St. Louis, MO), and a fraction subjected to SDS-PAGE and either stained with Coomassie brilliant blue or transferred to a PVDF membrane (Immobilon-P, Millipore, Billerica, MA, USA) and analyzed by Western blotting using mAb 3F4 (Dako, Glostrup, Denmark). The supernatant fraction is sPrPSc, and the pellet fraction, PK-resistant PrPSc (rPrPSc). The concentration of PrPSc was estimated in each of these samples and in brain homogenates. PrPSc was isolated by ultracentrifugation using the method of Bolton et al. [44], with slight modifications [20]. The resulting pellets were dissolved in 200 µL of 6 M guanidinium chloride and allowed to stand at RT for 24 hours to inactivate the PrPSc [45]. The inactivated prion solutions were precipitated with methanol and subjected to analysis by mass spectrometry using previously described methods [20], [21]. Briefly, pellets were dissolved in 0.01% aqueous beta-octylglucopyranoside (BOG) and [13C5,15N]-VVEQMCTTQYQK added as internal standard. The samples were then sequentially reduced, alkylated and digested with trypsin. NanoLC/MS/MS was carried out with an Applied Biosystems (ABI/MDS Sciex, Toronto, Canada) model 4000 Q-Trap instrument equipped with a nano-electrospray ionization source. The mass spectrometer was operated in multiple reaction monitoring (MRM) mode, alternating between detection of VVEQMCTTQYQK (precursor ion m/z of 757.8, product ion of m/z 171.1) and [13C5,15N]-VVEQMCTTQYQK (precursor ion m/z of 760.8, product ion of m/z 177.1). Quantitation was done with the Intelliquan quantitation algorithm of Analyst 1.4.1 software (Applied Biosystems) using default parameters [20], [21].
Bioassays
Four prion isolates, 0.1% brain homogenate, purified PrPSc, sPrPSc, and rPrPSc, were diluted to yield four corresponding solutions containing 1.5±0.4 ng of PrP per 50 µL of sample, except in the case of the 0.1% brain homogenate, which contained 0.7 ng of PrP per 50 µL of sample (vide supra). A portion of each of these normalized solutions was diluted 100-fold, 1,000-fold and 10,000-fold into sterile phosphate buffered saline (PBS) to yield a set of four dilutions (including the undiluted material) for each of the prion isolates; in the case of the 0.1% brain homogenate sample, dilutions were 10-fold, 100-fold, and 1,000-fold. Fifty microliters of each dilution were ic inoculated into the right cerebral hemisphere of a four week old anesthetized female hamster. Eight animals were used per isolate/dilution.
The 128 animals were placed in cages (2 per cage) and observed for clinical signs. The first clinical sign to be observed and its date of occurrence recorded was an exaggerated startle response. A progression of clinical signs followed, including an exaggerated startle response that became more pronounced over time, ataxic gait whose severity increased with time, limb rigidity, characteristic head bobbing, and progressive lethargy. When the animals could no longer be roused from their recumbency to feed and drink water, they were humanely euthanized. The incubation time was measured (in days) as the time between inoculation and euthanization.
After 240 days one of the inoculated animals (sPrPSc; 10,000-fold dilution) animals remained healthy. The animal was euthanized and its brain removed. The brain was analyzed for the presence of PrP 27–30 by mass spectrometry [20]. There was no evidence of PrP 27–30 present in the brain.
In a separate experiment, two groups of 6 animals were inoculated ic with either 50 µl of a sample containing ∼40 ng of sPrPSc or the same volume of sPrPSc treated with 50 µg/ml of PK for 1 h at 37°C, followed by quenching of PK with 2 mM Pefabloc. Infection progression was monitored as described above, and approximate infectivity titers of the PK-treated and untreated sPrPSc samples were estimated by incubation period assay according to the procedure of Prusiner et al. [22].
Finally, two groups of 6 animals were intraperitoneally (ip) inoculated with 150 µl volumes of sPrPSc and rPrPSc prion isolates containing ∼125 ng of PrP each, and the course of infection observed as described above. The amount of PrPSc in these two samples was calculated approximately by comparison with a recombinant Syrian hamster (rSha) PrP (90–231) standard (a generous gift of Giuseppe Legname) after deglycosylation with PNGase F (New England Biolabs, Ipswich, MA, USA) and Western blotting with mAb 3F4.
Histopathology and immunohistochemical stains
Four µm thick sections were cut onto positively charged silanized glass slides and stained with hematoxylin and eosin, or immunostained using an antibody for PrP (3F4). Immunohistochemical stains for PrP were performed entirely on an automated Discovery XT staining apparatus (Ventana Medical Systems, Oro Valley, AZ) using a DAB Map XT Detection Kit (Ventana Medical Systems). Sections were deparaffinised and then washed in distilled water for 5 min. Epitope exposure was performed by heating sections to 100°C in a citrate buffer for 12 minutes. After treatment with protease 2 (Ventana Medical Systems) for 24 minutes, sections were incubated with anti-PrP 3F4 for 60 min and PrP was detected using HRP-conjugated antibodies and a DAB substrate.
Lesion profiling
We selected 6 anatomic brain regions in accordance with previous strain typing protocols [46], [47] from 2 hamsters per group. We scored spongiosis on a scale of 0–4 (not detectable, mild, moderate, severe and status spongiosus) and PrP immunological reactivity on a 0–3 scale (not detectable, mild, moderate, severe). A sum of the two scores resulted in the value obtained for the lesion profile for the individual animal. The ‘radar plots’ depict the scores for spongiform changes and PrP deposition. Numbers correspond to the following brain regions: (1) cerebellum, (2) medial thalamus, (3) hippocampus, (4) medial cerebral cortex dorsal to hippocampus, (5) medial cerebral cortex dorsal to septum, (6) white matter at cerebellar peduncles. Investigators blinded to animal identification performed histological analyses.
Biochemical characterization and quantitation of PrPSc in brains of inoculated animals
PrPSc was detected in brain homogenates of inoculated animals by Western blot using mAb 3F4 (primary antibody) and goat anti-mouse Fc (secondary antibody) (Sigma-Aldrich, St. Louis, MO).
To assess the relative amount of sPrPSc and rPrPSc in animals inoculated with the four different inocula described, duplicate PrPSc samples were isolated from the brains of inoculated hamsters using the method of Bolton et al. [44] with slight modifications [20]. The resulting pellets were either dissolved in 200 µL of 6 M guanidinium chloride (Sigma-Aldrich, St. Louis, MO) or resuspended in 0.1% Z 3,14-T-8.5 (0.1% 3-(N,N-dimethylmyristyl-ammonium)propane sulfonate; 20 mM Tris pH 8.5). The 6 M guanidinium chloride solutions were allowed to stand for 24 hours at RT to inactivate the PrPSc [45]. The samples suspended in 0.1% Z 3,14-T-8.5 were sonicated. After sonication, PK was added to make a final concentration of 5 µg/mL. The PK was permitted to react for 1 hour at 37°C. The reaction was quenched by addition of a sufficient amount of phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich, St. Louis, MO) to achieve a final concentration of 1 mM. Enough solid guanidinium chloride was then added to make a 6 M solution, which was left to stand for 24 hours at RT to inactivate the prions. The inactivated prion solutions were precipitated with methanol and the amount of PrP present in the sample was determined by mass spectrometry (vide supra) [20], [21].
NTCB cleavage reaction of PrPSc samples
All buffers and reagents were prepared fresh for the 2-nitro-5-thiocyanatobenzoic acid (NTCB) reactions. Between 15–20 µg of purified PrPSc was reduced in 2 mM DTT for 1 h at 37°C under denaturing conditions (6 M guanidinium chloride and 100 mM Tris buffer pH 8). In the same buffer, samples were cyanylated in 10 mM 2-nitro-5-(thiocyanato)-benzoate (NTCB) for 30 minutes at RT. After methanol precipitation, samples were redissolved in 6 M guanidinium chloride, 100 mM Tris buffer pH 8. PrPSc, specifically cyanylated at the reactive thiols, was cleaved by alkaline hydrolysis with 150 mM NaOH for 15 minutes at 37°C. The NTCB reaction was terminated by addition of trifluoroacetic acid (TFA) to a final pH of 2–3. PrPSc peptides were isolated from the reaction mixture using C-18 ZipTips (Millipore) according to the manufacturer's instructions. Peptides were eluted from the tips in 10 µl of 50% acetonitrile, 0.1% TFA and used directly for MALDI analysis.
MALDI mass spectrometry
A 2 µL portion of the PrPSc peptide sample was mixed with an equal volume of a saturated solution of sinapinic acid in acetonitrile and 0.1% aqueous TFA (1∶2). One microliter of the mixture was spotted onto a Bruker sample plate, allowed to air-dry, and analyzed using a Bruker Autoflex MALDI instrument in linear mode. The laser frequency was 25 Hz; pulsed ion extraction was set at a value of 140–150 ns. Repeated laser shots, typically 25–30, were averaged.
Analysis of PK-resistant fragments of sPrPSc and total PrPSc by Western blot
PK-treated samples were deglycosylated with PNGase F (New England Biolabs, Ipswich, MA, USA) at 37°C for 48 h, according to the manufacturer's instructions, followed by precipitation with ice-cold 85% MetOH. Pellets were boiled in 10 µl of reducing tricine sample buffer (BioRad, Hercules, CA, USA). Tricine SDS-PAGE [48] was carried out using 10–20% Tris-Tricine/Peptide Precast gels (BioRad), in a Criterion electrophoresis system (BioRad). The cathode buffer was Tris-Tricine-SDS buffer (Sigma-Aldrich) and the anode buffer, 100 mM Tris-HCl, pH 8.9. Electrophoresis was performed at a constant voltage of 125 volts for 200 minutes, on ice. After electroblotting on PVDF membranes, these were probed with antibody R1 (a generous gift from Hanna Serban, UCSF), which recognizes residues 226–231. Peroxidase-labeled anti-human antibody was used as the secondary antibody.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. PrusinerSB 1982 Novel proteinaceous infectious particles cause scrapie. Science 216 136 144
2. PrusinerSB 1998 Prions. Proc Natl Acad Sci U S A 95 13363 13383
3. DetwilerLABaylisM 2003 The epidemiology of scrapie. Rev Sci Tech 22 121 143
4. HarmanJLSilvaCJ 2009 Bovine spongiform encephalopathy. J Am Vet Med Assoc 234 59 72
5. WilliamsES 2005 Chronic wasting disease. Vet Pathol 42 530 549
6. CaugheyBWDongABhatKSErnstDHayesSF 1991 Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 30 7672 7680
7. BaronGSHughsonAGRaymondGJOfferdahlDKBartonKA 2011 Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 50 4479 4490
8. AlperTHaigDAClarkeMC 1966 The exceptionally small size of the scrapie agent. Biochem Biophys Res Commun 22 278 284
9. Bellinger-KawaharaCGKempnerEGrothDGabizonRPrusinerSB 1988 Scrapie prion liposomes and rods exhibit target sizes of 55,000 Da. Virology 164 537 541
10. RiesnerD 2003 Biochemistry and structure of PrP(C) and PrP(Sc). Br Med Bull 66 21 33
11. StahlNBaldwinMATeplowDBHoodLGibsonBW 1993 Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 32 1991 2002
12. StahlNBaldwinMTeplowDBHoodLEBeavisR 1992 Cataloging post-translational modifications of the scrapie prion protein by mass spectrometry. PrusinerSBCollingeJPowellJAndertonB Prion diseases of humans and animals New York Ellis Horwood 361 379
13. RuddPMEndoTColominasCGrothDWheelerSF 1999 Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc Natl Acad Sci U S A 96 13044 13049
14. SafarJWilleHItriVGrothDSerbanH 1998 Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 4 1157 1165
15. TzabanSFriedlanderGSchonbergerOHoronchikLYedidiaY 2002 Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41 12868 12875
16. SafarJGGeschwindMDDeeringCDidorenkoSSattavatM 2005 Diagnosis of human prion disease. Proc Natl Acad Sci U S A 102 3501 3506
17. CaugheyBLansburyPT 2003 Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26 267 298
18. SilveiraJRRaymondGJHughsonAGRaceRESimVL 2005 The most infectious prion protein particles. Nature 437 257 261
19. PastranaMASajnaniGOniskoBCastillaJMoralesR 2006 Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry 45 15710 15717
20. SilvaCJOniskoBCDyninIEricksonMLRequenaJR 2011 Utility of Mass Spectrometry in the Diagnosis of Prion Diseases. Anal Chem 83 1609 1615
21. OniskoBCSilvaCJDyninIEricksonMVenselWH 2007 Sensitive, preclinical detection of prions in brain by nanospray liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 21 4023 4026
22. PrusinerSBCochranSPGrothDFDowneyDEBowmanKA 1982 Measurement of the scrapie agent using an incubation time interval assay. Ann Neurol 11 353 358
23. BessenRAMarshRF 1992 Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol 73 329 334
24. PrusinerSBMcKinleyMPGrothDFBowmanKAMockNI 1981 Scrapie agent contains a hydrophobic protein. Proc Natl Acad Sci U S A 78 6675 6679
25. PrusinerSBCochranSPAlpersMP 1985 Transmission of scrapie in hamsters. J Infect Dis 152 971 978
26. KimberlinRHWalkerCA 1986 Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J Gen Virol 67 Pt 2 255 263
27. SajnaniGPastranaMADyninIOniskoBRequenaJR 2008 Scrapie prion protein structural constraints obtained by limited proteolysis and mass spectrometry. J Mol Biol 382 88 98
28. WuJGageDAWatsonJT 1996 A strategy to locate cysteine residues in proteins by specific chemical cleavage followed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Biochem 235 161 174
29. KimberlinRHWalkerC 1977 Characteristics of a short incubation model of scrapie in the golden hamster. J Gen Virol 34 295 304
30. BessenRAMarshRF 1994 Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68 7859 7868
31. BarronRMCampbellSLKingDBellonAChapmanKE 2007 High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J Biol Chem 282 35878 35886
32. LasmezasCIDeslysJPRobainOJaeglyABeringueV 1997 Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 275 402 405
33. MansonJC 1999 Understanding transmission of the prion diseases. Trends Microbiol 7 465 467
34. RaceRMeade-WhiteKRainesARaymondGJCaugheyB 2002 Subclinical scrapie infection in a resistant species: persistence, replication, and adaptation of infectivity during four passages. J Infect Dis 186 Suppl 2 S166 170
35. TremblayPBallHLKanekoKGrothDHegdeRS 2004 Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J Virol 78 2088 2099
36. WeberPReznicekLMittereggerGKretzschmarHGieseA 2008 Differential effects of prion particle size on infectivity in vivo and in vitro. Biochem Biophys Res Commun 369 924 928
37. MaselJJansenVA 2001 The measured level of prion infectivity varies in a predictable way according to the aggregation state of the infectious agent. Biochim Biophys Acta 1535 164 173
38. CronierSGrosNTattumMHJacksonGSClarkeAR 2008 Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 416 297 305
39. WeberPGieseAPieningNMittereggerGThomzigA 2007 Generation of genuine prion infectivity by serial PMCA. Vet Microbiol 123 346 357
40. McKinleyMPBraunfeldMBBellingerCGPrusinerSB 1986 Molecular characteristics of prion rods purified from scrapie-infected hamster brains. J Infect Dis 154 110 120
41. GabizonRMcKinleyMPPrusinerSB 1988 Properties of scrapie prion proteins in liposomes and amyloid rods. Ciba Found Symp 135 182 196
42. DeleaultAMDeleaultNRHarrisBTReesJRSupattaponeS 2008 The effects of prion protein proteolysis and disaggregation on the strain properties of hamster scrapie. J Gen Virol 89 2642 2650
43. DiringerHBeekesMOzelMSimonDQueckI 1997 Highly infectious purified preparations of disease-specific amyloid of transmissible spongiform encephalopathies are not devoid of nucleic acids of viral size. Intervirology 40 238 246
44. BoltonDCRudelliRDCurrieJRBendheimPE 1991 Copurification of Sp33–37 and scrapie agent from hamster brain prior to detectable histopathology and clinical disease. J Gen Virol 72 2905 2913
45. PrusinerSBGrothDSerbanAStahlNGabizonR 1993 Attempts to restore scrapie prion infectivity after exposure to protein denaturants. Proc Natl Acad Sci U S A 90 2793 2797
46. BruceMEMcConnellIFraserHDickinsonAG 1991 The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol 72 Pt 3 595 603
47. FraserHDickinsonAG 1968 The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78 301 311
48. SchaggerH 2006 Tricine-SDS-PAGE. Nat Protoc 1 16 22
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- A Foot in the Door for Dermatophyte Research
- Structural Insights into a Unique Effector LidA Recognizing Both GDP and GTP Bound Rab1 in Their Active State
- An Entomopathogenic Nematode by Any Other Name
- New Insights into spp.: A Potential Link with Irritable Bowel Syndrome
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy