
Completion of Hepatitis C Virus Replication Cycle in Heterokaryons Excludes Dominant Restrictions in Human Non-liver and Mouse Liver Cell Lines
Hepatitis C virus (HCV) is hepatotropic and only infects humans and chimpanzees. Consequently, an immunocompetent small animal model is lacking. The restricted tropism of HCV likely reflects specific host factor requirements. We investigated if dominant restriction factors expressed in non-liver or non-human cell lines inhibit HCV propagation thus rendering these cells non-permissive. To this end we explored if HCV completes its replication cycle in heterokaryons between human liver cell lines and non-permissive cell lines from human non-liver or mouse liver origin. Despite functional viral pattern recognition pathways and responsiveness to interferon, virus production was observed in all fused cells and was only ablated when cells were treated with exogenous interferon. These results exclude that constitutive or virus-induced expression of dominant restriction factors prevents propagation of HCV in these cell types, which has important implications for HCV tissue and species tropism. In turn, these data strongly advocate transgenic approaches of crucial human HCV cofactors to establish an immunocompetent small animal model.
Published in the journal:
. PLoS Pathog 7(4): e32767. doi:10.1371/journal.ppat.1002029
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002029
Summary
Hepatitis C virus (HCV) is hepatotropic and only infects humans and chimpanzees. Consequently, an immunocompetent small animal model is lacking. The restricted tropism of HCV likely reflects specific host factor requirements. We investigated if dominant restriction factors expressed in non-liver or non-human cell lines inhibit HCV propagation thus rendering these cells non-permissive. To this end we explored if HCV completes its replication cycle in heterokaryons between human liver cell lines and non-permissive cell lines from human non-liver or mouse liver origin. Despite functional viral pattern recognition pathways and responsiveness to interferon, virus production was observed in all fused cells and was only ablated when cells were treated with exogenous interferon. These results exclude that constitutive or virus-induced expression of dominant restriction factors prevents propagation of HCV in these cell types, which has important implications for HCV tissue and species tropism. In turn, these data strongly advocate transgenic approaches of crucial human HCV cofactors to establish an immunocompetent small animal model.
Introduction
HCV is an enveloped virus that at present chronically infects about 130 million people worldwide [1]. It possesses a positive strand RNA genome of about 9.6 kb composed of non-translated regions (NTRs) at the 5′and 3′ termini required for translation and RNA replication and a single open reading frame encoding a large polyprotein [2]. One hallmark of HCV is its high degree of sequence variability which likely contributes to its ability to establish chronic infections. Different patient isolates are grouped into 6 major genotypes and more than 100 subtypes within the genus Hepacivirinae of the family Flaviviridae [3]. Persistent infection is associated with a variable degree of liver damage often progressing in severity over the course of decades. Accordingly, a large number of patients are at risk of severe sequelae including life threatening conditions like cirrhosis and hepatocellular carcinoma [4]. The best available treatment, a combination of polyethylene glycol (PEG)-conjugated interferon alpha (IFN-α) and ribavirin, is effective in only a fraction of patients and associated with severe side effects [5]. A prophylactic or therapeutic vaccine is not available.
HCV displays a distinct and narrow tissue and host species tropism and naturally infects only humans and chimpanzees. The latter therefore are the only reliable immunocompetent animal model. However, their use is limited due to ethical reasons, high costs and restricted availability [6]. Transgenic mice containing human liver xenografts have been described and are permissive for HCV infection [7], and summarized in [8]. However, these animals are immunodeficient and hence cannot be used to analyze HCV pathogenesis and immune control or for vaccine development [9].
The mechanisms underlying the restricted tropism of HCV are poorly characterized and likely reflect specific host factor requirements for viral entry, RNA replication, assembly and release. It has been shown that two of the four HCV receptors, namely CD81 and occludin (OCLN) are used in a highly species-specific manner and that the rodent orthologs limit viral entry [10], [11]. Furthermore, mouse scavenger receptor class B type I (SR-BI) and also mouse claudin-1 (CLDN1) were reported to support HCV infection with slightly lower efficiency compared to their human orthologs [12], [13]. Importantly, ectopic expression of all four human entry factors or a combination of mouse SR-BI and CLDN1 with human CD81 and OCLN rendered non-human cell lines permissive for HCV cell entry in tissue culture [10]. Moreover, very recently HCV was adapted in tissue culture to efficiently utilize mouse CD81 as receptor which permitted viral entry into mouse cells in the absence of human entry factors [14]. Together, these results indicate that transgenesis or viral adaptation may be sufficient to overcome the barrier to HCV cell entry also in vivo.
HCV RNA replication has been observed in human non-hepatic and murine cell lines. However, the efficiency was very low and required long-term selection procedures using HCV replicon constructs expressing dominant antibiotic-selectable markers [15], [16], [17], [18], [19]. Furthermore, assembly and release of new progeny viruses has so far only been described in cells of human liver origin and was not observed in mouse cells [20]. Presently, it is not clear if low replication efficiency and lack of virus production in these cell types are caused by the absence of crucial viral cofactors, by genetic incompatibility of essential host factors or alternatively by expression of dominant restriction factors interfering with replication and virus production in these cells.
Notably, a number of host-encoded restriction factors which protect mammalian cells from viral infections, particularly by retroviruses, have been recognized [21], [22]. Well characterized examples are APOBEC3G [23], TRIM5-α proteins [24], and tetherin [25] that inhibit human immunodeficiency virus 1 (HIV-1) propagation at various steps of its replication cycle. In case of HCV, EWI-2wint, a CD81-binding protein, impedes HCV entry by competing with the viral glycoproteins for interaction with CD81 in several non-hepatic cells [26]. Moreover, VAP-C which is highly expressed in various non-hepatic tissues but was not detected in human liver from chronically infected HCV patients negatively regulates HCV RNA replication thus possibly contributing to the hepatotropism of HCV [27].
Given these findings, we developed trans-complementation systems to investigate if constitutive or virus-induced expression of dominant restriction factors precludes HCV propagation in human non-hepatic tissues or non-human liver cells.
Results
Inefficient replication and virus production in human non-hepatic and mouse liver cell lines
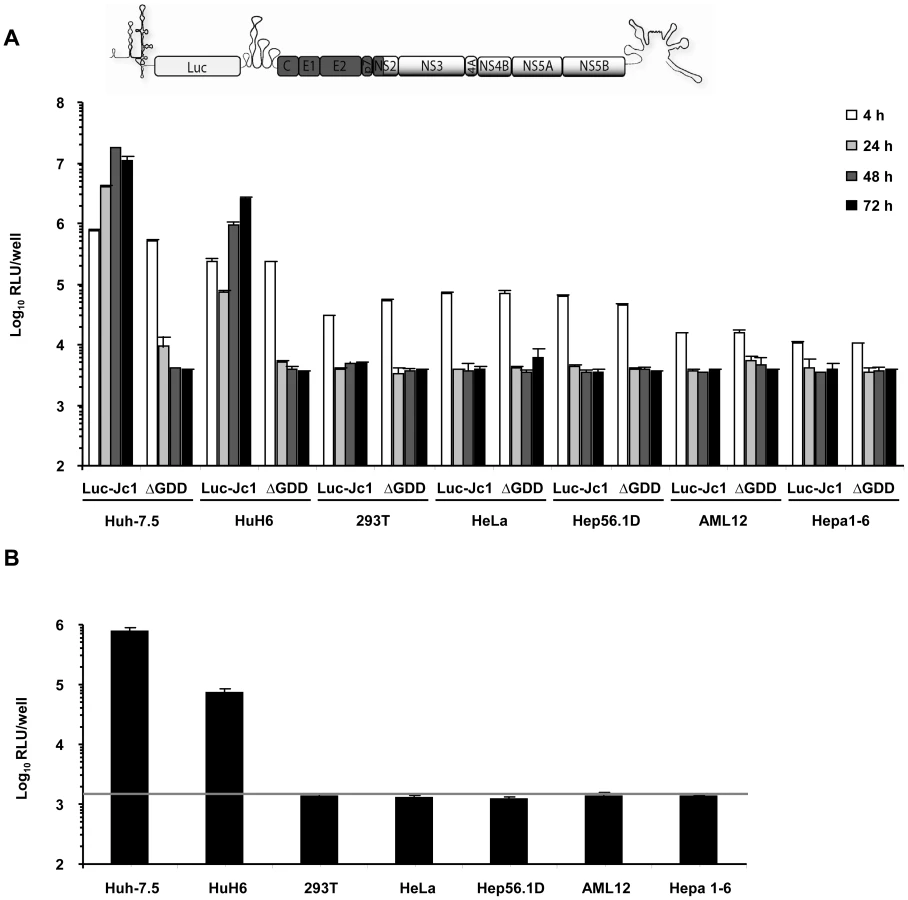
Given previous reports that HCV replication is inefficient in human non-liver and mouse liver cells and that virus production does not occur, we first quantitatively assessed the extent of HCV RNA replication and virus production in a number of cell lines of human non-liver origin and in mouse liver cell lines. To this end, we transfected 293T (human embryonic kidney), HeLa (human cervix carcinoma), and the mouse liver cell lines Hep56.1D, AML12, and Hepa1-6 with Luc-Jc1 RNA, a highly efficient genotype 2a chimeric genome containing a luciferase reporter gene [28]. Human liver cell lines Huh-7.5 [29] and HuH6 [30] were transfected as reference. Transient viral RNA replication was monitored by quantification of luciferase activity between 4 and 72 h post transfection (Figure 1A). A non-replicative mutant genome with a deletion of the conserved GDD motif within the NS5B RNA dependent RNA polymerase (ΔGDD) was used as control. In parallel we quantified production of infectious virions by inoculation of the highly permissive human hepatoma cell line Huh-7.5 with the culture fluid of the transfected cells and subsequent determination of luciferase activity (Figure 1B). When we transfected Huh-7.5 cells with Luc-Jc1, luciferase activity rose to more than 107 relative light units (RLU) per well at 48 h surpassing reporter activity measured in ΔGDD-transfected cells by more than three orders of magnitude. Similarly, transfection of HuH6 cell yielded high luciferase activity 72 h post transfection, thus confirming highly efficient RNA replication of this HCV genome in these human liver cell lines. In contrast, luciferase activity in transfected human non-liver and non-human cells was indistinguishable between replication-competent Luc-Jc1 and the ΔGDD mutant suggesting that RNA replication did not occur or was too inefficient for detection by this assay. Of note, reporter gene activity 4 h post transfection was lower in these latter cell lines compared to Huh-7.5 and HuH6 cells indicating that either transfection efficiency had been poor or that RNA translation was low in these cells. However, when delivering the viral RNA by lipofection rather than electroporation we reached comparable levels of luciferase activity between all cell lines and more than 1,000-fold above the background at 4 h post transfection. Nevertheless, at subsequent time points post transfection there was again no difference between Luc-Jc1 and ΔGDD (data not shown), thus excluding that merely low transfection or translation efficiency was responsible for absent or inefficient RNA replication. Release of infectious particles was only observed from Huh-7.5 and HuH6 cells transfected with Luc-Jc1, but not from the other cell lines tested (Figure 1B). Collectively, these results are in line with previous observations of a limited HCV RNA replication efficiency in human non-hepatic and mouse liver cell lines.
Efficient trans-complementation of HCV assembly and release in heterokaryons between human liver cell lines
To explore if one or more steps of the HCV life cycle are inhibited by expression of host factors that dominantly restrict HCV, we first developed a system to measure dominant restrictions of HCV assembly and release. To this end Huh-7.5 cells were transfected with an assembly-defective subgenomic JFH1 replicon carrying a luciferase reporter gene but lacking the essential viral assembly factors core, E1, E2, p7 and NS2. Subsequently, these cells were co-cultured with Huh-7.5 packaging cells constitutively expressing the aforementioned viral proteins [31]. After induction of cell fusion between these cell types by PEG, trans-complementation between replicon and constitutively expressed viral proteins should selectively restore virus production in heterokaryons resulting from fusion of replicon with packaging cells (Figure 2A). Heterokaryon formation was monitored by co-localization of NS5A and E2 proteins, expressed from the replicon and the packaging cell, respectively. Finally, virus production was quantified by collecting the culture fluid of the co-cultured cells 48 h after fusion induction and by inoculation of highly permissive Huh-7.5 cells and subsequent luciferase assays. In case of PBS-treated co-cultured cells we did not observe cells simultaneously expressing E2 and NS5A proteins indicating that this kind of treatment did not induce formation of heterokaryons (Figure 2B). In contrast, when co-cultured cells were treated with PEG we readily detected cells expressing both E2 and NS5A proteins thus indicating that cell fusion between the different cell types had occurred. Importantly, cell fusion between these cells efficiently restored virus production and resulted in transduction of luciferase activity to naïve Huh-7.5 cells inoculated with culture fluids collected from PEG-treated co-cultures of replicon and Huh-7.5[CE1][E2p7NS2] packaging cells (Figure 2C). Notably, PBS treatment of these co-cultures as well as PEG treatment of co-cultures between replicon and naïve Huh-7.5 cells did not permit transduction of luciferase activity. Together these results indicate that the trans-complementation assay described above permits rescue of HCV particle production in heterokaryons between human liver cells in a cell fusion-dependent fashion.

Production of infectious HCV from heterokaryons between human liver cell lines and human non-hepatic or mouse liver cell lines
Next, we applied this system to investigate if packaging cell lines from human non-hepatic or mouse liver cells would also rescue HCV particle production when fused with Huh-7.5 replicon cells. To this end we created several novel packaging cell lines by using two lentiviral vectors transducing core, E1 and E2, p7, NS2 proteins, respectively [31] (Table S1). Transgene expression was confirmed by western blot (data not shown) and by using a commercial core specific ELISA as well as an E2-specific ELISA for detection of representative proteins derived from the individual vectors (Figure 3A and 3B and Figure S1). Core protein expression was readily detected and was very similar between all individual packaging cell lines (Figure 3A). Similarly, E2 protein expression was well detectable (Figure 3B) with high E2 protein levels in HeLa and 293T, intermediate amounts in Huh-7.5 and Hep56.1D and comparatively low quantities in AML12, Hepa1-6 and HuH6 packaging cells. Importantly, after co-culture with Huh-7.5 cells transfected with the luciferase replicon and induction of heterokaryons by PEG all packaging cell lines tested efficiently rescued production of infectious HCV particles as is evident from transduction of high levels of luciferase activity to the inoculated naïve Huh-7.5 cells (Figure 3C). Since neither PBS treatment of these co-cultures, nor cell fusion between Huh-7.5 replicon cells and the naïve non-liver or non-human cell lines permitted transduction of luciferase, we concluded that virus production required cell fusion and expression of viral proteins core, E1, E2, p7 and NS2 in the packaging cell lines. Together, these data indicate that infectious HCV particles are produced upon fusion between human liver cells expressing a subgenomic HCV replicon with various human non-liver cells or mouse liver cells providing the remaining viral proteins in trans.

To firmly establish that infectious particles produced from these heterokaryons enter Huh-7.5 target cells in the same way as authentic HCV particles and that luciferase expression in the inoculated cells resulted from genuine infection rather than non-specific transfer of reporter gene activity, we investigated if the released particles infect target cells in a CD81-dependent manner. Huh-7.5 cells were inoculated with cell culture fluids harvested from heterokaryon-cultures in the presence of CD81-specific antibodies or control antibodies against CD13, an irrelevant protein expressed on the surface of Huh-7.5 target cells. As depicted in Figure 4, anti-CD81 antibodies reduced reporter activity in inoculated cells by more than 80% to almost background levels (Figure 4).

Taken together, these results show that formation of heterokaryons between Huh-7.5 replicon cells and human hepatic, human non-hepatic and mouse hepatoma packaging cell lines rescued virus production and that produced infectious particles utilize CD81 during infection. Therefore, these data indicate that HCV replication, assembly and release are not repressed in these non-permissive cell lines in a dominant negative fashion.
Notably, the efficiency of virus production between heterokaryons was substantially different (Figure 3C). This could be due to various reasons, including dissimilar expression levels of transgenes (HCV core to NS2 proteins), fusion efficiency, and abundance of replication co-factors or restriction factors. Regarding the latter, kinetic and extent of response to pathogen associated molecular patterns (PAMPs) and subsequent induction of antiviral mechanisms may substantially influence virus production. Therefore, to investigate why heterokaryons between human Huh-7.5 replicon cells and mouse AML12 packaging cells produced more than 20-fold lower infectious virus particles than heterokaryons between Huh-7.5 replicon cells and human HuH6 packaging cells or mouse Hep56.1D packaging cells (Figure 3C), we compared fusion efficiency and responsiveness to viral PAMPs between these packaging cell lines. The relative number of fused cells displaying both replicon-expressed NS5A and at the same time E2 from the lentiviral transduction was similar between these packaging cell lines (Figure S2). Therefore, we can exclude that dissimilar fusion was responsible for these marked differences in virus production. Responsiveness of packaging cell lines to viral PAMPs was measured by inoculation with a recombinant La Crosse virus lacking the non-structural protein NSs (rLACVdelNSs;[32]). Subsequently, mRNA levels of ISG56 and IFN-β were determined by quantitative RT-PCR as a measure of antiviral signaling. As expected, Huh-7.5 packaging cells which carry a defective RIG-I protein [33], that is essential for detection of viral PAMPs, did not upregulate ISG56 or IFN-β mRNA levels (Figure 5). Notably, also HuH6 packaging cells responded poorly and only marginally up-regulated ISG56 and IFN-βmRNA levels upon La Crosse virus challenge. In contrast both mouse packaging cell lines strongly and rapidly increased transcription of ISG56 and IFN-β thus indicating that they efficiently detect viral PAMPs. Given that Hep56.1D packaging cells fused with Huh-7.5 replicon cells readily produce infectious particles despite of vigorous responsiveness to viral PAMPS, these findings exclude that rapid and efficient recognition of viral PAMPs in mouse packaging cell lines precludes production of infectious virus particles in the heterokaryons.

Both human and mouse IFN-α inhibit virus production in human-mouse heterokaryons
HCV propagation in tissue culture is strongly inhibited by interferon, albeit by as far incompletely defined anti-viral mechanisms. To exclude that Hep56.1D packaging cells sustain virus production due to the inability to respond to IFN, we used mouse interferon alpha (IFN-α) to establish that these cells are not refractory to IFN and that our cell fusion assay efficiently detects dominant restriction factors – in this case IFN induced – that prevent efficient HCV virus production. First we confirmed that mouse IFN-α induces an antiviral state only in mouse cells and not in human cells incubated with recombinant mouse IFN-α. Since Hep56.1D cells cannot be infected with HCV, we chose an interferon-sensitive vesicular stomatitis reporter virus (Luc-VSV) which efficiently infects mouse cells to assess responsiveness of these cells to mouse and human IFN-α. To evaluate sensitivity of Huh-7.5 cells to IFNs of human or mouse origin we employed the HCV reporter virus Luc-Jc1. Importantly, infection of Huh-7.5 cells by Luc-Jc1 was only inhibited by human IFN but not affected by mouse IFN (Figure 6A). In turn infection of Hep56.1D cells by Luc-VSV was only inhibited by mouse IFN but not by human IFN (Figure 6B). Together these results confirm that both cell lines are responsive to IFNs and that induction of the antiviral state by IFN is species-specific in both cell lines. Next we explored if both mouse and human IFN induce restriction factors in Huh-7.5 replicon or Hep56.1D packaging cells which prevents virus production in fused heterokaryons. To this end, Huh-7.5 replicon or Hep56.1D packaging cells were pre-incubated with either human or mouse IFN-α, subsequently these cells were co-cultured and fused by PEG, before production of virus particles was measured by transduction of luciferase activity to naive Huh-7.5 cells (Figure 6C). In this context production of infectious HCV particles was strongly repressed by both mouse and human IFN-α suggesting that both mouse and human IFN-α induced antiviral effector mechanisms that efficiently control HCV particle production.

Finally, to explore the importance of the time point of IFN-treatment for virus production in the heterokaryon fusion assay between Hep56.1D packaging cells and Huh-7.5 replicon cells, we applied mouse IFN-α for 4 h at different time points before co-culture, during co-culture or after induction of cell fusion by PEG (Figure 6D). Interestingly, mouse IFN-α repressed virus production with comparable efficiency irrespective of the time point of addition arguing that the antiviral response induced via mouse IFN-α is relatively sustained thus suppressing virus production at least up to 80 h post treatment.
Completion of entire virus replication cycle in human-mouse heterokaryons
It is well established that a high burden of viruses can saturate and ultimately overcome endogenous host cell restrictions. To exclude that a high viral load in transfected Huh-7.5 cells may have precluded detection of restrictions present in human non-liver or mouse liver cells simply by saturation of such factors, we wished to confirm our findings using a second independent approach based on HCV infection rather than transfection. To this end we used a Huh-7-derived cell line designated Lunet N which lacks endogenous CD81 expression [34]. As a consequence, Lunet N cells cannot be infected by HCV unless human CD81 is reintroduced [14]. When these cells are fused with cells expressing human CD81 but not the complete set of viral entry factors, only fused cells display all necessary entry factors thus rendering only the heterokaryons susceptible. Following this rationale, we co-cultured Lunet N cells either with 293T or HeLa cells which both express human CD81 but which are non-permissive to HCV cell entry due to lack of at least one essential HCV entry factor [10], [35] (Table S2). In parallel we fused Lunet N cells with naïve Hep56.1D, AML12 or Hepa1-6 cells or derivates of these cell lines expressing human CD81. Prior to co-cultivation, cell lines were stained by two different fluorescent CellTrackers to allow discrimination of cell types and detection of successful cell fusion [36]. Formation of heterokaryons between cells was subsequently induced by a transient transfection of a plasmid encoding a fusogenic viral glycoprotein of the prototype foamy virus [37] (Figure 7A). This method of cell fusion induction was chosen to increase sensitivity of the assay due to increased fusion rates. Fluorescence microscopy revealed that the expression of the highly fusogenic glycoprotein led to the accumulation of syncytia stained with both CellTrackers (Figure 7B). Thirty hours after transfection of the fusogenic envelope protein into the co-cultured cells, the cell population was inoculated with the HCV Jc1 chimera (MOI 2.3) which grows to high virus titers in tissue culture [38]. Completion of the viral replication cycle including productive entry, RNA replication as well as assembly and release of infectious progeny virus particles was measured by titration of cell free infectivity released from these cells 48 h post inoculation. To rule out that residual infectivity of the inoculum or low numbers of directly infected susceptible Lunet N cells are solely responsible for the detection of infectious virus at this time point, we included Lunet N cells fused with Lunet N cells as control. Importantly, in this control we observed only very low levels of infectious HCV ranging between 5 and 10 TCID50/mL which is close to the detection limit of the limiting dilution assay. In contrast, when Lunet N cells were fused with 293T cells and challenged with the same viral inoculum, viral titers as high as 4×103 were detected providing strong evidence that in this case relatively high numbers of progeny infectious particles had been produced (Figure 7C). Similarly, when using HeLa cells for heterokaryon formation with Lunet N cells, also substantial numbers of infectious particles were produced, reaching levels approximately 10-fold higher than the background infectivity observed with Lunet N cells. Importantly, HCV inoculation of Lunet N cells fused with mouse liver cells expressing human CD81 also resulted in production of well detectable quantities of infectious HCV about 5 - to 20-fold higher than after fusion of naïve mouse liver cells with Lunet N cells (Figure 7C). In conclusion these results indicate that fusion of Lunet N cells with cells expressing human CD81 complements the lacking surface receptor, thus allows HCV cell entry and de novo production of infectious particles from the resulting heterokaryons. Importantly, completion of the HCV replication cycle was observed when human liver cells (Lunet N cells) were fused with human non-liver cells (293T, HeLa) and three different mouse liver cell lines. It is worth mentioning here that all three mouse liver cell lines express functional viral pattern recognition pathways as is evident from efficient induction of IFN-β and ISG56 gene transcription as well as IFN secretion upon stimulation with viral RNA, poly(I:C) or upon infection with a Bunyavirus mutant (Figure S3). Together these findings provide strong evidence that these cell types do not express dominant restriction factors precluding HCV entry, RNA replication, virus assembly and release and that the endogenous pattern recognition pathways are insufficient to fully control HCV propagation.

Discussion
HCV is a highly tissue - and species-specific virus which efficiently replicates only in human and chimpanzee liver cells. Three principal mechanisms may prevent HCV from infecting other species. These are lack of essential host cell factors, their genetic incompatibility, or alternatively the expression of dominant restriction factors. Previous reports have highlighted that indeed inefficient usage of HCV entry factors from mouse origin precludes infection of mouse cells [10], [11], [12], [13]. Moreover, ectopic expression of a liver-specific miRNA (miR-122) which is important for efficient HCV RNA replication [39] has been shown to facilitate HCV replication in human non-hepatic cell lines or mouse fibroblasts [40], [41]. Finally, it was noted that HCV cell entry is inhibited by expression of a CD81-binding protein in non-liver cells [26]. However, comprehensive studies investigating dominant cellular restriction factors which may limit HCV replication have been lacking.
In case of retroviruses, most notably HIV-1, it is well established that host factors can potently repress completion of the retroviral replication cycle: For instance pro-virus formation of HIV-1 is impeded by APOBEC3G [23], [42], TRIM5-α inhibits HIV-1 by acting before reverse transcription [24], and tetherin prevents release of virions from infected cells [25], [43]. Notably, HIV-1 encodes viral factors like Vif and Vpu that efficiently suppress these innate host cell defense mechanisms by inducing degradation of APOBEC3G and tetherin, respectively [25], [44], [45]. Importantly, viral interference with these factors is species-specific, since for instance Vpu fails to protect HIV-1 from non-human tetherin [45], [46] and as HIV-1 Vif does not interfere with murine APOBEC3G [47], [48]. Consequently, the interaction of HIV-1 with restriction factors plays a key role in determining species tropism (summarized in [49]). Notably, also the hepatitis B virus is targeted by APOBEC3G [50] and tetherin prevents release of diverse enveloped viruses from divergent virus families including retroviruses, arenaviruses, filoviruses and herpesviruses [51], [52], [53], [54].
Given these circumstances we investigated if restriction factors limit HCV propagation in non-permissive cell lines by using somatic cell fusion between permissive and non-permissive cells. Employing this approach we provide strong evidence that HCV efficiently completes its replication cycle in heterokaryons between human liver cells and human non-hepatic or mouse liver cell lines. In turn, these results suggest that HCV propagation in these cell types, and possibly also in primary human non-liver tissue or in mouse liver, is not ablated by constitutive or virus-induced expression of restriction factors. This conclusion is based on two independent experimental systems employing in total five cell lines. We ensured that virus production can only occur in true heterokaryons between human liver cells and non-permissive cell lines by choosing two different trans-complementation systems. On one hand, virus production of a subgenomic HCV replicon transfected into highly permissive Huh-7.5 cells was complemented by fusion of these cells with packaging cells constitutively expressing the lacking viral assembly factors. Alternatively, we took advantage of a Huh-7-derived cell clone lacking endogenous CD81 expression and fusion with cell lines expressing human CD81. When challenged with HCV, only heterokaryons between these cell types express all necessary factors rendering only these permissive for viral entry.
Notably, in both systems productive complementation of the viral replication cycle likely depends on a number of factors including transgene expression, efficiency of cell fusion as well as abundance of essential (in part unknown) viral co-factors and viral restriction factors. In part these factors cannot be quantified; therefore it is not possible to establish at this point why virus production efficiency differs between heterokaryons. Notably, we observed much lower virus production in heterokaryons between Huh-7.5 replicon cells and AML12 packaging cells compared to heterokaryons between Huh-7.5 replicon cells and HuH6, or Hep56.1D packaging cells (Figure 3C). Since the fusion efficiency was similar between these cells, we can exclude that this accounts for these dissimilarities in virus production. Moreover, we can rule out that efficient detection of viral PAMPs precludes virus production in these heterokaryons as for instance Hep56.1D cells readily respond to viral triggers of antiviral signaling (Figure 5) but nevertheless produce comparatively high levels of infectious particles in the heterokaryon assay (Figure 3C). It is worth mentioning though that virus production in heterokaryons involving either Huh-7.5 packaging cells or HuH6 packaging cells was consistently 3 to 4 fold more efficient than in those with Hep56.1D packaging cells. Since Hep56.1D packaging cells express at least as much viral transgenes and fuse similarly well as the human liver Huh-7.5 - and HuH6-derived packaging cells this difference could be due to either lower abundance of important viral cofactors in the mouse liver cell line or due to partial control of virus production by cellular antiviral defenses in Hep56.1D packaging cells. In any case, our data firmly exclude that dominant restriction factors fully ablate HCV propagation in these cell types. In turn, it is reasonable to assume that lack of essential human co-factors is a primary limitation for efficient HCV propagation in mouse cells. Such species-specific host factors could for instance be identified by introduction of human cDNA libraries into mouse cells and subsequent screening for those cDNAs which confer increased HCV replication capacity to mouse cells.
Our results also provide first evidence that virus-induced antiviral programs in mouse cells may be insufficient to fully control HCV infection and replication: It is unlikely that high viral load, as present after transfection, simply overrides endogenous antiviral pathways since we observed completion of the HCV replication cycle not only upon fusion of HCV transfected cells carrying a high dose of replication-competent viral RNA, but also after infection with HCV. Second, we can exclude that the mouse cells are unable to sense viral infection due to lesions in the pathways triggering interferons since ligands of MDA5 [poly(I:C)] and RIG-I (5′triphosphorylated VSV RNA), as well as Bunyavirus infection efficiently elicited ISG56 and IFN-β gene expression as well as IFN secretion. Finally, we can also rule out that the cells tested by us are non-responsive to IFN, since stimulation of antiviral effectors in Hep56.1D mouse cells via mouse IFN-α strongly inhibited Luc-VSV infection or HCV particle production in mouse-human heterokaryons. Importantly, treatment of either Huh-7.5 replicon cells with human IFN-α or likewise Hep56.1D packaging cell lines with mouse IFN-α at different time points prior to co-culture efficiently reduced virus production in the heterokaryons (Figure 6). These results suggest that not only human factors induced by human IFN but also mouse factors stimulated by mouse IFN are able to control HCV particle production. However, in the context of mouse-human heterokaryons these IFN-dependent antiviral effectors are either not sufficiently activated by HCV or not capable to ablate production of infectious particles upon HCV infection for instance due to incompatibility of mouse antiviral effectors for control of HCV infection/replication. Notably, HCV naturally induces cellular innate immunity through RNA composition-dependent activation of RIG-I [55], [56] and subsequent signaling via IRF-3. However, at least in human liver cells HCV interferes with RIG-I dependent signaling to IRF-3 by NS3-4A-dependent cleavage of the crucial adaptor protein Cardif [55]. Likewise, TLR-3 dependent signaling to IRF-3 is ablated by cleavage of Trif through the viral NS3-4A protease [57]. Since infection of mouse-human heterokaryons with HCV particles apparently did not induce innate defenses to an extent sufficient to control HCV propagation, we speculate that HCV's ability to interfere with the induction of innate immunity may be sufficient to prevent adequate recognition and clearance of virus also by mouse innate immune factors. In this regard it is worth mentioning that Ahlén et al. recently reported that the HCV NS3-4A protease is capable of cleaving mouse RIG-I which may be an essential prerequisite for (partial) viral evasion of innate immune control in mouse liver cells [58]. On the other hand, Lin et al. observed that poor replication of HCV in murine embryonic fibroblasts was facilitated by deletion of interferon regulatory factor 3 (IRF-3) suggesting that IRF-3-dependent mechanisms may partially restrict HCV replication in mouse cells [41]. Clearly more work is needed to fully define the interplay between HCV and mouse innate immunity and to appreciate by which extent it contributes to low replication of HCV in these cells.
Despite successful development of cell culture systems to study the complete HCV replication cycle [59], [60], [61], analysis of mechanisms of virus pathogenesis and immune control as well as vaccine development are severely hampered by the lack of immunocompetent small animal models. The results presented in this study provide strong evidence that dominant restriction factors are unlikely to preclude propagation of HCV in mouse liver cells. This raises the hope that permissive small animal models could be developed without manipulation of innate, intrinsic or adaptive immunity factors. Adaptation of HCV to mouse cells or transgenic expressions of human viral co-factors are promising approaches to ultimately establish urgently needed small animal models for HCV.
Materials and Methods
Plasmids
The plasmids pFK-Jc1, pFK-Luc-Jc1, pFKi389Luc-EI/NS3-3′_JFH1_dg have been described earlier [28], [31], [38], The construct Luc-Jc1ΔGDD has an in-frame deletion of 10 amino acids (MLVCGDDLVV) encompassing the GDD motif of NS5B [62]. Lentiviral plasmids pWPI-CE1-BSD, pWPI-E2p7NS2-BSD have been described previously [31]. pWPI-CE1-GUN was created by restriction digest of pWPI-CE1-BSD with BamHI and SpeI and transfer of the insert in the digested vector pWPI-GUN [14]. The plasmids pczHFVenvEM066 and pWPIhCD81BLR have been described previously [14], [63]. Exact cloning strategies are available on request. IFN reporter plasmid Mx1-Luc was described elsewhere.
A recombinant, glycoprotein G gene-deleted VSV replicon expressing firefly luciferase, VSV*ΔG(Luc) has been generated by reverse genetics according to published protocols [64]. A detailed characterization of this vector will be published elsewhere.
Cell culture
Huh-7.5, HuH6, 293T, HeLa, Hep56.1D, and Hepa1-6 cell lines were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Karlsruhe, Germany) supplemented with 2 mM L-glutamine, non-essential amino-acids, 100 U/mL penicillin, 100 µg/mL streptomycin and 10% fetal calf serum (DMEM complete). AML12 cells were maintained in DMEM/Ham's F-12 medium supplemented with 10% FCS, 40 ng/mL dexamethasone, 5 µg/mL insulin, 5 µg/mL transferrin, 5 ng/mL selenium, 100 U/mL penicillin and 100 µg/mL streptomycin as described before [65].
Cell lines expressing viral proteins C, E1, and E2, p7, and NS2 from two independent genetic cassettes were created by lentiviral gene transfer as described previously [31]. Lentiviral particles were produced using the plasmids pCMV-ΔR8.74 [66], pcz-VSV-G [67], and derivatives of pWPI, (encoding the genes of interest and in addition either the blasticidin S deaminase of Aspergillus terreus or a GFP-ubiquitin-neomycin fusion protein (GUN), conferring resistance against blasticidin or G418, respectively [14]. Parental cell lines were transduced and selected in the presence of either blasticidin (Invivo Gen, San Diego, USA) alone (2.5 µg/mL or 5 µg/mL), or together with geneticin (G418; 750 µg/mL, Life Technologies), as indicated in Table S1. Expression of viral proteins was confirmed by Western blot analysis using α-E2 (AP33), α-core (C7-50) and α-mouse-peroxidase antibodies as reported elsewhere [31]. Murine cell lines expressing human CD81 were created by lentiviral gene transfer as described above using a pWPI derivative [14] and selected using blasticidin. Expression of CD81 was confirmed by FACS analysis using α-CD81 (1.3.3.22, Ancell) and goat α-mouse antibody conjugated to APC (eBioscience, data not shown).
HCV replication and infection assays
HCV particles and firefly luciferase HCV reporter viruses were generated as reported previously [28]. In brief, plasmid DNA was linearized and transcribed into RNA, which was then electroporated into Huh-7.5 cells. Virus-containing culture fluids of transfected cells were harvested 48 h and 72 h after transfection. Luciferase reporter virus infection assays were carried out and analyzed as described [28]. Wildtype HCV particles were titrated by using a limiting dilution assay [31]. The 50% tissue culture infectious dose (TCID50) was calculated based on the methods described by Spearman and Kärber [68], [69].
Polyethylene glycol-mediated fusion and infectivity assay
Formation of heterokaryons between permissive and non-permissive cell lines was induced by PEG. In detail, 10 µg in-vitro RNA transcript of the luciferase reporter HCV replicon Luc-NS3-5B was transfected into naïve Huh-7.5 cells (6×106 cells) by electroporation; the cells were resuspended in 10 mL DMEM and seeded onto a 10 cm dish. The next day cells were washed with PBS and 5×105 cells were co-cultured with 5×105 Huh-7.5 naïve or packaging cells (cell numbers adjusted to growth efficiency) on a 6-well culture plate. After 24 h of incubation, at a cell density of ∼80%, fusion was induced by treating co-cultivated cells with 40% PEG-1500 in PBS (Roche, Mannheim, Germany) or PBS as a control for 5 min at 37°C, followed by extensive washing, addition of 2 mL DMEM/well and 48 h of incubation. Production of infectious trans-complemented particles was determined as described [31]. In case of heterokaryons formed with AML12 and Hepa1-6 naïve and the respective packaging cells, assay sensitivity was enhanced by concentration of the supernatants from 6-well plates by 20-fold using Amicon ultrafiltration devices (Millipore) according to the manufacturer's protocol.
Induction of antiviral responses by Interferon treatment
To test if IFN-α treatment induces cellular restrictions, Huh-7.5 replicon and Hep56.1D packaging cells were pre-incubated with either human IFN-α-2b (0–100 U/mL, IntronA, Essex Pharma, München, Germany) or mouse IFN-α-1 (0–100 U/mL, PBL InterferonSource, Piscataway, USA) for 4 h prior to co-cultivation. For the time kinetic Hep56.1D packaging cells were pre-incubated with mouse IFN-α-1 (100 U/mL) at indicated time points prior to co-cultivation, 4 h prior to PEG-mediated cell fusion, or 4 h post fusion.
Fluorescence microscopy
Staining of NS5A was performed by using the 9E10 hybridoma supernatant [59] at a dilution of 1∶2000. Immunostaining of E2 was performed using an E2-specific human monoclonal antibody CBH-23 (kindly provided by Steven Foung, unpublished) at a dilution of 1∶250 in PBS supplemented with 5% normal goat serum. After three times washing with PBS, bound primary antibodies were visualized using goat anti-human or goat anti-mouse IgG–specific secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 546 (Invitrogen, Karlsruhe, Germany) at a dilution of 1∶1,000. DNA was stained with DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride; Invitrogen) for 1 min. Finally, cells were washed three times with PBS, once with water and mounted on glass slides with Fluoromount G (Southern Biotechnology Associates, Birmingham, USA).
Staining of cells using CellTracker (CellTracker Green CMFDA, Cell Tracker Orange CMTMR, Invitrogen) was performed as recommended by the manufacturer. Briefly, cells were incubated with 5 µM or 10 µM of CellTracker Green or Orange, respectively in serum-free media. Medium was changed after 45 minutes and cells were incubated in standard DMEM complete medium for 6 hours.
Determination of core and E2 expression
Viral transgene expression of the two lentiviral vectors encoding HCV core, E1 and E2, p7, NS2, respectively was determined by quantitative measurement of core and E2 proteins by enzyme-linked immunoassay (ELISA). Packaging cells (4.5×106) were lysed in 1.5 mL of PBS supplemented with 1% Triton X-100 and core protein abundance was determined using the ARCHITECT HCV Core AG Test (Abbott, Wiesbaden, Germany) according to the instructions of the manufacturer. For detection of HCV E2, packaging cells (4.5×106) were lysed in 1.5 mL Ripa buffer (0.3 M NaCl, 20 mM Tris-HCl (pH 8), 1% sodium desoxycholate, 0.1% sodium dodecyl sulphate, 1% Triton X-100 and protease inhibitors (Roche, Mannheim). Total protein content of cell lysates was determined by a standard Bradford assay. Subsequently equal quantities of cell lysates were analyzed as described elsewhere [70]. In brief, Maxisorb mircrotiter plates (Nunc, Langenselbold, Germany) were coated with 500 ng/well Galanthus nivalis (GNA, Sigma-Aldrich, Steinheim, Germany) and blocked in blocking buffer consisting of 2.5% non-fat dry milk, 2.5% normal goat serum, 20 mM Tris-HCl (pH 7.5), 150 mM NaCl and 0.1% Tween-20. Cell lysates were bound on the GNA coated plates prior to incubation with the primary antibody (AP-33 [71]) at concentrations of 10 µg/well. After extensive washing with TBST (20 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.1% Tween-20) the bound primary antibodies were detected using an AP-conjugated secondary antibody and 4-nitrophenyl phosphate disodium salt hexahydrate (Sigma-Aldrich, Steinheim, Germany). Absorbance was measured at 405 nm and 570 nm using a BioTek Synergy 2 plate reader (BioTek, Winooski, Vermont, USA) and the Gen5 1.08 software (BioTek).
To directly compare E2 expression between individual packaging cell lines, we performed a linear regression analysis of the relation between the Log OD value and the Log of the reciprocal dilution of the cell lysate. Based on this analysis we calculated the reciprocal dilution of the cell lysate required for an OD value of 0.5 in the ELISA assay, which reflects the relative expression of E2 between the given cell lysates.
CD81-mediated neutralization of HCV infection
Infectivity of HCV particles produced from heterokaryons was inhibited with monoclonal antibodies against CD81. Huh-7.5 cells were pre-incubated with either α-CD81 specific antibodies (JS-81, Becton-Dickinson, Heidelberg, Germany) or α-CD13 antibodies (Becton-Dickinson, Heidelberg, Germany) as a negative control at a concentration of 5 µg/mL in DMEM for 30 min prior to infection as previously described [28]. Luciferase activity was determined 72 h post inoculation as described above.
Virus replication und influence of human or mouse IFN-α
Huh-7.5 cells or Hep56.1D cells were seeded onto a 12-well plate. The next day cells were pre-incubated with either recombinant human (0–100 U/mL, hIFN-α-2b, IntronA, Essex Pharma, München, Germany) or mouse IFN-α (0–100 U/mL, mIFN-α-1, PBL Interferon Source, Piscataway, USA) for 4 h prior to infection with Luc-Jc1 or Luc-VSV, respectively. Four hours later medium was changed and IFN-α treatment resumed. Replication was determined 72 h post-infection by analyzing luciferase reporter activity in infected cells as described above.
Heterokaryon formation induced by foamy virus glycoprotein expression and virus titration
Huh-7 Lunet N cells lacking CD81 [34] were co-cultured with the indicated naive cell lines or the transduced cell lines expressing human CD81 at a cell density of 1.5×105 cells in a 12-well culture plate. The ratio of the different cell lines to Huh-7 Lunet N cells was adjusted according to the respective growth efficiency and ranged between 1∶1 and 1∶2. Fusion between these cells was initiated the next day by transfection of the co-cultures with a highly fusogenic variant of the prototype foamy virus envelope protein pczHFVenvEM066 [63] using lipofectamine 2000 (Invitrogen). Cells were challenged 30 hours later with infectious HCV particles produced in cell culture at a multiplicity of infection (MOI) of 2.3. To remove any unbound HCV particles left from inoculation, cells were washed with PBS 12 hours after inoculation. Cell entry, RNA replication and virus production was quantified by determining infectivity titers (TCID50/mL) in the culture fluids of these cells 48 h post inoculation as described above.
Assessment of functional viral pattern recognition pathways in mouse liver cells
Given mouse liver cell lines were mock treated or transfected with 500 ng 5′triphosphorylated genomic RNA of VSV or with 10 µg poly(I:C) per well of a 6-well plate using Metafectene (Biontex, Martinsried, Germany) according to the instructions of the manufacturer. Alternatively, cells were challenged with recombinant La Crosse virus lacking the non-structural protein NSs (rLACVdelNSs;[32]) at a MOI equal to 5. Cells were incubated for 16 h at 37°C and harvested. Total cellular RNA was extracted using the RNeasy Mini kit (Qiagen, Hilden, Germany) according to the instruction of the manufacturer. Transcript levels of mouse ISG56 and IFN-β were analyzed using the QuantiTect SYBR Green PCR Kit in combination with QuantiTect Primer Assay (Qiagen) and the following primers: Qiagen Mm_Ifit1_1_SG = QT01161286 (mouse ISG56), Qiagen Mm_Ifnb1_1_SG = GT00249662 (mouse IFN-β). In total 100 ng of cDNA was used for real time PCR in a final reaction volume of 25 µL using an initial 15 minute denaturation at 95°C and 40 cycles consisting of 15 seconds denaturation at 94°C, 30 seconds annealing at 55°C and 30 seconds extension at 72°C. Signals of inducible mRNAs were normalized to the GAPDH mRNA (mouse GAPDH: Qiagen Mm_Gapdh_3_SG = GT01658692) using the ΔΔCt method [72]. Values of mock treated cells were set to 1. Secretion of IFN was monitored using a luciferase-based reporter gene assay. Briefly, supernatants were treated with benzonase for 2 h at 37°C to destroy inducer RNA. Alternatively supernatant was incubated with β-propiolactone (Acros Organics, Geel, Belgium) at 4°C over night followed by 2 h at 37°C for inactivation of the rLACdelNSs inducer virus. These inducer-cleared supernatants were incubated for 16 h with murine embryonic fibroblasts that had been transfected with 250 ng Mx1-Luc [73] and 25 ng pRL SV40 (Promega, Mannheim, Germany) reporter plasmids using Nanofectin (PAA, Cölbe, Germany). Cells were lysed and luciferase activity was determined using the Promega Dual Luciferase assay according to the instruction of the manufacturer. Recombinant, pan-specific IFN-α (IFN-α B/D BgIII, PBL Biomedical Laboratories, Piscataway, USA) served as standard.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AlterMJ 2007 Epidemiology of hepatitis C virus infection. World J Gastroenterol 13 2436 2441
2. BartenschlagerRFreseMPietschmannT 2004 Novel insights into hepatitis C virus replication and persistence. Adv Virus Res 63 71 180
3. SimmondsPBukhJCombetCDeleageGEnomotoN 2005 Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42 962 973
4. HoofnagleJH 1997 Hepatitis C: the clinical spectrum of disease. Hepatology 26 15S 20S
5. MannsMPWedemeyerHCornbergM 2006 Treating viral hepatitis C: efficacy, side effects, and complications. Gut 55 1350 1359
6. BukhJ 2004 A critical role for the chimpanzee model in the study of hepatitis C. Hepatology 39 1469 1475
7. MercerDFSchillerDEElliottJFDouglasDNHaoC 2001 Hepatitis C virus replication in mice with chimeric human livers. Nat Med 7 927 933
8. MeulemanPLeroux-RoelsG 2008 The human liver-uPA-SCID mouse: a model for the evaluation of antiviral compounds against HBV and HCV. Antiviral Res 80 231 238
9. PlossARiceCM 2009 Towards a small animal model for hepatitis C. EMBO Rep 10 1220 1227
10. PlossAEvansMJGaysinskayaVAPanisMYouH 2009 Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457 882 886
11. FlintMvon HahnTZhangJFarquharMJonesCT 2006 Diverse CD81 proteins support hepatitis C virus infection. J Virol 80 11331 11342
12. CataneseMTAnsuiniHGrazianiRHubyTMoreauM 2010 Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol 84 34 43
13. HaidSWindischMPBartenschlagerRPietschmannT 2010 Mouse-specific residues of claudin-1 limit hepatitis C virus genotype 2a infection in a human hepatocyte cell line. J Virol 84 964 975
14. BitzegeioJBankwitzDHuegingKHaidSBrohmC 2010 Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog 6 e1000978
15. ZhuQGuoJTSeegerC 2003 Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J Virol 77 9204 9210
16. KatoTDateTMiyamotoMZhaoZMizokamiM 2005 Nonhepatic cell lines HeLa and 293 support efficient replication of the hepatitis C virus genotype 2a subgenomic replicon. J Virol 79 592 596
17. AliSPellerinCLamarreDKukoljG 2004 Hepatitis C virus subgenomic replicons in the human embryonic kidney 293 cell line. J Virol 78 491 501
18. DateTKatoTMiyamotoMZhaoZYasuiK 2004 Genotype 2a hepatitis C virus subgenomic replicon can replicate in HepG2 and IMY-N9 cells. J Biol Chem 279 22371 22376
19. ChangKSCaiZZhangCSenGCWilliamsBR 2006 Replication of hepatitis C virus (HCV) RNA in mouse embryonic fibroblasts: protein kinase R (PKR)-dependent and PKR-independent mechanisms for controlling HCV RNA replication and mediating interferon activities. J Virol 80 7364 7374
20. UprichardSLChungJChisariFVWakitaT 2006 Replication of a hepatitis C virus replicon clone in mouse cells. Virol J 3 89
21. StrebelKLubanJJeangKT 2009 Human cellular restriction factors that target HIV-1 replication. BMC Med 7 48
22. GoffSP 2004 Retrovirus restriction factors. Mol Cell 16 849 859
23. SheehyAMGaddisNCChoiJDMalimMH 2002 Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418 646 650
24. StremlauMOwensCMPerronMJKiesslingMAutissierP 2004 The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427 848 853
25. NeilSJZangTBieniaszPD 2008 Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451 425 430
26. Rocha-PeruginiVMontpellierCDelgrangeDWychowskiCHelleF 2008 The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One 3 e1866
27. KukiharaHMoriishiKTaguwaSTaniHAbeT 2009 Human VAP-C negatively regulates hepatitis C virus propagation. J Virol 83 7959 7969
28. KoutsoudakisGKaulASteinmannEKallisSLohmannV 2006 Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80 5308 5320
29. BlightKJMcKeatingJARiceCM 2002 Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76 13001 13014
30. WindischMPFreseMKaulATripplerMLohmannV 2005 Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J Virol 79 13778 13793
31. SteinmannEBrohmCKallisSBartenschlagerRPietschmannT 2008 Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J Virol 82 7034 7046
32. BlakqoriGDelhayeSHabjanMBlairCDSanchez-VargasI 2007 La Crosse bunyavirus nonstructural protein NSs serves to suppress the type I interferon system of mammalian hosts. J Virol 81 4991 4999
33. SumpterRJrLooYMFoyELiKYoneyamaM 2005 Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79 2689 2699
34. WitteveldtJEvansMJBitzegeioJKoutsoudakisGOwsiankaAM 2009 CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J Gen Virol 90 48 58
35. EvansMJvon HahnTTscherneDMSyderAJPanisM 2007 Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446 801 805
36. SkelleyAMKirakOSuhHJaenischRVoldmanJ 2009 Microfluidic control of cell pairing and fusion. Nat Methods 6 147 152
37. LindemannDGoepfertPA 2003 The foamy virus envelope glycoproteins. Curr Top Microbiol Immunol 277 111 129
38. PietschmannTKaulAKoutsoudakisGShavinskayaAKallisS 2006 Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103 7408 7413
39. JoplingCLYiMLancasterAMLemonSMSarnowP 2005 Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309 1577 1581
40. ChangJGuoJTJiangDGuoHTaylorJM 2008 Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J Virol 82 8215 8223
41. LinLTNoyceRSPhamTNWilsonJASissonGR 2010 Replication of subgenomic hepatitis C virus replicons in mouse fibroblasts is facilitated by deletion of interferon regulatory factor 3 and expression of liver-specific microRNA 122. J Virol 84 9170 9180
42. FriewYNBoykoVHuWSPathakVK 2009 Intracellular interactions between APOBEC3G, RNA, and HIV-1 Gag: APOBEC3G multimerization is dependent on its association with RNA. Retrovirology 6 56
43. VarthakaviVSmithRMBourSPStrebelKSpearmanP 2003 Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc Natl Acad Sci U S A 100 15154 15159
44. YuXYuYLiuBLuoKKongW 2003 Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302 1056 1060
45. GoffinetCAllespachIHomannSTervoHMHabermannA 2009 HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5 285 297
46. McNattMWZangTHatziioannouTBartlettMFofanaIB 2009 Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog 5 e1000300
47. MarianiRChenDSchrofelbauerBNavarroFKonigR 2003 Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114 21 31
48. BishopKNHolmesRKSheehyAMMalimMH 2004 APOBEC-mediated editing of viral RNA. Science 305 645
49. KirchhoffF 2010 Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8 55 67
50. TurelliPMangeatBJostSVianinSTronoD 2004 Inhibition of hepatitis B virus replication by APOBEC3G. Science 303 1829
51. JouvenetNNeilSJZhadinaMZangTKratovacZ 2009 Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol 83 1837 1844
52. KaletskyRLFrancicaJRAgrawal-GamseCBatesP 2009 Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A 106 2886 2891
53. SakumaTNodaTUrataSKawaokaYYasudaJ 2009 Inhibition of Lassa and Marburg virus production by tetherin. J Virol 83 2382 2385
54. PardieuCViganRWilsonSJCalviAZangT 2010 The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog 6 e1000843
55. MeylanECurranJHofmannKMoradpourDBinderM 2005 Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437 1167 1172
56. SaitoTOwenDMJiangFMarcotrigianoJGaleMJr 2008 Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454 523 527
57. LiKFoyEFerreonJCNakamuraMFerreonAC 2005 Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A 102 2992 2997
58. AhlenGDerkEWeilandMJiaoJRahbinN 2009 Cleavage of the IPS-1/Cardif/MAVS/VISA does not inhibit T cell-mediated elimination of hepatitis C virus non-structural 3/4A-expressing hepatocytes. Gut 58 560 569
59. LindenbachBDEvansMJSyderAJWolkBTellinghuisenTL 2005 Complete replication of hepatitis C virus in cell culture. Science 309 623 626
60. ZhongJGastaminzaPChengGKapadiaSKatoT 2005 Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102 9294 9299
61. WakitaTPietschmannTKatoTDateTMiyamotoM 2005 Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11 791 796
62. BurgelBFrieslandMKochAMannsMPWedemeyerH 2010 Hepatitis C virus enters human peripheral neuroblastoma cells - evidence for extra-hepatic cells sustaining hepatitis C virus penetration. J Viral Hepat doi:10.1111/j.1365-2893.2010.01339.x
63. LindemannDPietschmannTPicard-MaureauMBergAHeinkeleinM 2001 A particle-associated glycoprotein signal peptide essential for virus maturation and infectivity. J Virol 75 5762 5771
64. HanikaALarischBSteinmannESchwegmann-WesselsCHerrlerG 2005 Use of influenza C virus glycoprotein HEF for generation of vesicular stomatitis virus pseudotypes. J Gen Virol 86 1455 1465
65. SahaiAMalladiPMelin-AldanaHGreenRMWhitingtonPF 2004 Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am J Physiol Gastrointest Liver Physiol 287 G264 273
66. DullTZuffereyRKellyMMandelRJNguyenM 1998 A third-generation lentivirus vector with a conditional packaging system. J Virol 72 8463 8471
67. KalajzicIStoverMLLiuPKalajzicZRoweDW 2001 Use of VSV-G pseudotyped retroviral vectors to target murine osteoprogenitor cells. Virology 284 37 45
68. SpearmanC 1908 The method of “right and wrong cases” (“constant stimuli”) without Gauss's formulae. British Journal of Psychology 2 227 242
69. KärberG 1931 Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Archiv für experimentelle Pathologie und Pharmakologie 162 480 487
70. KeckZYLiSHXiaJvon HahnTBalfeP 2009 Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J Virol 83 6149 6160
71. OwsiankaAClaytonRFLoomis-PriceLDMcKeatingJAPatelAH 2001 Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J Gen Virol 82 1877 1883
72. LivakKJSchmittgenTD 2001 Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 402 408
73. JornsCHolzingerDThimmeRSpangenbergHCWeidmannM 2006 Rapid and simple detection of IFN-neutralizing antibodies in chronic hepatitis C non-responsive to IFN-alpha. J Med Virol 78 74 82
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- NF-κB Hyper-Activation by HTLV-1 Tax Induces Cellular Senescence, but Can Be Alleviated by the Viral Anti-Sense Protein HBZ
- Bacterial and Host Determinants of MAL Activation upon EPEC Infection: The Roles of Tir, ABRA, and FLRT3
- : Reservoir Hosts and Tracking the Emergence in Humans and Macaques
- On Being the Right Size: The Impact of Population Size and Stochastic Effects on the Evolution of Drug Resistance in Hospitals and the Community
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy