
Engineering HIV-Resistant Human CD4+ T Cells with CXCR4-Specific Zinc-Finger Nucleases
HIV-1 entry requires the cell surface expression of CD4 and either the CCR5 or CXCR4 coreceptors on host cells. Individuals homozygous for the ccr5Δ32 polymorphism do not express CCR5 and are protected from infection by CCR5-tropic (R5) virus strains. As an approach to inactivating CCR5, we introduced CCR5-specific zinc-finger nucleases into human CD4+ T cells prior to adoptive transfer, but the need to protect cells from virus strains that use CXCR4 (X4) in place of or in addition to CCR5 (R5X4) remains. Here we describe engineering a pair of zinc finger nucleases that, when introduced into human T cells, efficiently disrupt cxcr4 by cleavage and error-prone non-homologous DNA end-joining. The resulting cells proliferated normally and were resistant to infection by X4-tropic HIV-1 strains. CXCR4 could also be inactivated in ccr5Δ32 CD4+ T cells, and we show that such cells were resistant to all strains of HIV-1 tested. Loss of CXCR4 also provided protection from X4 HIV-1 in a humanized mouse model, though this protection was lost over time due to the emergence of R5-tropic viral mutants. These data suggest that CXCR4-specific ZFNs may prove useful in establishing resistance to CXCR4-tropic HIV for autologous transplant in HIV-infected individuals.
Published in the journal:
. PLoS Pathog 7(4): e32767. doi:10.1371/journal.ppat.1002020
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002020
Summary
HIV-1 entry requires the cell surface expression of CD4 and either the CCR5 or CXCR4 coreceptors on host cells. Individuals homozygous for the ccr5Δ32 polymorphism do not express CCR5 and are protected from infection by CCR5-tropic (R5) virus strains. As an approach to inactivating CCR5, we introduced CCR5-specific zinc-finger nucleases into human CD4+ T cells prior to adoptive transfer, but the need to protect cells from virus strains that use CXCR4 (X4) in place of or in addition to CCR5 (R5X4) remains. Here we describe engineering a pair of zinc finger nucleases that, when introduced into human T cells, efficiently disrupt cxcr4 by cleavage and error-prone non-homologous DNA end-joining. The resulting cells proliferated normally and were resistant to infection by X4-tropic HIV-1 strains. CXCR4 could also be inactivated in ccr5Δ32 CD4+ T cells, and we show that such cells were resistant to all strains of HIV-1 tested. Loss of CXCR4 also provided protection from X4 HIV-1 in a humanized mouse model, though this protection was lost over time due to the emergence of R5-tropic viral mutants. These data suggest that CXCR4-specific ZFNs may prove useful in establishing resistance to CXCR4-tropic HIV for autologous transplant in HIV-infected individuals.
Introduction
For HIV to infect cells, the viral envelope (Env) protein must bind to the host protein CD4 and then to a coreceptor, most commonly CCR5 (R5 HIV) (reviewed in [1]). The importance of CCR5 for HIV-1 pathogenesis is shown by the fact that individuals who are homozygous for an inactivating 32 base pair deletion in ccr5 (ccr5Δ32) are highly resistant to HIV infection [2], [3], while heterozygotes typically live longer after HIV infection due to reduced CCR5 expression levels [4], [5]. Recently, an HIV infected patient with acute myelogenous leukemia received a bone marrow transplant from a ccr5Δ32 homozygous donor [6]. This patient's viral load remains undetectable even in the absence of anti-retroviral therapy more than three years post-transplant, suggesting that this individual's HIV infection has been eradicated. In theory, the success of this approach could be recapitulated by inhibiting CCR5 with an orally bioavailable small molecule such as maraviroc, which binds to CCR5 and prevents its use by most R5 HIV-1 strains. However, virus strains that can utilize CXCR4 either in place of (X4 HIV) or in addition to CCR5 (R5X4 HIV) are found at significant levels in roughly 50% of late-stage infected individuals [7], [8], supporting the need for therapies targeted to CXCR4 [9]. Ideally, an approach to target CXCR4 would complement CCR5-specific therapy, but the broad expression pattern of CXCR4 has made systemic inhibition of this coreceptor by small molecules problematic [10], [11]. In addition, resistance to CCR5 and CXCR4 antagonists can arise in patients by mutations in the viral envelope protein that enable it to utilize the drug-bound forms of these coreceptors [12]–[16]. The ability of HIV-1 to adapt to new selective pressures and the plasticity with which Env interacts with its coreceptors argues for approaches that reduce or eliminate coreceptor expression rather than simply altering coreceptor conformation. If approaches could be developed that specifically target expression of both CCR5 and CXCR4 on CD4+ T cells, virus entry should be inhibited more effectively.
Several genetic approaches have been taken to reduce or eliminate CCR5 expression in human cells, including the use of ribozymes [17], [18], single-chain intracellular antibodies [19], trans-dominant coreceptor mutants [20], and RNAi [21], [22]. However, these studies are limited by the requirement for stable expression of an exogenous gene. To circumvent this, a CCR5 specific zinc-finger nuclease pair (R5-ZFNs) has been developed [23]. Zinc finger proteins that recognize a specific 24 bp DNA sequence are fused with a monomeric cleavage domain from FokI endonuclease that functions only as a dimer (Figure 1). For DNA cleavage to occur, two zinc finger proteins must bind, each to specific, adjoining sequences in the CCR5 gene, leading to FokI dimerization and subsequent DNA cleavage resulting in a double strand break [24]–[26]. The double strand break then can be repaired by error-prone non-homologous end joining (NHEJ) often introducing insertions and deletions leading to a non-functional gene product when this break is placed within the coding region of the targeted gene [27]. Following introduction into human CD4+ T cells [23] or hematopoietic stem cells [28] via an adenovirus vector or DNA nucleofection, respectively, the ccr5 gene was efficiently and specifically disrupted. This confers protection in vitro and in humanized mice to infection by HIV-1 isolates that require CCR5 (but not CXCR4). Several early stage clinical trials using autologous infusions of ZFN-generated CCR5-modified CD4+ T cells are currently underway (clinicaltrials.gov identifiers NCT00842634, NCT01252641, NCT01044654).
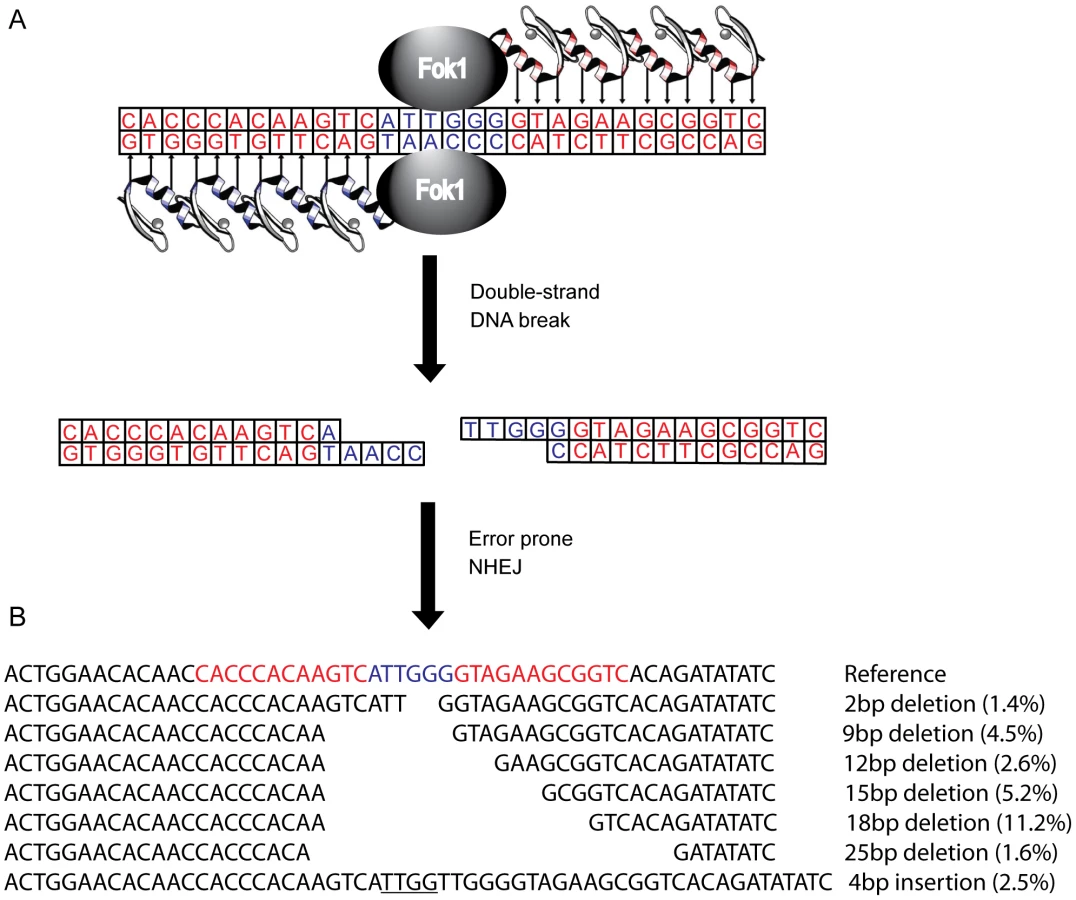
In this study we describe the design and pre-clinical evaluation of a CXCR4-specific ZFN pair (X4-ZFNs) that specifically and efficiently disrupts cxcr4, rendering human CD4+ T cells permanently resistant to HIV-1 strains that require CXCR4 for infection. We also demonstrate that cxcr4 can be safely and efficiently disrupted in CD4+ T cells obtained from ccr5Δ32 homozygotes resulting in cells resistant to all strains of HIV-1 tested. This suggests that combined treatment of mature CD4+ T cells with X4-ZFNs and R5-ZFNs can provide permanent protection against HIV-1 infection.
Methods
Zinc-finger nuclease constructs
We designed ZFNs specific to the human and rhesus CXCR4 and CCR5 genes using a previously described approach [29]. One ZFN pair was used to target both the human and rhesus macaque CXCR4 genes since the 24 bp target sequences are identical. Zinc-finger proteins were optimized against the target gene sequence and assembled as described [30] from an archive of in-vitro-selected modules [31], [32]. The ZFP moieties (target gene; ZFP name; target sequence (5′→3′); recognition α-helices (finger number)) are as follows: CXCR4; X4-ZFN-L; GTAGAAGCGGTC, DRSALSR (1), RSDDLTR (2), QSGNLAR (3), QSGSLTR (4); CXCR4; X4-ZFN-R; GACTTGTGGGTG, RSDSLLR (1), RSDHLTT (2), RSDSLSA (3), DRSNLTR (4). Rhesus CCR5; rhR5-ZFN-L; GATGAGGACGAC, RSDNLAR (1), TSGNLTR (2), RSDNLAR (3), TSGNLTR (4); Rhesus CCR5; rhR5-ZFN-R; AAACTGCAAAAG; RSDNLSV (1), QKINLQV (2), RSDVLSE (3), QRNHRTT (4)., The human CCR5-specific ZFNs are described in Perez et al [23]. The Ad5/F35 adenoviral vectors were generated on an E1/E3 deleted backbone. The ZFNs targeting either the cxcr4 or ccr5 genes were linked via a 2A peptide sequence and cloned into the pAdEasy-1/F35 vector under control of the CMV TetO promoter, and the Ad5/F35 virus for each construct was generated using TREx 293T cells as described [33]. The Ad5/F35 vector encoding the X4-ZFNs is identical to that use by Nilsson, et al. [33] except for the ZFN inserts, promoter, polyA and linker sequences.
Cel1 (surveyor nuclease) assay
Genomic DNA was extracted with the MasterPure kit (Epicentre Biotechnologies) according to manufacturer's instructions. Frequency of gene modification by NHEJ was evaluated as described previously [23], [25], [28]. Briefly, the purified genomic DNA was used as a template to amplify a fragment of the cxcr4 gene using the specific primers (human CXCR4 : 5′-CAACCTCTACAGCAGTGTCCTCATC -3′and 5′ - GGAGTGTGACAGCTTGGAGATG -3′; rhesus CXCR4 : 5′ - GGTGGTCTATGTTGGAGTCTGG -3′and 5′ - GGAGTGTGACAGCTTGGAGATG -3′) in the presence of a 32P-dATP and dCTP. The PCR products were then heated, allowed to re-anneal followed by treatment with the mismatch-sensitive Surveyor nuclease as described in order to detect insertions and deletions caused by NHEJ. For humanized mice samples, whole genome amplification using the REPLI-g Mini Kit (Qiagen) was conducted prior to the surveyor nuclease assay due to limiting cell numbers.
Human CD4+ T cell stimulation and transduction
Fresh CD4+ T cells from normal human donors, purified by negative selection, were obtained from the Center for AIDS Research Human Immunology Core at the University of Pennsylvania. 2.5 million CD4+ T cells were seeded at a density of 0.8×106 cells/ml in RPMI containing 10% fetal calf serum, 1% penicillin/streptomycin, and 100 U/ml interleukin-2 (IL-2). The cells were stimulated with anti-CD3/anti-CD28 coated magnetic beads at a 3∶1 bead to cell ratio [34]. Approximately 18 hrs post-stimulation, the cells were transduced with an Ad5/F35 vector encoding either the X4-ZFNs or R5-ZFNs at a multiplicity of infection (MOI) of 600. Beginning 72 hours post-stimulation, cells were counted every 48 hours using trypan blue dye exclusion on an automated hemocytometer (Countess, Invitrogen) and split to 0.8×106 with fresh media containing 100 U/ml IL-2. Five days post-stimulation, the magnetic beads were removed and washed twice in fresh media. Cells were counted and split until cell growth plateaued 10–14 days post stimulation. For longer experiments, cells were restimulated with beads and cultured for an additional 10–14 days.
In vitro HIV-1 challenge of CD4+ T cells treated with AdX4-ZFNs
Five days post-stimulation the anti-CD3/anti-CD28 coated magnetic beads were removed from each of the three cultures (non-transduced (NTD), AdX4-ZFNs, and AdR5-ZFNs) and 2.5 million cells were seeded in each of four cultures that were subsequently infected with either Bk132 (primary X4 isolate), HxB2 (lab-adapted X4 isolate), R3A (R5X4 primary isolate), or media only (mock). 100 ng p24 of HIV-1 was used per million cells.
Flow cytometry
All staining was done at room temperature in FACS Wash Buffer (1 mM EDTA, 2.5% fetal calf serum in PBS) and all antibodies were from BD Biosciences unless otherwise noted. 0.5–1.0×106 cells were washed in PBS and stained with Live/Dead Aqua (Invitrogen) for 10 min. Then, anti-CD4 PE Cy5.5 and anti-CXCR4 APC (clone 12G5) were added and cells were stained for 20–30 minutes. Cells were then washed and permeabilized per manufacturer's protocol using Cytofix/cytoperm (BD) and stained intracellularly for HIV gag with KC57-RD1 (Beckman Coulter). For compensation, ArC beads (Invitrogen) were used for live/dead, and CompBeads (BD) were used for all other fluorochromes. To detect wtCXCR4 and CXCR4Δ18 in 293T transient transfection experiments, anti-CXCR4 APC (clone 12G5) and anti-CXCR4 PE (clone 4G10) (Santa Cruz Biotechnologies) were used. All samples were run on an LSRII (BD) and analyzed using FlowJo 8.8.6 (Treestar Inc).
Events were gated as follows: singlets (FSC-A by FSC-H), live cells (SSC-A by Live/Dead), lymphocytes (FSC-A by SSC-A), CD3+CD4+ (CD3 by CD4), and then events were divided into CXCR4+ and CXCR4 - populations based upon a fluorescence minus one (FMO) control.
454 deep sequencing and cxcr4 analysis
Genomic DNA was isolated from CD4+ T cells using the QIAamp DNA Micro Kit (Qiagen). For each condition, 200 ng genomic DNA was then PCR amplified using Platinum Taq High Fidelity (Invitrogen) using the following primers plus 454 adaptor sequences and 8 letter DNA barcodes: CAACCTCTACAGCAGTGTCCTCATC (forward) and GGAGTGTGACAGCTTGGAGATG (reverse). Cycle conditions were 95° for 5 min, then 30 cycles of 95° for 30 sec, 55° for 3 sec, 68° for 30 sec, followed by 68° for 2 min. Following PCR amplification the PCR product was analyzed on a 2% agarose gel and then extracted and gel purified using Wizard SV Gel and PCR Clean-Up System (Promega). Quant-iT dsDNA High-Sensitivity Assay Kit (Invitrogen) was then used to determine the concentration of each bar-coded amplicon. DNA samples were then pooled at an equimolar ratio and run on a Roche/454 GS FLX using standard chemistries at the University of Pennsylvania's DNA Sequencing Facility. Approximately 30,000–100,000 reads were obtained for each experiment. CXCR4 pyrosequencing data were assigned to samples by DNA barcode. Any reads containing ambiguous base calls or without a perfect match to barcode and primer were discarded. All remaining reads were aligned to the CXCR4 reference sequence using Mosaik (http://bioinformatics.bc.edu/marthlab/Mosaik). All deviations from the CXCR4 consensus sequence 40 base pairs up or downstream from the ZFN binding site were determined. Any reads that did not extend across this region or that failed to align were discarded. Reads containing only two or fewer substitutions were not classified as mutations as these likely represent sequencing artifacts. Next, background pyrosequencing error, identified by an untransduced control sample, was subtracted from each group of reads. For frameshift analysis, the sequencing error was determined and subtracted for each individual insertion or deletion size.
To ensure sufficient sampling of diverse amplicons, at least 200 ng gDNA was used for CXCR4 analysis and at least 400 ng gDNA was used for off-target site amplification, representing the genomic DNA content of approximately 70,000 and 140,000 alleles, respectively. Determining genetic disruption frequency by both the Cel1 and 454 assays require the assumption that wild type and disrupted alleles are not differentially amplified.
Systemic evolution of ligands by exponential enrichment (SELEX) and determination of off-target sites
To empirically determine the DNA binding preference of the X4-ZFNs, we employed SELEX as previously described [23]. Briefly, each ZFP was HA-tagged and incubated with randomized DNA oligonucleotides and anti-HA Fab fragments. Any DNA bound to the ZFPs was then isolated and amplified. The newly amplified DNA was then used to repeat this process for a total of four rounds of enrichment. The DNA pool was then sequenced at approximately 50× coverage to generate a positional-weighted matrix. This matrix was then aligned to the human genome with the following criteria: putative off-target sites could have up to six mismatches compared to the SELEX consensus sequence, the ZFP pairs must be separated by either 5 or 6 bps, and both ZFP homo - and heterodimers were considered. Off-target sites were ranked and scored by multiplying the probability of each nucleotide at each of the 12 positions of the positional-weighted matrix. The highest scores were then deemed most likely to be disrupted. 454 off-target site data was analyzed as discussed previously [23].
NSG mice
NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/Szj) mice, 8–9 weeks old at time of initial injection, were derived from breeders purchased from The Jackson Laboratory (Bar Harbor, ME). Animals were maintained in a defined flora animal barrier facility at the University of Pennsylvania's Stem Cell and Xenograft Core.
Human CD4+ T cells were isolated and stimulated as previously described and then transduced with an Ad5/F35 vector expressing either the R5-ZFNs or the X4-ZFNs at an MOI of 600. Cells were maintained as previously described. Ten days post stimulation 107 modified cells resuspended in 100 µL PBS were injected intravenously into the tail vein of each mouse. 23 animals received cells treated with X4-ZFNs and 22 mice received cells treated with R5-ZFNs. Animals were randomized by age, sex, and cage. Mice were maintained on the antibiotic Baytril (Bayer) for 24 hours post-injection.
To infect the mice with HIV-1, 105 autologous CD4+ T cells previously infected with X4 HIV-1 strain Bk132 were injected into the tail vein of each mouse. Autologous cells used to infect mice that were not transduced were obtained and stimulated simultaneously as the initially engrafted cells. Five days post-stimulation cells were infected with 100 ng p24/million cells and then were cryopreserved four days post-infection. Cell engraftment was assessed 27 days post injection, and mice were infected with HIV-1 the following day.
To obtain whole blood, mice were anesthetized with isoflurane and a capillary tube was used to drain the retroorbital vein. Human CD4+ T cell counts were determined by staining 50 µl of whole blood in Trucount tubes (BD) with anti-CD45 FITC (Biolegend), anti-CD3 Qdot 655 (Invitrogen), anti-CD4 Alexa Fluor 700, anti-CD8 Pacific Blue (Biolegend), and anti-CXCR4 PE-Cy5. Human CD4+ T cells were defined as CD45+CD3+CD4+CD8-.
At the time of sacrifice, a cardiac puncture was performed to obtain maximal blood volume and then the spleen was harvested. Spleens were homogenized and erythrocytes were lysed with ACK lysis buffer (Invitrogen) before cell purification. Human CD4+ T cells were then isolated with the Human CD4 Positive Selection Kit using the Robosep robotic cell separator (Stem Cell Technologies).
Rhesus macaque CD4+ T cell modification
Whole blood from rhesus macaques (Macaca mulatta) housed at the Tulane National Primate Research Center was used for CD4+ T cell isolation and ZFN treatment. Peripheral blood mononuclear cells were isolated by centrifugation with 96% Ficoll (BD), followed by erythrocyte lysis with ACK lysis buffer. CD4+ T cells were then isolated by negative selection with a non-human primate CD4+ T cell selection kit (Miltenyi). Cells were then stimulated with 1∶4 anti-CD3 (clone FN-18)/anti-CD28 (clone L293) M-450 tosylactivated beads (Invitrogen) at a ratio of 1 bead per cell [35], [36].
Approximately 18 hours post-transduction, cells were transduced with an Ad5/F35 vector expressing either the X4-ZFNs or rhesus specific R5-ZFNs. Cells were maintained in culture as human CD4+ T cells. Surveyor nuclease assay was performed six-ten days post transduction to assess disruption efficiency.
Ethics statement
Human CD4+ T cells were obtained after written informed consent and approval by the University of Pennsylvania's institutional review board. All humanized mouse experiments were approved by the University of Pennsylvania's Institutional Animal Care and Use Committee (Protocol 802436), and were carried out in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All rhesus macaque experiments were approved by the Tulane Institutional Animal Care and Use Committee approval (Protocol P0085; Project 3520) The Tulane National Primate Research Center (TNPRC) is an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility (AAALAC #000594). The NIH Office of Laboratory Animal Welfare assurance number for the TNPRC is A3071-01. All clinical procedures, including administration of anesthesia and analgesics, are carried out under the direction of a veterinarian. Blood was collected while the animals were anesthetized with Tiletamine-zolazepam with Burprenorphine given as an analgesic. All possible measures are taken to minimize discomfort of all the animals used in this study. The University of Pennsylvania and Tulane comply with NIH policy on animal welfare, the Animal Welfare Act, and all other applicable federal, state and local laws.
Results
Design and characterization of X4-ZFNs
To genetically disrupt the CXCR4 allele, we designed a pair of zinc-finger proteins (ZFPs) targeting the region of the cxcr4 gene that encodes residues Asp 187 to Val 196 in the second extracellular loop (ECL2) of this seven-transmembrane domain receptor using methods previously described [29]–[32] (Figure 1). The ECL2 was chosen because this region is less well conserved amongst the CXC family of chemokine receptors, which should reduce the frequency with which other CXC receptors might be targeted, and because ECL2 is important in supporting interactions with the HIV-1 Env protein [37], [38]. Two ZFPs were designed to bind each of two 12 bp targets separated by 6 bp in this region of CXCR4. Each ZFP was then fused to a modified FokI cleavage domain, active preferentially as a dimer to reduce nonspecific DNA cleavage, resulting in zinc-finger nucleases (ZFNs) [25]. Upon binding of both X4-ZFNs, the FokI nuclease cleavage domains dimerize and then generate a double strand break that can subsequently be repaired by error-prone NHEJ resulting in mutations targeted to the cleavage site that can include missense mutations, deletions and insertions (Figure 1).
Efficiency of CXCR4 allele disruption in human CD4+ T cells
To determine the efficiency and specificity with which the cxcr4 genes could be disrupted in human T cells, we produced a bicistronic Ad5/F35 vector to deliver the X4-ZFNs (AdX4-ZFNs). The Ad5/F35 vector is a serotype 5 virus with the fiber protein from a serotype 35 adenovirus that utilizes CD46 for entry as opposed to the coxsackie and adenovirus receptor (CAR), which is poorly expressed on human CD4+ T cells [39]. Primary human CD4+ T cells were stimulated with anti-CD3/anti-CD28 coated magnetic beads and transduced 18 hours later with AdX4-ZFNs, AdR5-ZFNs which expresses previously described CCR5-specific ZFNs [23], or an Ad5/F35 vector that expresses green fluorescent protein (AdGFP). To identify optimal disruption conditions, multiplicities of infection ranging from 100 to 1000 were employed. Cell growth was monitored every 48 hours post-stimulation for approximately two weeks and the efficiency of CXCR4 disruption was assessed at day five post-transduction by both the Surveyor nuclease assay and by deep-sequencing of the CXCR4 target site. As shown in Figure 2A, the Ad5/F35 vectors had a slight dose-dependent impact on cell growth at higher multiplicities of infection that was similar with the AdX4-ZFNs and AdGFP vectors.

Cxcr4 allelic disruption efficiencies as determined by either deep sequencing or the Surveyor nuclease assay were comparable, and were approximately 10% at an MOI of 100, 20% at an MOI of 300, 34% at an MOI of 600, and 38% at an MOI of 1000 (Figure 2B). For subsequent experiments we used an MOI of 600 as this provided near-maximal disruption efficiency with limited impact on cell growth. Notably, this is also the MOI being used in an adoptive therapy phase I clinical trial with R5-ZFNs. Importantly, the level of cxcr4 disruption in cells from multiple donors was stable over nearly four weeks in culture (Table S1), indicating that CXCR4-disrupted cells continued to grow normally. Cell proliferation remained dependent on stimulation, and transformation has not been observed after treatment with ZFNs (data not shown).
Mutations introduced by cleavage with X4-ZFNs
Deep sequencing of the ZFNs target site 10 days after transduction made it possible to assess the mutations introduced by NHEJ reactions following cleavage with X4-ZFNs. Of the nearly 50,000 modified cxcr4 alleles analyzed across five independent experiments, 81.1% (range 75.3–81.7%) contained pure deletions from 1–64 bp in size with the most common deletions being 2, 9, 12, 15, 18, and 25 bp, while 13.5% (range 12.8–16.9%) of cxcr4 alleles contained pure insertions ranging from 1 to 69 bp with more than 90% being 7 bp or less (Figure 1B). The remaining 5.3% (range 4.3–7.4%) of disruption events contained multiple insertions and deletions that may be due to more extensive DNA end-processing or multiple cycles of ZFN-mediated cleavage and subsequent NHEJ. Surprisingly, frameshift mutations occurred at a ratio of 0.90 in-frame per out-of-frame mutation as opposed to the expected frequency of 0.50 (1 in-frame per 2 out-of-frame mutations; Table S1). This unexpected bias likely resulted from microhomology-mediated joining that produced in-frame deletions. To our knowledge, preferential in-frame repair has not been reported or seen with other ZFNs [23], [40], [41].
To further characterize the consequences of disruption mediated by X4-ZFNs, we analyzed an unusually common lesion, an in-frame 18 bp deletion (CXCR4Δ18) that results in the deletion of DNA encoding amino acids R188 to D193 (Figure 1B). This deletion comprised 11.2% (range 9.8 and 11.9%) of all cxcr4 disruptions across five independent experiments with cells from five different donors. The resulting CXCR4Δ18 protein, containing a six-residue deletion in ECL2, could potentially be expressed at the cell surface and support HIV infection. To examine this, we transiently expressed CXCR4Δ18 or wt CXCR4 as a control in 293T cells, which have low endogenous CXCR4 expression. CXCR4 cell surface and intracellular expression was detected by flow cytometry after co-staining with the N-terminal specific CXCR4 antibody 4G10 and the extracellular loop (ECL) specific antibody 12G5 whose epitope includes the CXCR4Δ18 deleted residues [42]. As expected, CXCR4 could be detected on the surface of control cells by both the N-terminal and ECL antibodies. However, CXCR4Δ18 was not detected at the cell surface, though it was detected intracellularly by the N-terminal antibody (Figure 3). In addition, cells expressing CXCR4Δ18 along with CD4 did not support HIV-1 infection. These findings indicate that CXCR4Δ18, the most common in-frame deletion resulting from the X4-ZFNs, does not readily traffic to the cell surface and does not function as an HIV-1 coreceptor.

Specificity of cleavage by X4-ZFNs
Potential off-target genome modification comprises the predominant safety concern with ZFNs. Although ultra-deep full genome sequencing could best identify off-target effects, it is impractical and cost-prohibitive with current technology. Instead, we took a more targeted approach that used an experimentally derived binding site for each X4-ZFP to guide the identification of potential off-target cleavage sites. We conducted in vitro selection, or SELEX (systemic evolution of ligands by exponential enrichment) to determine the actual binding site preference of each X4-ZFP (Figure S1) [43], [44]. A positional-weighted matrix was then generated of the 12 bp binding site and 1 bp flanking region for each ZFP. A BLAST search against the human genome was then used to determine the top 15 off-target binding sites by allowing up to six mismatches per ZFP binding site, a 5 or 6 bp gap between ZFPs, and formation of hetero or homodimers (Table S2) [23]. To assess low frequency disruption events, we conducted 454 deep sequencing on all 15 sites in both control CD4+ T cells and those treated with X4-ZFNs, yielding approximately 7,500–26,000 reads per site in the ZFN-treated samples (Table S2). In a sample with 26.9% of CXCR4 alleles disrupted, NHEJ events were detected at a frequency of 2.3% (170/7531 reads) in an extragenic region on chromosome 12 and 0.8% (84/10531) in ADAMTS17, a metalloprotease of unknown function [45]. The four mutations out of 20,312 reads found in DEC1 (a putative tumor suppressor [46]) and the single mutation out of 21,139 reads found in an extragenic region of chromosome 11 could be due to PCR and sequencing errors or to very low levels (<0.02%) of ZFN-mediated cleavage events. Overall, the X4-ZFNs are highly specific for cxcr4 with low frequency disruption clearly seen at 2 of 15 putative off-target sites with the highest homology to the intended target.
X4-ZFNs confer in vitro protection to human CD4+ T cells from HIV challenge
Disruption of both cxcr4 alleles should render human CD4+ T cells resistant to X4 - and perhaps some R5X4 - viruses as well, while cells harboring a single disrupted allele might express lower levels of CXCR4 and so be more resistant to virus entry. To determine whether ZFN-mediated disruption of cxcr4 indeed protects CD4+ T cells from an in vitro HIV challenge, human CD4+ T cells from three different ccr5 wild type donors were stimulated and transduced with AdX4-ZFNs or an AdR5-ZFNs control. Four days post-transduction, the cells were infected with three diverse HIV-1 strains: BK132 (primary X4 HIV), HxB2 (lab-adapted X4 HIV), or R3A (primary R5X4 HIV). Approximately two weeks post-transduction the cells were restimulated with anti-CD3/anti-CD28 beads, and cultures were maintained for an additional two weeks.
In the absence of HIV infection, there was no detectable growth difference between the X4-ZFNs treated, R5-ZFNs treated, and non-transduced controls over the course of the experiment. However, upon infection with the X4 - or R5X4 - HIV-1 strains, X4-ZFNs treated cells maintained exponential growth compared to profound cell death seen in the R5-ZFNs and untransduced controls. Despite the ability of R3A to utilize both CCR5 and CXCR4 to infect cell lines, in human CD4+ T cells stimulated with anti-CD3/anti-CD28 coated magnetic beads, CCR5 is downregulated causing transient resistance to R5 HIV [47]. Thus, R5X4 HIV strains are likely to function predominantly as X4 HIV strains under these conditions [47]. The growth advantage conferred by treatment with X4-ZFNs in the presence of HIV was magnified upon restimulation. (Figure 4A). This likely resulted from increased cell activation, which increases the ability of HIV to infect and replicate in CXCR4 positive cells.

To determine whether the growth advantage conferred by X4-ZFNs treatment in the presence of X4 - and R5X4 - HIV resulted from a survival advantage of CXCR4 disrupted cells, we performed flow cytometry at various time points post infection as well as deep sequencing of the X4-ZFNs target site on HIV-infected and uninfected cultures. In the absence of HIV infection, the cxcr4 disruption frequency remained stable over time in four independent experiments testing four different ccr5 wild type donors as measured by deep sequencing. A representative experiment is shown in Figure 4B and CXCR4 disruption data from all experiments is shown in Tables S1 and S3. While CXCR4 gene disruption remained stable over time at approximately 30%, CXCR4 gene disruption in HIV-infected cultures increased to 87%, 91%, and 88% in the presence of BK132, HxB2, and R3A respectively after 21 days of infection. FACS analysis showed that at day 19 post-HIV challenge, the frequency of CXCR4 negative cells amongst all live mock HIV-infected CD4+ lymphocytes was 13.0% in untransduced cells, 14.1% in cells transduced with R5-ZFNs, and 35.0% in cells transduced with X4-ZFNs compared to greater than 98%, 97%, and 99% of Bk132, HxB2, and R3A infected cultures transduced with the X4-ZFNs, (Figure 4C). We also found that after 19 days post-HIV infection, reduced but significant cell growth was detectable in several of the HIV-infected control cultures, untransduced and treated with R5-ZFNs. However, greater than 95% of these cells, compared to approximately 10% of cells treated with X4-ZFNs, were CD3+CD4 - suggesting that the surviving cell population was protected from HIV infection by down-regulating CD4 (Figure S2). Thus, CXCR4 disruption had no impact on cell viability, but conferred a significant survival advantage in the presence of HIV strains that can use CXCR4 to infect cells. Furthermore, in control cultures that were untransduced or treated with R5-ZFNs, viral titers exponentially increased until extensive cell death began approximately 8–10 days post infection. In contrast, in cultures treated with X4-ZFNs viral titers steadily decreased after peak viremia while cell growth remained exponential suggesting there was not significant viral production (data not shown).
Ccr5Δ32 CD4+ T cells treated with X4-ZFNs are resistant to R5 and X4 HIV
Given the ongoing adoptive therapy trial of CD4+ T cells treated with R5-ZFNs and the anti-viral success of the recent ccr5Δ32 bone marrow transplant in an HIV-infected patient [6], we sought to determine if cxcr4 could be genetically disrupted simultaneously with ccr5. Human CD4+ T cells from a ccr5Δ32 homozygote were transduced with AdX4-ZFNs or AdR5-ZFNs and subsequently infected with HIV-1 strains Bk132, HxB2, and R3A as described above. Representative data from one of two independent experiments conducted in cells from the same donor is shown in Figure 5 and data from both experiments is shown in Tables S1 and S3. As seen in ccr5 wild type CD4+ T cells, exponential cell growth was preserved in cultures treated with X4-ZFNs compared to control cultures that were untransduced or treated with R5-ZFNs (Figure 5A). In addition, disruption frequency in cultures treated with X4-ZFNs as determined by deep sequencing remained remarkably stable between 32–33% from day 5 to day 26 post-transduction in the absence of HIV, which suggests that simultaneous disruption of ccr5 and cxcr4 does not adversely affect cell growth. However, in the presence of Bk132, HxB2, and R3A, cxcr4 disruption increased after 21 days of HIV challenge to 89%, 83%, and 90%, respectively (Figure 5B), and was associated with markedly diminished virus replication (data not shown), again consistent with significant protection conferred by cxcr4 disruption. Thus, treatment with X4-ZFNs of both wild-type and ccr5Δ32 CD4+ T cells confers stable cxcr4 disruption and a marked survival advantage in the presence of R5X4-HIV and X4-HIV in vitro without any detectable effect on cell growth or viability in the absence of HIV. This suggests that both ccr5 and cxcr4 can be genetically targeted simultaneously for the treatment of HIV infection, while preserving the replicative capacity of the CD4+ T cells.

X4-ZFNs confer partial protection in NSG humanized mouse model
As a first step in evaluating the safety and efficacy of the X4-ZFNs in vivo, we employed a NSG humanized mouse model. Briefly, human CD4+ T cells were stimulated with anti-CD3/anti-CD28 beads and transduced with either AdX4-ZFNs or an AdR5-ZFNs control at an MOI of 600. Cells were then expanded in vitro for ten days after which 107 CD4+ T cells treated with X4-ZFNs (n = 23) or R5-ZFNs (n = 22) were injected intravenously into each mouse. Engraftment was assessed by peripheral blood CD4+ T cell counts 27 days post-injection. All 45 animals successfully engrafted; however, one animal that received cells treated with the X4-ZFNs had a significantly higher but stable CD4+ T cell count and was thus excluded as an outlier from the remainder of the study. On day 28 post-engraftment, mice were intravenously injected with 105 autologous CD4+ T cells that were previously infected with the highly cytopathic X4 HIV-1 strain Bk132 or a mock control. CD4 counts, viral load, and CXCR4 disruption were then monitored to determine the effect of treatment with X4-ZFNs.
To determine if X4-ZFNs impacted cell growth or viability in the absence of HIV, we first compared CD4 counts over time between the uninfected X4-ZFN and R5-ZFN control mice. There was no significant difference in CD4 counts between the two groups over the course of the 61 day experiment as determined by a generalized estimating equation (GEE) method (p = .88) (Figure 6A). Next, we examined the frequency of CXCR4 DNA disruption over time with the surveyor nuclease assay. At the time of injection the percentage of cxcr4 alleles disrupted was 24.3%. This remained constant in both the blood (p = .32) and spleen (p = .70) over the course of the experiment suggesting that CXCR4 disruption did not significantly impact trafficking between these two compartments (Figure 6B). Next, we characterized CXCR4 cell surface expression over time by FACS. In the R5-ZFN control group, with intact cxcr4 genes, 88% of CD4+ T cells expressed CXCR4 protein at day 27 post engraftment, compared to 84% of cells in the X4-ZFN mice (∼24% cxcr4 gene disruption) as determined by a fluorescence minus-one (FMO) control. This difference persisted over time in the absence of HIV-1 infection (p <0.001) (data not shown). Together the stable disruption of CXCR4 as determined by both the surveyor nuclease assay and flow cytometry suggests that CXCR4 disruption did not negatively impact cell viability or growth in humanized NSG mice over a two-month period. As expected, xenogeneic graft versus host disease (GVHD), assessed clinically by dermatitis and hair loss, was observed in mice receiving cells treated with both R5-ZFNs and X4-ZFNs in the absence of HIV challenge. The development of GVHD was equivalent between the two groups (data not shown), suggesting that treatment with X4-ZFNs did not affect CD4+ T cell effector functionality.
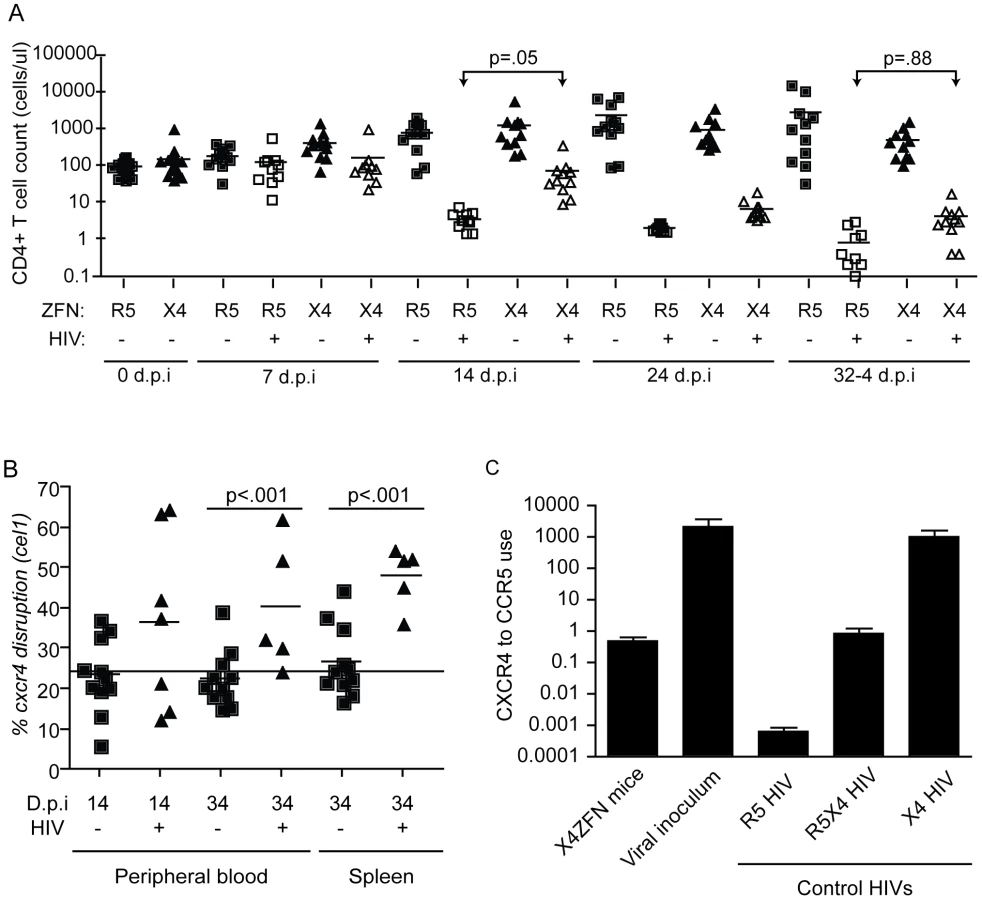
In response to X4 HIV challenge with HIV-1 Bk132, CD4 counts decreased in both X4-ZFN and R5-ZFN mice. However, this rate of decline was slower in the X4-ZFN mice. The X4-ZFN group exhibited a mean 1.1 log CD4 count protection by day 14 post infection (p = .05 for a parametric t-test). However, this protective effect waned over time and there was no significant difference in CD4 counts by day 33 post infection (p = .88) suggesting that treatment with X4-ZFNs conferred only transient protection (Figure 6A).
One mechanism that could account for this would be if mutations arose in the viral Env protein to enable it to use CCR5. To explore this possibility, we bulk cloned and sequenced the V3 loop of Env, the main determinant of coreceptor tropism [48], from plasma isolated from three R5-ZFN mice and three X4-ZFN mice at the time of sacrifice. We identified a single amino acid substitution (Y302N) present in Env isolated from X4-ZFN mice but not R5-ZFN mice or the viral innoculum. Next, we cloned six distinct, functional Envs from the X4-ZFN mice and three distinct, functional Envs from the viral innoculum. As full length Bk132 Env would not pseudotype on an NL43 HIV core we truncated the cytoplasmic tail of the Envs [49], [50], and conducted tropism testing on NP2 cell lines expressing CD4 with either CCR5 or CXCR4. Of the six functional Envs from X4-ZFN mice, four contained the Y302N mutation. Interestingly, these four Envs were able to utilize CCR5 and CXCR4 equivalently, similar to the R5X4-tropic control R3A. All clones with the wild type Tyr302, including the Envs from the viral innoculum and two Envs from X4-ZFN mice utilized CXCR4 approximately 1000-fold more efficiently than CCR5 and comparably to the X4-tropic control TYBE (Figure 6C). Thus, in an NSG humanized mouse model of HIV infection, the cells treated with X4-ZFNs engrafted, trafficked, and persisted comparably to control cells. In addition, treatment with X4-ZFNs resulted in significant transient protection of CD4+ T cell counts in response to X4-tropic HIV challenge, and HIV challenge provided cxcr4 disrupted cells with a survival advantage as determined by increase of cxcr4 disruption in the presence but not the absence of HIV. However, the extent of the protection conferred by the X4-ZFNs was mitigated by evolution or outgrowth of preexisting R5X4-tropic HIV.
ZFN-mediated coreceptor disruption is feasible in rhesus macaque CD4+ T cells
While humanized mouse models for HIV infection have utility, the model is limited due to incomplete immune reconstitution, development of xenogeneic graft versus host disease (GVHD), and the absence of normal T cell homeostasis. For these reasons and others, the NSG model is suboptimal compared to non-human primate models to further elucidate the safety and efficacy of treatment with X4-ZFNs and R5-ZFNs. As a proof of concept for future clinical adoptive therapy studies, we attempted to disrupt the ccr5 and cxcr4 genes with ZFNs in rhesus macaque CD4+ T cells. Briefly, rhesus CD4+ T cells were isolated from whole blood, purified by magnetic bead negative selection, and then stimulated with anti-CD3/anti-CD28 coated beads as previously described [35], [36]. As the 24 bp X4-ZFPs' binding site is identical between rhesus and humans, we were able to utilize the same ZFN pair. However, in order to target rhesus CCR5, rhesus specific R5-ZFNs were developed. As for human cells, the ZFNs were delivered with an Ad5/F35 vector and disruption was assessed by the surveyor nuclease assay. Utilizing a range of MOIs of 600, 1000, and 2000 we observed mean ccr5 and cxcr4 disruption levels of 19.6% and 14.0%, respectively (Figure 7), which suggests that adoptive therapy of cells modified with ZFNs is feasible to model in rhesus macaques.

Discussion
The apparent eradication of HIV resulting from a ccr5Δ32 homozygous allogeneic bone marrow transplant into an HIV-infected patient represents the first reported “cure” of HIV [6]. While an important proof-of-principle, few individuals could benefit from allogeneic ccr5Δ32 homozygous transplants due to toxicities of allogeneic rejection and limitations of finding sufficient HLA-matched ccr5Δ32 homozygous donors. However, coreceptor-specific ZFNs represent a novel therapeutic approach to recapitulate this success via autologous transplantation of gene-modified hematopoietic stem cells and mature CD4+ T cells. Ccr5 can be efficiently disrupted in both human CD4+ T cells and hematopoietic stem cells, conferring protection to HIV challenge in vitro and in humanized mice [23], [28]. In addition, transgenic autologous hematopoietic stem cells can be successfully transplanted in HIV-infected individuals [18] and several phase I adoptive transfer trials of CD4+ T cells treated with R5-ZFNs in HIV infected individuals are currently underway. By design, this strategy addresses only viruses that require CCR5 to infect cells. Our long-term goal, therefore, is to explore the potential to genetically disrupt both ccr5 and cxcr4 for cell replacement therapies in HIV infected individuals, and in the case of cxcr4 do so in a way that specifically targets CXCR4 on T cells and not the many other cell types on which it is expressed.
Unlike for ccr5, there are no known humans with loss of function cxcr4 mutations that would provide insight into the safety and viability of cxcr4 disruption in mature CD4+ T cells. A concern associated with targeting CXCR4 is that it is broadly expressed, while CCR5 expression is largely limited to hematopoietic cells. CXCR4, along with its natural ligand CXCL12, plays a critical role in normal B cell, cardiovascular, and cerebellar development, though T lymphocytes appear to develop normally in cxcr4−/− mice [51]. Thus, it is possible that the selective disruption of cxcr4 in mature post-thymic CD4+ T cells may be tolerable. In addition to its role in development, the CXCR4-CXCL12 axis is a potent CD4+ T cell chemoattractant, and the broad expression of both proteins suggests that this axis may play a fundamental role in basal chemotaxis as opposed to a response to inflammation [52]. Indeed, inhibiting CXCR4 function systemically with the small molecule antagonist plerixafor results in the peripheral mobilization of hematopoetic stem cells, thus mitigating the potential of such therapy for long-term anti-retroviral therapy. However, plerixafor, which has not been reported to have adverse immunologic consequences resulting from inhibiting CXCR4 function in mature CD4+ T cells, provides proof of principle that inhibiting CXCR4 in mature CD4+ T cells may prove to be safe and viable [10], [53]. This suggests that this essential gene can be targeted in a cell-type specific manner with CXCR4-specific ZFNs that limits the toxicities of systemic disruption. While we have demonstrated that CXCR4 is not essential for CD4+ T cell viability and function in vitro and in humanized mice in vivo, the redundancy of lymphocyte chemokine receptors and their ligands makes predicting the in vivo consequences of cxcr4 disruption in a normal host on CD4+ T cell function and trafficking difficult. We conclude that a logical next step will be to study the consequences of cxcr4 disruption in a non-human primate model of HIV infection, which will simultaneously permit the assessment of the consequences of this approach on T cell function and trafficking.
A significant advantage of ZFN gene modification, compared to retrovirus based approaches, is that only transient transgene expression is required to permanently engineer an HIV resistant cell. As a result, adenovirus or other delivery mechanisms such as RNA transfection can be employed that avoid toxicities that can be associated with retroviral integration, such as cellular expansion or transformation. This “hit-and-run” approach limits the requirement of chronic transgene expression and the potential leakiness of other approaches including siRNA [21], [22], intrabodies [19], and ribozymes [17]. However, like most gene transfer approaches a major concern with ZFN technology is the potential for oncogenesis due to off-target effects. While additional study is clearly needed, our current studies have clearly identified off-target disruption in two of the top 15 putative off-target sites: an extragenic site on chromosome 12 and in the metalloprotease ADAMTS17, which is not expressed in CD4+ T cells. In addition, mature CD4+ T cells appear to be resistant to malignant transformation [54], thus mitigating the potential concerns of off-target disruption. Consistent with this, more than 200 people have safely undergone adoptive transfer of genetically engineered lymphocytes with no reported cases of therapy-induced oncogenesis [55]. Reasons for resistance to transformation of mature lymphocytes are unclear, but may involve an unknown mechanism that ensures the diversity of the TCR repertoire and thus limits clonal outgrowth [54]. In contrast, the safety record of hematopoietic stem cell gene therapy is less clear, with a significant frequency of gene-therapy induced oncogenesis or clonal outgrowth reported in several hematopoietic stem cell trials [56], [57].
One unexpected finding reported here is the predominance of in-frame mutations, particularly in-frame deletions, resulting from ZFN mediated cleavage of cxcr4. This has not been observed in other ZFN studies reported thus far. The deep-sequencing approach we have taken makes it possible to comprehensively and accurately assess the types and frequencies of mutations that result from ZFN cleavage followed by DNA repair. The striking preponderance of in-frame deletions may have resulted from toxicities of frameshift mutations shortly after treatment with X4-ZFNs leading to decreased survival relative to in-frame mutants. However, this is unlikely given that the frequency of in-frame mutations remained stable over nearly four weeks in culture, that there was no significant increase in cell death between control cultures and those treated with X4-ZFNs, and that the most common in-frame mutant was not expressed on the cell surface and thus cannot maintain functionality. Rather, the preference for in-frame deletions is likely due to preferential in-frame DNA repair. The deletion in the most common X4-ZFN-induced lesion, cxcr4Δ18, is flanked by a GTCA microhomology domain at the 5′ and 3′ ends consistent with a repair mechanism of microhomology-mediated NHEJ [58]. Similar microhomology sites are present in other common ZFN-induced cxcr4 mutants that we identified. Thus, it appears that the nucleotide sequence of the X4-ZFN binding site directs a preference for an in-frame repair mechanism.
Our studies provide a fundamental demonstration that inactivation of cxcr4 by treatment with X4-ZFNs rendered human CD4+ T cells resistant to infection by X4 virus strains, while CXCR4 inactivation in the context of a ccr5Δ32 homozygous background rendered cells resistant to infection by both R5 and R5X4 strains. Genetic ablation of both CCR5 and CXCR4 will likely make CD4+ T cells entirely resistant to HIV-1. Dual-disruption of CCR5 and CXCR4 will be needed for maximal therapeutic benefit since 46% of treatment-experienced individuals harbor R5X4 strains of HIV compared to 4% with only X4-HIV strains [59]. While virus strains have been identified that can infect cells in the absence of CD4 (reviewed in [60]), none have been identified that can infect cells in the absence of a suitable coreceptor. In addition, virus strains that can use coreceptors other than CCR5 or CXCR4 to infect primary human cells are exceedingly rare. However, targeting CXCR4 alone could provide a selective advantage to CCR5-tropic virus strains. Suppression of CXCR4 by plerixafor in vitro can lead to the emergence of CCR5-tropic virus strains [61], and highly active antiretroviral therapy can sometimes result in enhanced prevalence of R5 relative to R5/X4 virus strains in infected patients [62]. In the humanized mouse model under the conditions studied here, partial loss of cxcr4 in human T cells due to treatment with X4-ZFNs provided selective pressure for either the evolution or emergence of a pre-existing single amino acid mutation in the V3 loop of the infecting X4 HIV-1 strain that enabled it to use CCR5 as efficiently as CXCR4. Thus, just as either genetic or therapeutic suppression of CCR5 can provide an advantage to virus strains that use CXCR4, deletion of CXCR4 is expected to provide an advantage to CCR5-tropic viruses. However, this could provide a clinical benefit given the increased in vitro pathogenicity and correlation with progression to AIDS of X4-tropic HIV.
While humanized mouse models provided a logical first approach to examine in vivo efficacy of CXCR4 disruption, this system does not make it possible to fully assess the functional impact of CXCR4 loss on CD4+ T cell function. To study this in the most rigorous way possible, we have explored the possibility of targeting CCR5 and CXCR4 in CD4+ T cells derived from rhesus macaques. Following re-design of the R5-ZFNs to account for sequence differences between the human and macaque alleles, we found that ZFNs could disrupt both alleles with reasonable efficiency in macaque CD4+ T cells. By inactivating CXCR4 singly and in combination with CCR5, it will be possible to study the effects of CXCR4 loss on T cell function as well as virus infection in a more relevant animal model.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. TiltonJCDomsRW 2010 Entry inhibitors in the treatment of HIV-1 infection. Antiviral Res 85 91 100
2. LiuRPaxtonWAChoeSCeradiniDMartinSR 1996 Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86 367 77
3. SamsonMLibertFDoranzBJRuckerJLiesnardC 1996 Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382 722 5
4. DeanMCarringtonMWinklerCHuttleyGASmithMW 1996 Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273 1856 62
5. HuangYPaxtonWAWolinskySMNeumannAUZhangL 1996 The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med 2 1240 3
6. HutterGNowakDMossnerMGanepolaSMussigA 2009 Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med 360 692 8
7. ScarlattiGTresoldiEBjorndalAFredrikssonRColognesiC 1997 In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat Med 3 1259 65
8. ConnorRISheridanKECeradiniDChoeSLandauNR 1997 Change in coreceptor use correlates with disease progression in HIV-1–infected individuals. J Exp Med 185 621 8
9. WestbyMLewisMWhitcombJYouleMPozniakAL 2006 Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J Virol 80 4909 20
10. HendrixCWFlexnerCMacFarlandRTGiandomenicoCFuchsEJ 2000 Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother 44 1667 73
11. HendrixCWCollierACLedermanMMScholsDPollardRB 2004 Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr 37 1253 62
12. TiltonJCWilenCBDidiguCASinhaRHarrisonJE 2010 A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J Virol 84 10863 76
13. TiltonJCAmrine-MadsenHMiamidianJLKitrinosKMPfaffJ 2010 HIV type 1 from a patient with baseline resistance to CCR5 antagonists uses drug-bound receptor for entry. AIDS Res Hum Retroviruses 26 13 24
14. WestbyMSmith-BurchnellCMoriJLewisMMosleyM 2007 Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol 81 2359 71
15. OgertRAWojcikLBuontempoCBaLBuontempoP 2008 Mapping resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2-V5 domain of gp120. Virology 373 387 99
16. TsibrisAMSagarMGulickRMSuZHughesM 2008 In vivo emergence of vicriviroc resistance in a human immunodeficiency virus type 1 subtype C-infected subject. J Virol 82 8210 4
17. BaiJGorantlaSBandaNCagnonLRossiJ 2000 Characterization of anti-CCR5 ribozyme-transduced CD34+ hematopoietic progenitor cells in vitro and in a SCID-hu mouse model in vivo. Mol Ther 1 244 54
18. DiGiustoDLKrishnanALiLLiHLiS 2010 RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med 2 36ra43
19. SwanCHBuhlerBSteinbergerPTschanMPBarbasCF 2006 T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther 13 1480 92
20. Luis AbadJGonzalezMAdel RealGMiraEManesS 2003 Novel interfering bifunctional molecules against the CCR5 coreceptor are efficient inhibitors of HIV-1 infection. Mol Ther 8 475 84
21. AndersonJBanerjeaAAkkinaR 2003 Bispecific short hairpin siRNA constructs targeted to CD4, CXCR4, and CCR5 confer HIV-1 resistance. Oligonucleotides 13 303 12
22. AndersonJBanerjeaAPlanellesVAkkinaR 2003 Potent suppression of HIV type 1 infection by a short hairpin anti-CXCR4 siRNA. AIDS Res Hum Retroviruses 19 699 706
23. PerezEEWangJMillerJCJouvenotYKimKA 2008 Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26 808 16
24. ManiMKandavelouKDyFJDuraiSChandrasegaranS 2005 Design, engineering, and characterization of zinc finger nucleases. Biochem Biophys Res Commun 335 447 57
25. MillerJCHolmesMCWangJGuschinDYLeeYL 2007 An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25 778 85
26. PodhajskaAJSzybalskiW 1985 Conversion of the FokI endonuclease to a universal restriction enzyme: cleavage of phage M13mp7 DNA at predetermined sites. Gene 40 175 82
27. DuraiSManiMKandavelouKWuJPorteusMH 2005 Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res 33 5978 90
28. HoltNWangJKimKFriedmanGWangX 2010 Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol 28 839 47
29. UrnovFDMillerJCLeeYLBeausejourCMRockJM 2005 Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435 646 51
30. IsalanMChooY 2001 Rapid, high-throughput engineering of sequence-specific zinc finger DNA-binding proteins. Methods Enzymol 340 593 609
31. JamiesonACMillerJCPaboCO 2003 Drug discovery with engineered zinc-finger proteins. Nat Rev Drug Discov 2 361 8
32. IsalanMKlugAChooY 2001 A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat Biotechnol 19 656 60
33. NilssonMLjungbergJRichterJKieferTMagnussonM 2004 Development of an adenoviral vector system with adenovirus serotype 35 tropism; efficient transient gene transfer into primary malignant hematopoietic cells. J Gene Med 6 631 41
34. LevineBLMoscaJDRileyJLCarrollRGVaheyMT 1996 Antiviral effect and ex vivo CD4+ T cell proliferation in HIV-positive patients as a result of CD28 costimulation. Science 272 1939 43
35. OnlamoonNHudsonKBryanPMayneAEBonyhadiM 2006 Optimization of in vitro expansion of macaque CD4 T cells using anti-CD3 and co-stimulation for autotransfusion therapy. J Med Primatol 35 178 93
36. OnlamoonNPlagmanNRogersKAMayneAEBostikP 2007 Anti-CD3/28 mediated expansion of macaque CD4+ T cells is polyclonal and provides extended survival after adoptive transfer. J Med Primatol 36 206 18
37. BrelotAHevekerNAdemaKHosieMJWillettB 1999 Effect of mutations in the second extracellular loop of CXCR4 on its utilization by human and feline immunodeficiency viruses. J Virol 73 2576 86
38. LuZBersonJFChenYTurnerJDZhangT 1997 Evolution of HIV-1 coreceptor usage through interactions with distinct CCR5 and CXCR4 domains. Proc Natl Acad Sci U S A 94 6426 31
39. LeonRPHedlundTMeechSJLiSSchaackJ 1998 Adenoviral-mediated gene transfer in lymphocytes. Proc Natl Acad Sci USA 95 13159 64
40. DoyonYMcCammonJMMillerJCFarajiFNgoC 2008 Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 26 702 8
41. LloydAPlaisierCLCarrollDDrewsGN 2005 Targeted mutagenesis using zinc-finger nucleases in Arabidopsis. Proc Natl Acad Sci USA 102 2232 7
42. BrelotAHevekerNPleskoffOSolNAlizonM 1997 Role of the first and third extracellular domains of CXCR-4 in human immunodeficiency virus coreceptor activity. J Virol 71 4744 51
43. RouletEBussoSCamargoAASimpsonAJMermodN 2002 High-throughput SELEX SAGE method for quantitative modeling of transcription-factor binding sites. Nat Biotechnol 20 831 5
44. FamulokMHuttenhoferA 1996 In vitro selection analysis of neomycin binding RNAs with a mutagenized pool of variants of the 16S rRNA decoding region. Biochemistry 35 4265 70
45. MoralesJAl-SharifLKhalilDSShinwariJMBaviP 2009 Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet 85 558 68
46. LeungACWongVCYangLCChanPLDaigoY 2008 Frequent decreased expression of candidate tumor suppressor gene, DEC1, and its anchorage-independent growth properties and impact on global gene expression in esophageal carcinoma. Int J Cancer 122 587 94
47. CarrollRGRileyJLLevineBLFengYKaushalS 1997 Differential regulation of HIV-1 fusion cofactor expression by CD28 costimulation of CD4+ T cells. Science 276 273 6
48. HartleyOKlassePJSattentauQJMooreJP 2005 V3: HIV's switch-hitter. AIDS Res Hum Retroviruses 21 171 89
49. WilkTPfeifferTBoschV 1992 Retained in vitro infectivity and cytopathogenicity of HIV-1 despite truncation of the C-terminal tail of the env gene product. Virology 189 167 77
50. HarrisonJELynchJBSierraLJBlackburnLARayN 2008 Baseline resistance of primary human immunodeficiency virus type 1 strains to the CXCR4 inhibitor AMD3100. J Virol 82 11695 704
51. NagasawaTHirotaSTachibanaKTakakuraNNishikawaS 1996 Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382 635 8
52. BleulCCFuhlbriggeRCCasasnovasJMAiutiASpringerTA 1996 A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J Exp Med 184 1101 9
53. BraveMFarrellAChing LinSOcheltreeTPope MiksinskiS 2010 FDA review summary: Mozobil in combination with granulocyte colony-stimulating factor to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation. Oncology 78 282 8
54. NewrzelaSCornilsKLiZBaumCBrugmanMH 2008 Resistance of mature T cells to oncogene transformation. Blood 112 2278 86
55. JuneCHBlazarBRRileyJL 2009 Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol 9 704 16
56. Hacein-Bey-AbinaSHauerJLimAPicardCWangGP 2010 Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 363 355 64
57. Cavazzana-CalvoMPayenENegreOWangGHehirK 2010 Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 467 318 22
58. BoboilaCYanCWesemannDRJankovicMWangJH 2010 Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med 207 417 27
59. WilkinTJSuZKuritzkesDRHughesMFlexnerC 2007 HIV type 1 chemokine coreceptor use among antiretroviral-experienced patients screened for a clinical trial of a CCR5 inhibitor: AIDS Clinical Trial Group A5211. Clin Infect Dis 44 591 5
60. HoxieJALaBrancheCCEndresMJTurnerJDBersonJF 1998 CD4-independent utilization of the CXCR4 chemokine receptor by HIV-1 and HIV-2. J Reprod Immunol 41 197 211
61. EsteJACabreraCBlancoJGutierrezABridgerG 1999 Shift of clinical human immunodeficiency virus type 1 isolates from X4 to R5 and prevention of emergence of the syncytium-inducing phenotype by blockade of CXCR4. J Virol 73 5577 85
62. PhilpottSWeiserBAnastosKKitchenCMRobisonE 2001 Preferential suppression of CXCR4-specific strains of HIV-1 by antiviral therapy. J Clin Invest 107 431 8
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- NF-κB Hyper-Activation by HTLV-1 Tax Induces Cellular Senescence, but Can Be Alleviated by the Viral Anti-Sense Protein HBZ
- Bacterial and Host Determinants of MAL Activation upon EPEC Infection: The Roles of Tir, ABRA, and FLRT3
- : Reservoir Hosts and Tracking the Emergence in Humans and Macaques
- On Being the Right Size: The Impact of Population Size and Stochastic Effects on the Evolution of Drug Resistance in Hospitals and the Community
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy