
Evolution of Extensively Drug-Resistant Tuberculosis over Four Decades: Whole Genome Sequencing and Dating Analysis of Isolates from KwaZulu-Natal
Background:
The continued advance of antibiotic resistance threatens the treatment and control of many infectious diseases. This is exemplified by the largest global outbreak of extensively drug-resistant (XDR) tuberculosis (TB) identified in Tugela Ferry, KwaZulu-Natal, South Africa, in 2005 that continues today. It is unclear whether the emergence of XDR-TB in KwaZulu-Natal was due to recent inadequacies in TB control in conjunction with HIV or other factors. Understanding the origins of drug resistance in this fatal outbreak of XDR will inform the control and prevention of drug-resistant TB in other settings. In this study, we used whole genome sequencing and dating analysis to determine if XDR-TB had emerged recently or had ancient antecedents.
Methods and Findings:
We performed whole genome sequencing and drug susceptibility testing on 337 clinical isolates of Mycobacterium tuberculosis collected in KwaZulu-Natal from 2008 to 2013, in addition to three historical isolates, collected from patients in the same province and including an isolate from the 2005 Tugela Ferry XDR outbreak, a multidrug-resistant (MDR) isolate from 1994, and a pansusceptible isolate from 1995. We utilized an array of whole genome comparative techniques to assess the relatedness among strains, to establish the order of acquisition of drug resistance mutations, including the timing of acquisitions leading to XDR-TB in the LAM4 spoligotype, and to calculate the number of independent evolutionary emergences of MDR and XDR. Our sequencing and analysis revealed a 50-member clone of XDR M. tuberculosis that was highly related to the Tugela Ferry XDR outbreak strain. We estimated that mutations conferring isoniazid and streptomycin resistance in this clone were acquired 50 y prior to the Tugela Ferry outbreak (katG S315T [isoniazid]; gidB 130 bp deletion [streptomycin]; 1957 [95% highest posterior density (HPD): 1937–1971]), with the subsequent emergence of MDR and XDR occurring 20 y (rpoB L452P [rifampicin]; pncA 1 bp insertion [pyrazinamide]; 1984 [95% HPD: 1974–1992]) and 10 y (rpoB D435G [rifampicin]; rrs 1400 [kanamycin]; gyrA A90V [ofloxacin]; 1995 [95% HPD: 1988–1999]) prior to the outbreak, respectively. We observed frequent de novo evolution of MDR and XDR, with 56 and nine independent evolutionary events, respectively. Isoniazid resistance evolved before rifampicin resistance 46 times, whereas rifampicin resistance evolved prior to isoniazid only twice. We identified additional putative compensatory mutations to rifampicin in this dataset. One major limitation of this study is that the conclusions with respect to ordering and timing of acquisition of mutations may not represent universal patterns of drug resistance emergence in other areas of the globe.
Conclusions:
In the first whole genome-based analysis of the emergence of drug resistance among clinical isolates of M. tuberculosis, we show that the ancestral precursor of the LAM4 XDR outbreak strain in Tugela Ferry gained mutations to first-line drugs at the beginning of the antibiotic era. Subsequent accumulation of stepwise resistance mutations, occurring over decades and prior to the explosion of HIV in this region, yielded MDR and XDR, permitting the emergence of compensatory mutations. Our results suggest that drug-resistant strains circulating today reflect not only vulnerabilities of current TB control efforts but also those that date back 50 y. In drug-resistant TB, isoniazid resistance was overwhelmingly the initial resistance mutation to be acquired, which would not be detected by current rapid molecular diagnostics employed in South Africa that assess only rifampicin resistance.
Published in the journal:
. PLoS Med 12(9): e32767. doi:10.1371/journal.pmed.1001880
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1001880
Summary
Background:
The continued advance of antibiotic resistance threatens the treatment and control of many infectious diseases. This is exemplified by the largest global outbreak of extensively drug-resistant (XDR) tuberculosis (TB) identified in Tugela Ferry, KwaZulu-Natal, South Africa, in 2005 that continues today. It is unclear whether the emergence of XDR-TB in KwaZulu-Natal was due to recent inadequacies in TB control in conjunction with HIV or other factors. Understanding the origins of drug resistance in this fatal outbreak of XDR will inform the control and prevention of drug-resistant TB in other settings. In this study, we used whole genome sequencing and dating analysis to determine if XDR-TB had emerged recently or had ancient antecedents.
Methods and Findings:
We performed whole genome sequencing and drug susceptibility testing on 337 clinical isolates of Mycobacterium tuberculosis collected in KwaZulu-Natal from 2008 to 2013, in addition to three historical isolates, collected from patients in the same province and including an isolate from the 2005 Tugela Ferry XDR outbreak, a multidrug-resistant (MDR) isolate from 1994, and a pansusceptible isolate from 1995. We utilized an array of whole genome comparative techniques to assess the relatedness among strains, to establish the order of acquisition of drug resistance mutations, including the timing of acquisitions leading to XDR-TB in the LAM4 spoligotype, and to calculate the number of independent evolutionary emergences of MDR and XDR. Our sequencing and analysis revealed a 50-member clone of XDR M. tuberculosis that was highly related to the Tugela Ferry XDR outbreak strain. We estimated that mutations conferring isoniazid and streptomycin resistance in this clone were acquired 50 y prior to the Tugela Ferry outbreak (katG S315T [isoniazid]; gidB 130 bp deletion [streptomycin]; 1957 [95% highest posterior density (HPD): 1937–1971]), with the subsequent emergence of MDR and XDR occurring 20 y (rpoB L452P [rifampicin]; pncA 1 bp insertion [pyrazinamide]; 1984 [95% HPD: 1974–1992]) and 10 y (rpoB D435G [rifampicin]; rrs 1400 [kanamycin]; gyrA A90V [ofloxacin]; 1995 [95% HPD: 1988–1999]) prior to the outbreak, respectively. We observed frequent de novo evolution of MDR and XDR, with 56 and nine independent evolutionary events, respectively. Isoniazid resistance evolved before rifampicin resistance 46 times, whereas rifampicin resistance evolved prior to isoniazid only twice. We identified additional putative compensatory mutations to rifampicin in this dataset. One major limitation of this study is that the conclusions with respect to ordering and timing of acquisition of mutations may not represent universal patterns of drug resistance emergence in other areas of the globe.
Conclusions:
In the first whole genome-based analysis of the emergence of drug resistance among clinical isolates of M. tuberculosis, we show that the ancestral precursor of the LAM4 XDR outbreak strain in Tugela Ferry gained mutations to first-line drugs at the beginning of the antibiotic era. Subsequent accumulation of stepwise resistance mutations, occurring over decades and prior to the explosion of HIV in this region, yielded MDR and XDR, permitting the emergence of compensatory mutations. Our results suggest that drug-resistant strains circulating today reflect not only vulnerabilities of current TB control efforts but also those that date back 50 y. In drug-resistant TB, isoniazid resistance was overwhelmingly the initial resistance mutation to be acquired, which would not be detected by current rapid molecular diagnostics employed in South Africa that assess only rifampicin resistance.
Introduction
The global burden of tuberculosis (TB) remains high, with an estimated 9 million active disease cases and 1.5 million deaths in 2013 [1]. Multidrug-resistant (MDR) TB, defined as Mycobacterium tuberculosis with in vitro resistance to both isoniazid and rifampicin, accounted for at least 480,000 incident cases and 210,000 attributed deaths in 2013 [1]. Extensively drug-resistant (XDR) TB, which is MDR with additional resistance to both quinolones and second-line injectable agents [2], has been reported in 100 countries [1]. With high morbidity, XDR poses a dire threat to public health, particularly in populations with high HIV prevalence [1,3].
The incidence of TB in South Africa is estimated by the WHO to be 860 (776–980) per 100,000 population, which is among the highest in the world [1]. With a population of approximately 10 million, KwaZulu-Natal is the easternmost of South Africa’s nine provinces. While its provincial TB incidence is similar to that of the rest of South Africa (889 per 100,000 in 2012, based on treatment initiation data) [4], KwaZulu-Natal has been notable for disproportionately high rates of drug-resistant TB [4,5]. Compounding this epidemic, South Africa has seen a dramatic increase in HIV prevalence in the last 25 y. The Joint United Nations Programme on HIV and AIDS (UNAIDS) estimates that national adult HIV prevalence was only 0.3% in 1990 but rose to 19.1% in 2013 [6]. In KwaZulu-Natal, HIV rates are particularly high, with 37.4% HIV prevalence documented among pregnant women in 2011 [4].
In 2005, the identification of an outbreak of XDR-TB at the Church of Scotland Hospital in KwaZulu-Natal, Tugela Ferry, raised global alarm and called attention to the prospect of dissemination of potentially untreatable TB [7]. Not only was resistance to four or more classes of antibiotics noted in these strains, but also the disease, in the context of HIV coinfection, was rapidly fatal, with 98% mortality [7]. Traditional genotyping by IS6110 fingerprinting and spoligotyping identified a predominant strain of global M. tuberculosis lineage 4 and spoligotype, ST60, later termed LAM4/F15/KZN (henceforth referred to as LAM4), suggestive of a clonal outbreak of a single drug-resistant strain [7–11]. Targeted sequencing of resistance mutations in a subset of these XDR strains revealed identical mutations [9], further supporting the theory of acquisition of XDR-level resistance and subsequent transmission. Nosocomial spread was deemed likely by a social network analysis [9].
Since the events at Church of Scotland Hospital, which still stands as the largest outbreak of XDR-TB ever reported, XDR-TB has been reported in the majority of hospitals across KwaZulu-Natal [12], and more than 516 XDR-TB cases have been reported within Tugela Ferry alone [13]. In addition, XDR-TB caused by strains not falling within the LAM4 spoligotype have been seen, indicating repeated evolutionary emergences of XDR among strains circulating within the region [10,14]; however, the relative contribution of de novo versus vertically inherited resistance of XDR-TB is unknown. It is also unknown how and when XDR-level drug resistance developed, information that could be exploited to detect and prevent higher-level resistances from emerging in South Africa and elsewhere in the world.
While accumulation of drug-resistance mutations can confer a fitness cost to bacteria, subsequent development of compensatory mutations can ameliorate these costs by restoring certain affected physiological functions while maintaining drug resistance [15–17]. Identification of compensatory mutations among clinical strains of M. tuberculosis [18–20] has improved our understanding of drug resistance and fitness, but this area remains incompletely studied.
Whole genome sequencing efforts that target large collections of M. tuberculosis have provided critical insights into M. tuberculosis population dynamics, including M. tuberculosis transmission and the molecular causes of drug resistance [20–23]. Although some strains from KwaZulu-Natal have been sequenced [24,25], there has been no large-scale sequencing project from this province or studies that have systematically addressed the molecular evolution of XDR. In the largest compilation of whole genome sequences from clinical isolates of M. tuberculosis from South Africa, we used a combination of comparative genomic techniques to elucidate when and how epidemic XDR drug resistance emerged. With knowledge of a strain’s date of collection, determination of the number of single nucleotide polymorphism (SNP) differences between sequenced strains, and the estimated mutation rate of M. tuberculosis, we were able to utilize Bayesian [26] inference to estimate the dates of acquisition of resistance mutations within the Tugela Ferry ancestor. We discuss the implications of these findings with respect to current and future TB control.
Methods
Specimen Collection and Characterization
We selected 337 clinical isolates of M. tuberculosis with diverse drug susceptibility patterns. Strains were collected both retrospectively and prospectively from 2008 to 2013 from all 11 districts of KwaZulu-Natal (Table 1). Strains were chosen for study inclusion on the basis of a predetermined drug resistance pattern so that the dataset was heavily weighted toward drug-resistant strains rather than accurately reflecting the epidemiology of the region. Written informed consent was obtained from study participants prior to cohort enrollment. Biomedical Research Ethics Council (BREC) approval from the University of KwaZulu-Natal was granted for whole genome sequencing of clinical strains. On all study isolates, drug susceptibility testing (DST) was performed by the critical concentration method, using the WHO recommended concentrations [27]. The following drugs were assayed in all strains, with their respective critical concentration in parentheses (in μg/mL): rifampicin (1.0), isoniazid (0.2 and/or 1.0), streptomycin (2.0), kanamycin (6.0), and ofloxacin (2.0). Extended DST was performed for key isolates (Table 1) with the following drugs: capreomycin (10.0), ethambutol (7.5), and ethionamide (10.0). Pyrazinamide resistance testing was performed using PZA MGIT (100.0) or Nicotinamide (500.0). Subject data included age, gender, AFB smear, and HIV status, when available. Study participants were assigned GPS coordinates corresponding to their home provincial district or site of sputum collection.

We also selected for sequencing three historical strains previously collected in KwaZulu-Natal for resequencing [25,24]: KZN4207 (drug susceptible, collected in Durban in 1995), KZN1435 (MDR, collected in Durban in 1994), and KZN605 (XDR, collected in Tugela Ferry in 2005).
Whole Genome Sequencing
Genomic DNA was extracted using published methods [31]. The majority of strains were single colony selected prior to DNA isolation (S1 Methods and S1 Table). Library preparation and whole genome sequencing (WGS) were performed as previously described on the Illumina HiSeq 2000 at the Broad Institute [32]. The median depth of sequencing was 143x, and coverage of the H37Rv genome was 99.9%. Sequencing data were submitted to the Sequence Read Archive NCBI under the following umbrella BioProject identifiers: PRJNA183624 and PRJNA235615.
Bioinformatic Analysis
Primary analysis
Reads were mapped onto a reference strain of H37Rv (GenBank accession number CP003248.2) using BWA version 0.5.9 [33]. In cases in which read coverage of the reference was greater than 200x, reads were down-sampled using Picard [34] prior to mapping. Positions that varied relative to the reference were identified using Pilon version 1.5 as described [32].
Strain diversity and biogeography
We conducted phylogenetic analyses for both the entire set of 340 strains, as well as for a subset of 111 strains belonging to the LAM4 spoligotype. For each set, all sites with unambiguous SNPs in at least one strain were combined into a concatenated alignment. Ambiguous positions were treated as missing data. The concatenated alignment was then used to generate a midpoint rooted phylogenetic tree in RAxML (version 7.3.3) [35] under a GTRCAT substitution model with 1,000 bootstrap replicates. Global M. tuberculosis lineage designations were assigned based on phylogeny and regions of difference [36]. Each strain’s “digital” spoligotype was predicted by statistically testing for the presence of each of 43 unique spacer sequences used in classical spoligotyping from sequence reads. Results were matched to spacer pattern profiles at SITVITWEB to generate a named spoligotype (S1 Methods) [37]. Clonal strains were identified using a density-based clustering algorithm [38] that grouped strains that differ by no more than ten SNPs to at least one other member within a clone (S1 Methods) [39–41].
Mantel tests were performed to evaluate the relationship between genetic and geographic distances among strains using the ZT software v1.1 [42]. Pairwise genetic distances were calculated as the number of SNP differences between strains, and geographic distances were calculated using the haversine formula [43] and points of origin for strain pairs.
Ordering and dating evolution of drug resistance
A curated list of genomic polymorphisms associated with drug resistance was defined for each tested drug based on a literature review (S1 Methods). Polymorphisms associated with compensatory mechanisms to isoniazid, rifampicin, and ethambutol were also defined (S1 Methods). Strains with predicted resistance were identified based on the carriage of mutations from the curated list. We used PAUP [44] to reconstruct the patterns of drug resistance mutation gains and losses throughout the phylogenetic tree representing all 340 strains. PAUP was run using a cost matrix that assigned a 10x greater cost for a loss event relative to a gain event. We used BEAST [26] to estimate a mutation rate and to determine dates for the acquisition of mutations within the LAM4 spoligotype.
BEAST was run for 50 million iterations, sampling every 1,000 iterations, using the relaxed lognormal clock (uncorrelated) model. The relaxed molecular clock model assumes independent rates on different branches, which was consistent with previously published reports [45], as well as initial BEAST analyses that we conducted involving lineages 2 and 4, indicating that there may be substantial variation in evolutionary rates within M. tuberculosis. In addition, since the BEAST statistic “ucld.stdev” was greater than zero (0.189) for our dataset, this indicated that our data did exhibit rate heterogeneity within the LAM4 spoligotype. The first 5 million iterations were excluded as “burn-in.” We used the GTR + Gamma substitution model, estimated base frequencies, and the “Gamma + invariant sites” site heterogeneity model. We enforced the topology of the SNP-based tree determined using RAxML [35]. We used a starting value for the mean mutation rate of 0.35 SNPs/genome/year [39,41,46–48]. We assayed a range of values for the starting mean mutation rate, covering the range of values previously reported in the literature, with little difference in the output. BEAti was used to construct the BEAST input file, and default values were used for all other priors. The program Tracer was used to examine mixing and effective sample size (ESS) in order to assess chain length and model convergence. ESS indicates the number of effectively independent draws from the posterior distribution to which the Markov chain is equivalent. A low ESS for a particular parameter (ESS < 100) would indicate that the trace contained a lot of correlated samples and thus may not well represent the posterior distribution. In our analysis, all statistics had an ESS greater than 150. The results were consistent across several runs of the same model. Estimated dates are given with 95% highest posterior density (HPD) intervals.
Results
Our study included 337 participants with an average age of 33.8 y and a standard deviation of 10.7 y, of whom 165 (49%) were male (Table 2). Overall, 140 patients were HIV positive, 51 were HIV negative, and 146 had unknown HIV status. Baseline characteristics were similar among HIV-positive and HIV-negative individuals, with the exception that HIV-negative individuals were younger (p = 0.0030), more likely to be smear positive (p = 0.0139), and more likely to live outside of eThekwini, the provincial capital (p = 0.0132).

A clinical sample was obtained from each patient; M. tuberculosis was isolated using standard approaches and phenotypic DST was performed on each isolate using standard methodology (S1 Methods). Phenotypic DST revealed 88 susceptible, 23 monodrug-resistant (defined as phenotypic resistance to only one drug), 19 polydrug-resistant (defined as phenotypic resistance to two drugs that does not meet criteria for MDR), 140 MDR sensu stricto, and 67 XDR M. tuberculosis strains (Table 2). Phenotypic MDR and XDR-TB cases were identified in all 11 districts of KwaZulu-Natal. While we observed a trend toward HIV-negative individuals harboring more drug-susceptible TB, this observation did not meet statistical significance (p = 0.0542).
We performed WGS on all 337 clinical strains, as well as on three historical strains isolated prior to the study collection period. We assessed the diversity and phylogenetic relatedness among strains using information from 17,232 variable sites with SNPs relative to the H37Rv reference genome (Fig 1; S1 Fig). The resulting phylogenetic tree revealed four of the seven main global lineages of M. tuberculosis [36,49–51] to be circulating in KwaZulu-Natal during the sampling time frame. The vast majority of isolates (95%) belonged to lineages 2 and 4, with lesser representation from lineages 1 and 3.

A computational or digital spoligotype prediction was performed, and 17 unique spoligotypes were identified (S1 Table) [37]. Spoligotype diversity was well represented in all districts of KwaZulu-Natal (Fig 2, panel A). Using a Mantel test, we determined that there was very low correlation between geographic and genetic distances among strains (r = -0.067906, p = 0.001760), indicating that strains did not cluster geographically. Older transmission events and/or high patient mobility between districts likely account for this pattern.

We defined a “clone” as a set of strains in which each member differs by no more than ten SNPs to at least one other member, which is similar to definitions used in previous genomic studies of M. tuberculosis transmission (S2 Fig, S1 Methods) [39–41]. Nearly one-third of the strains (107 of 340, 31%) belonged to 11 such clones (S2 Table), which were distributed across six spoligotypes and three lineages (S3 Fig). All clones were phenotypically drug resistant, indicating recent person-to-person spread of a diverse set of drug-resistant strains that included both HIV-positive and HIV-negative individuals.
The “historical” Tugela Ferry XDR strain, KZN605, was nested phylogenetically within a large clone of 50 LAM4 strains with predominantly phenotypic XDR (Fig 3). All of the strains within this clone (henceforth referred to as the Tugela Ferry XDR Clone) possessed the characteristic drug resistance mutations that were previously identified in XDR-TB strains circulating in Tugela Ferry during the outbreak [9,24], further indicating this clone’s continued prevalence within KwaZulu-Natal. Patients in whom the Tugela Ferry XDR Clone was isolated were from ten of the 11 districts within the province (Fig 2, panel B). In addition, the Tugela Ferry XDR Clone was not overrepresented among HIV-positive patients (p = 0.6750) (Table 2). This suggests that strains within this clone were neither geographically constrained nor restricted to immunodeficient hosts.

Many of the sequenced LAM4 strains were closely related to the Tugela Ferry XDR Clone but had different DST profiles (Fig 3 and S2 Fig), giving us an opportunity to finely dissect the order of acquisition of mutations giving rise to the Tugela Ferry XDR Clone [9,24]. LAM4 strain phylogeny was recalculated using data from only LAM4 strains, and parsimony was used to place the origin of known resistance-conferring mutations on the tree. The recalculated LAM4 tree was consistent with our previous tree containing data from all strains with all key internal nodes involved in the evolution of drug resistance having bootstrap values greater than 89%. This enabled us to confidently assign evolutionary ordering of drug resistance mutation acquisition (Fig 3 and S5 Fig).
As shown in Fig 3, the first step towards XDR-level resistance in this epidemic clone was the acquisition of isoniazid and streptomycin resistance-conferring mutations in katG and gidB, respectively, which were gained at node A of the phylogenetic tree (100% bootstrap support). With accumulation of successive mutations, the ancestral strain (and its descendants) gained (i) additional polydrug resistance to ethionamide and ethambutol via mutations in the inhA promoter and embB (nodes B and C, respectively, 100% and 89% bootstrap support), (ii) MDR via mutations in rpoB and pncA that conferred resistance to rifampicin and pyrazinamide (node D, 100% bootstrap support), and (iii) XDR via mutations in rrs and gyrA, which conferred resistance to kanamycin and ofloxacin, respectively, and an additional rpoB mutation (node E, 97% bootstrap support). This ordering was highly supported by bootstrapping (all key nodes had bootstrap values ≥89%) in the phylogenetic reconstruction. Thus, the first step towards XDR-level drug resistance in this epidemic clone was the acquisition of isoniazid and streptomycin resistance followed by ethambutol and ethionamide resistance, then rifampicin and pyrazinamide resistance, and, ultimately, kanamycin and ofloxacin resistance.
Because we had dates of isolation for all sequenced strains—including strains that were isolated more than 20 y ago—we applied a Bayesian statistical approach to estimate when mutations leading to the Tugela Ferry XDR Clone emerged. Using this approach, which takes into account the phylogeny of LAM4 strains, the dates of their isolation, and published mutation rates for M. tuberculosis [39,41,46–48], we calculated that LAM4 in KZN mutated at a rate of 0.61 SNPs/genome/year. This mutation rate was higher than other previously published mutation rates, regardless of which rate from the literature was used as the starting mean value. Applying this rate, we estimated that drug resistance mutations at node A were acquired in 1957 (95% HPD: 1937–1971), soon after streptomycin and isoniazid were developed. MDR-level resistance was acquired in 1984 (95% HPD: 1974–1992; node D), and XDR-level resistance was acquired in 1995 (95% HPD: 1988–1999; node E), 10 y prior to its acute recognition in 2005 in Tugela Ferry (Fig 3). The dating analysis within the LAM4 spoligotype consistently assigned drug resistance gains after the drug discovery date, indicating that drug resistance emergence in the region mirrored the dates of drug discovery.
We also observed multiple drug resistance mutations within LAM4 that emerged outside the Tugela Ferry XDR Clone (Fig 3). Many of these mutations were acquired at leaf nodes, which implied very recent gains of resistance. Including the Tugela Ferry ancestor, we calculated that genotypic MDR sensu stricto—defined as both isoniazid and rifampicin resistance-conferring mutations—independently arose a minimum of 13 times. Within LAM4, the Tugela Ferry XDR Clone represented the single and only evolutionary gain of genotypic XDR—as defined by acquisition of resistance-conferring mutations to the four XDR-defining drugs: isoniazid, rifampicin, ofloxacin, and kanamycin. However, within LAM4 we also observed ten independent gains of either a kanamycin or an ofloxacin resistance-conferring mutation in a background of genotypic MDR sensu stricto. As such, 13 LAM4 strains identified in this study would be considered genotypic “pre-XDR” and only one SNP away from XDR-level resistance.
Beyond LAM4, we observed many other independent evolutionary emergences of MDR and XDR across this dataset. Twelve and seven spoligotypes contained strains with phenotypic MDR and XDR, respectively (S3 Table), suggesting that these resistance patterns emerged no fewer than 12 and 7 times. However, when we quantified the total number of independent evolutionary emergences of genotypic MDR and XDR across our entire dataset, we estimated that MDR sensu stricto and XDR evolved no less than 56 and nine independent times, respectively (S3 Table).
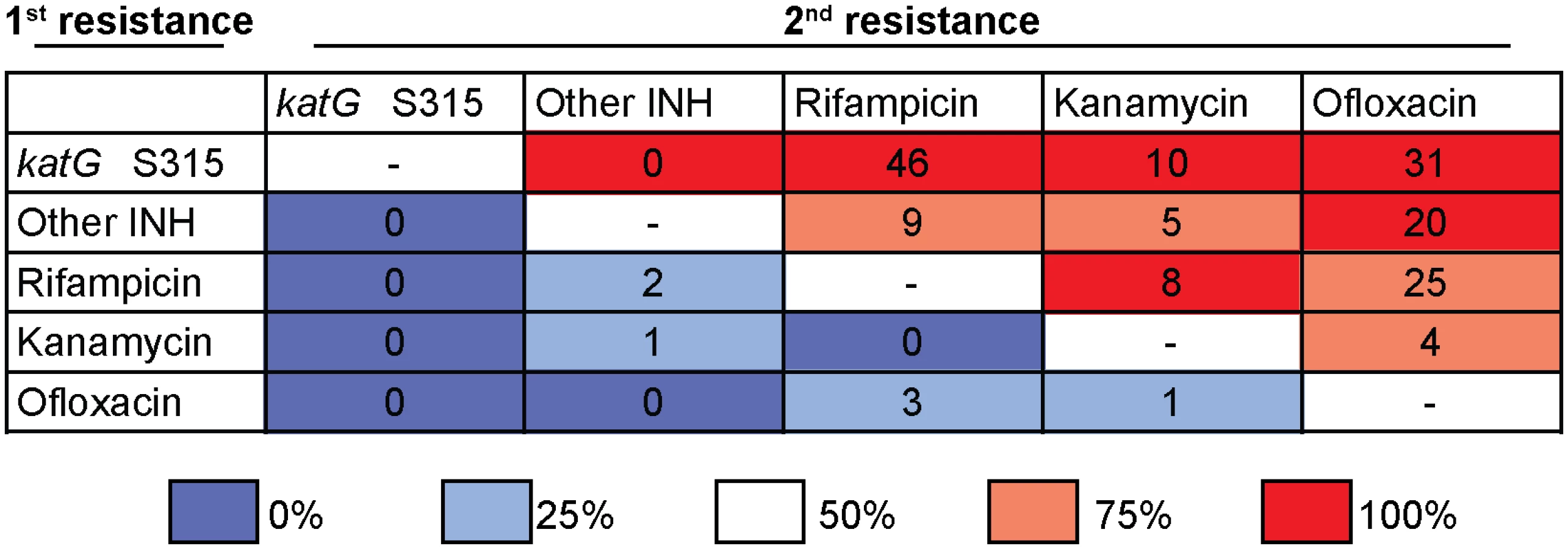
Remarkably, the first drug resistance acquisition in the Tugela Ferry XDR Clone was consistent with other emergences of MDR and XDR across the entire dataset. For the 214 strains with genotypic resistance to two or more of the MDR and XDR defining drugs, we quantified the number of evolutions in which a specific drug resistance mutation was gained before a second resistance mutation. We observed that isoniazid resistance via nonsynonymous mutation at the katG S315 codon was gained before rifampicin resistance in 46 unique evolutionary events, whereas rifampicin resistance was never acquired before the katG S315 mutation (Fig 4). When we repeated this for all pairwise comparisons, we found that isoniazid resistance, conferred by mutation of the katG S315 codon, preceded or co-occurred with resistance mutations to all other drugs in our dataset. Mutations other than the katG S315 mutations that confer isoniazid resistance (i.e., inhA promoter mutations or katG deletions) occurred before rifampicin resistance mutations in nine unique events, whereas we only observed the reverse ordering twice. These data indicate that, beyond the Tugela Ferry XDR Clone, isoniazid resistance, and in particular the S315 codon mutation in katG, has been the initial resistance-conferring mutation leading to polydrug resistance, including MDR and XDR, among strains from KwaZulu-Natal.
As in other organisms, in vitro studies have suggested that drug resistance in M. tuberculosis may be associated with a variable fitness cost that can be offset by compensatory evolution [18,15]. Nonsynonymous mutations in the α and β’ subunits of RNA polymerase have been postulated to compensate for fitness costs associated with rifampicin resistance [15,19]. Among our 226 strains with phenotypic rifampicin resistance, 76 strains had mutations known to compensate for rifampicin resistance (S4 Table) [20,19,54,55]. Using the phylogenetic framework and parsimony, we determined that 23 of the 27 previously described compensatory mutations had an evolutionary pattern consistent with rifampicin compensation, i.e., mutations that evolved only after or concurrent with mutations that conferred rifampicin resistance.
We also attempted to identify novel rifampicin compensatory mutations with this approach. In addition to the 27 previously described mutations, we detected an additional 38 nonsynonymous polymorphisms in rpoA, rpoC, and the non-rifampicin resistance-determining regions (RRDR) regions of rpoB (S4 Table). By parsimony analysis, we established the acquisition order of these rpoA, rpoC, and non-RRDR rpoB mutations in relation to genotypic rifampicin resistance. An additional 26 of these previously uncharacterized mutations also evolved in a pattern consistent with a role in compensation, which suggests that they may also function in this capacity. While there were ten unique RRDR mutations with subsequent or concurrent evolutionary gain of a putative rifampicin compensatory polymorphism, the vast majority of putative compensatory mutations occurred in association with rpoB S450L (p < 0.001) (S5 Table). This pattern was observed regardless of whether the compensatory mutation was previously known or uncharacterized.
Beyond rifampicin compensation, we also applied our combined phylogenetic and parsimony approach to known isoniazid and ethambutol compensatory mutations. With respect to isoniazid compensation, only a single evolution of the ahpC promoter mutation was observed in our dataset (S6 Table). It was gained after genotypic isoniazid resistance, which supports its compensatory role for certain isoniazid resistance mechanisms [41]. Nonsynonymous mutations in ubiA (Rv3086c) have previously been implicated in ethambutol resistance [56]. In our dataset, there were at least two occasions in which these mutations unambiguously arose prior to the acquisition of genotypic ethambutol resistance, suggesting that these are more likely to be stepping-stone mutations rather than compensatory (S6 Table).
Discussion
We report on the WGS and comparative analysis of the largest collection of drug-resistant M. tuberculosis sequenced to date from South Africa. From analysis of these genomes, we determined the molecular antecedents of the Tugela Ferry XDR Clone and dated the emergence of genotypic resistance to eight drugs. We showed that the development of XDR in KwaZulu-Natal had its roots in first-line drug resistance that arose in the late 1950s and MDR that emerged in the 1980s. Our dating analysis indicated that the Tugela Ferry XDR Clone took nearly four decades to evolve from its initial isoniazid and streptomycin resistances to full-blown XDR. Although our data confirmed that the XDR outbreak in KwaZulu-Natal was indeed a clonal event, we showed that drug resistance in this region is driven by both the development of de novo drug resistance and clonal spread. We elucidated common evolutionary patterns of drug resistance acquisition and determined that isoniazid was overwhelmingly the first drug resistance to be acquired. Lastly, we validated that certain previously described rifampicin compensatory mutations do indeed evolve in a pattern consistent with compensation and have identified 26 novel polymorphisms that may also function in this capacity. Collectively, these data have important implications for the public health control of TB in sub-Saharan Africa and elsewhere.
Using a combination of likelihood, parsimony, and Bayesian computational approaches, we observed a decades-long evolutionary trajectory toward XDR-level drug resistance in LAM4 that mirrored the order and timing of introduction of antitubercular drugs into clinical practice [57]. Though parsimony-based approaches can interpret rapid independent evolution of an identical polymorphism in multiple strains as a single evolutionary event (occurring at a single node), our predictions indicated that resistance-conferring mutations evolved only after each drug’s clinical introduction and not before, as might be expected if homoplasy were a major contributor to pattern predictions (Fig 3). In addition, one of the oldest acquired mutations toward XDR-level resistance was a specific 130 bp deletion in gidB, which is extremely unlikely to arise many times independently and supports accurate reconstruction in our evolutionary analysis. Furthermore, though our calculated mutation rate for LAM4 was slightly higher than previous reports [39,41,46–48], our estimate was within the reported 95% HPD interval and was based on a larger fraction of the H37Rv genome than previous studies (99.9% versus <90% H37Rv mapping coverage) [39]. This was due to the inclusion of sequence data generated from both PCR-free short fragment and jumping libraries and analysis with improved bioinformatics tools that enabled us to examine SNPs within more variable and high guanine-cytosine (GC) content regions of the genome, including proline-glutamic acid (PE) and proline-proline-glutamic acid (PPE) genes that have been reported to have a higher mutation rate [58]. Thus, because we are including data from more of the genome, our estimation of the M. tuberculosis mutation rate may more closely approximate the actual mutation rate of the organism as compared to previously published studies.
Importantly, from the pattern of drug resistance evolution within LAM4, it is clear that the precursors to XDR evolved well before the explosive South African HIV epidemic of the 1990s, indicating that the selection of transmissible XDR strains can occur in low-prevalence HIV settings. While recent failures in TB and infection control and the current high HIV prevalence rates, combined, undoubtedly contributed to the spread of XDR, they were not the sole causes of XDR in this setting. Indeed, strains that evolved first-line drug resistance soon after the introduction of chemotherapy were a critical entry point to today’s drug-resistant epidemic. Drug-resistant strains that emerged from the mid-20th century were evidently maintained within the population of M. tuberculosis, presenting the opportunity for the acquisition of successive resistance and compensatory mutations that culminated in transmissible XDR and the Tugela Ferry outbreak. Drug-resistant strains may have been maintained within a population over time either by ongoing cycles of infection and transmission or through reactivation of latent disease. It is unclear which of these may have been the most important in this setting, but it suggests that fitness costs due to first-line drug resistance may not be severe. Reactivation was recently shown to be important in the transcontinental spread of MDR-TB from Thailand to California over a 22-y period [59], but it is unclear whether this factor was also critical in KwaZulu-Natal.
Beyond LAM4, and as has been shown in other studies [10,14], drug resistance emerged de novo repeatedly in KwaZulu-Natal, as evidenced by our identification of numerous independent evolutionary events of MDR and XDR across multiple lineages and spoligotypes. Of particular note was the detection of multiple independent evolutions of MDR to pre-XDR within LAM4, which may herald a new wave of XDR in the near future. Thus, the repeated emergence of de novo high-level drug resistance underscores the reality that, even in middle-income sub-Saharan African countries, the current approach to TB control is failing to stem the ongoing emergence of drug resistance. In fact, results from our analyses suggest this was not due to infrequent poor adherence to TB drugs but instead to decades of inadequate TB control that has driven resistance development in a stepwise fashion, multiple times over. Given that our estimates of resistance evolution were based on identification of known resistance-conferring mutations and that the majority of sequenced strains were single colony purified, our calculations are likely an underestimation due to incomplete understanding of all mutations that confer drug resistance and the possibility of mixed infections, respectively. Thus, the state of drug resistance emergence is likely more dire than we have described.
Recent studies from KwaZulu-Natal have emphasized transmission of a limited number of strains as a driving force behind the emergence of drug resistance [10,14]. Our data also confirm that once drug resistance develops, clonal spread of resistant strains can and does occur in this context. We found that recent person-to-person spread of resistant strains is apparent in KwaZulu-Natal, as evidenced by identification of multiple drug-resistant clones. Importantly, in contrast to initial reports from Tugela Ferry in which nearly all XDR cases were TB/HIV coinfected [7,9], eight patients in our study in whom the Tugela Ferry XDR Clone was identified were HIV negative. This reemphasizes that even XDR drug-resistant strains are sufficiently fit to transmit person to person and cause morbidity in both immunocompetent and immunosuppressed persons. Improved infection control and rapid case finding will be necessary to prevent further spread of drug-resistant strains and to detect such cases in the community as well as in hospital settings [28].
Our genomic analysis uncovered a common initial pattern of drug resistance that is not optimally detected by current diagnostic algorithms. Isoniazid resistance was overwhelmingly the first drug resistance to occur along the pathway to multiple drug resistances. However, current TB control strategies in South Africa focus on early detection of rifampicin resistance as a surrogate marker of MDR and do not include the detection of isoniazid resistance. Clinical diagnostic policies that rely on Xpert MTB/RIF (a WHO-endorsed and widely deployed molecular diagnostic) [60] without more extensive drug resistance testing allow isoniazid resistance to go undetected and unchecked. Moreover, under current short course treatment guidelines that utilize 4 mo of isoniazid and rifampicin in the continuation phase [61], failure to recognize isoniazid monoresistance is tantamount to provision of unopposed rifampicin therapy and may rapidly select for rifampicin resistance. This phenomenon may be underappreciated and incompletely accounted for in mathematical models that recommend continued use of screening tools that identify only rifampicin resistance [62]. Furthermore, if rifampicin resistance is indeed detected by Xpert, failure to implement confirmatory secondary molecular testing for dual rifampin and isoniazid resistance, as is mandated by South African policy, occurs at unacceptably high rates [63]. Our ordering of drug resistance acquisition provides strong evidence that isoniazid monoresistance is a common pathway toward development of MDR and highlights the importance of prompt identification and treatment of isoniazid monoresistance. Failure to do so would be recapitulating the scenario that led to the current XDR problem.
Beyond detection, identification of the initial drivers of isoniazid monoresistance is also critical to the prevention of successive resistances. Isoniazid preventive therapy (IPT) has previously been implicated as a potential source of isoniazid monoresistance [64,65]. Our work highlights the need to understand the true risks of mass IPT implementation [66] in high-burden settings.
We were able to verify that the evolutionary patterns of select previously described rifampicin and isoniazid compensatory mutations do indeed appear to be consistent with compensation to their respective drug. Similarly, ubiA was observed to evolve in a stepping-stone pattern rather than a compensatory pattern with respect to ethambutol resistance [56]. Furthermore, we have identified novel putative rifampicin compensatory mutations that may have acted to restore bacterial fitness and facilitate transmission of drug-resistant strains. While the majority of the previously described rifampicin compensatory mutations had an evolutionary pattern consistent with this role, four polymorphisms previously associated with rifampicin compensation (rpoB I491F, rpoC G594E and N826K, and rpoA E319K) were not observed to evolve concurrently or subsequent to genotypic rifampicin resistance (S4 Table). These mutations may (i) not be compensatory mutations in the classic sense (i.e., mutations that evolve following gain of genotypic drug resistance to mitigate a fitness cost) but instead serve as stepping-stone mutations, (ii) evolve in concert with non-RRDR genotypic rifampicin resistance, or (iii) have no association with rifampicin resistance. We have proposed 26 novel mutations whose evolutionary patterns are consistent with rifampicin compensation, and these should be investigated in future studies.
The most commonly observed genotypic rifampicin resistance mutation among our sequenced strains was rpoB S450L (often referred to as S531L using the Escherichia coli codon numbering scheme), which is known to be the most prevalent RRDR mutation. Laboratory-derived strains carrying the S450L were previously shown to have relatively high fitness in in vitro growth assays [15], supporting the hypothesis that high prevalence of the S450L mutation among clinical strains was due to it imparting few fitness consequences. However, as shown in our study and in several others [20,54], rpoB S450L was the most likely RRDR polymorphism to evolve putative compensatory mutations, which calls into question the low fitness cost of S450L in vivo. Song et al. assessed rifampicin fitness by transcriptional efficiency (rather than growth) and showed that the S450L mutation has half the transcriptional efficiency of WT rpoB [54], which is likely to impart fitness consequences if not compensated.
This study has two main limitations. First, as our study isolates derived from only one geographic region, our conclusions regarding the timing and dating of the emergence of resistance may not be universal. However, two recent studies [67,68] have reported results compatible with ours from different settings. Using a similar approach, Eldholm et al. were able to date the first emergence of resistance in an MDR-TB outbreak from Argentina to the early 1970s and found that isoniazid and streptomycin resistance-conferring mutations were the first to be acquired. In a study of the global spread of the Beijing lineage [23], isoniazid and streptomycin resistances were also found to be common to all drug-resistant strains in two clonal complexes that resulted in the epidemic spread of two MDR clones in Russia and Central Asia 20 to 30 y ago. Another limitation, as discussed above, is that parsimony-based dating approaches may fail to distinguish rapid independent evolutions of a commonly occurring resistance mutation as two unique evolutionary events. This could lead to erroneous assignment of a mutation to a more basal part of the phylogenetic tree. While a theoretical risk, we believe the effect was minimal in our dataset since, as described above, our predictions were consistent with the timing of drug introduction, and we included a specific large deletion that is extremely unlikely to arise many times independently.
Here, we present the largest WGS study conducted to date of drug-resistant clinical isolates of M. tuberculosis from South Africa. Our dating analysis highlights the dire repercussions of failure to control first-line drug resistance. As acquisition of isoniazid resistance is the key initiation event for progression to MDR and beyond, TB control efforts that focus on the identification of isoniazid as well as rifampicin resistance will result in earlier detection of drug-resistant TB cases. Prudent antibiotic stewardship during the introduction of new antitubercular drugs will be critical to prevent the early fixation of resistance and protect the lifespan of novel agents.
Supporting Information
Zdroje
1. WHO. Global Tuberculosis Report. 2014. http://www.who.int/tb/publications/global_report/en/
2. Jassal M, Bishai WR. Extensively drug-resistant tuberculosis. Lancet Infect Dis. 2009;9 : 19–30. doi: 10.1016/S1473-3099(08)70260-3 18990610
3. WHO. Multidrug-resistant tuberculosis (MDR-TB) Update. 2013. http://www.who.int/tb/challenges/mdr?MDR_TB_FactSheet.pdf
4. Department of Health Province of KwaZulu-Natal Annual Report 2012/2013: Part B Performance Information. 2013. http://www.kznhealth.gov.za/1213report/partB.pdf
5. Wallengren K, Scano F, Nunn P, Margot B, Buthelezi SSS, Williams B, et al. Drug-Resistant tuberculosis, KwaZulu-Natal, South Africa, 2001–2007. Emerg Infect Dis. 2011;17 : 1913–6. doi: 10.3201/eid1710.100952 22000370
6. UNAIDS. The Gap Report: HIV estimates with uncertainty bounds 1990–2013. 2014. http://www.unaids.org/en/resources/campaigns/2014/2014gapreport/gapreport
7. Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, et al. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 2006;368 : 1575–80. 17084757
8. Pillay M, Sturm AW. Evolution of the extensively drug-resistant F15/LAM4/KZN strain of Mycobacterium tuberculosis in KwaZulu-Natal, South Africa. Clin Infect Dis. 2007;45 : 1409–14. 17990220
9. Gandhi NR, Weissman D, Moodley P, Ramathal M, Elson I, Kreiswirth BN, et al. Nosocomial transmission of extensively drug-resistant tuberculosis in a rural hospital in South Africa. J Infect Dis. 2013;207 : 9–17. doi: 10.1093/infdis/jis631 23166374
10. Gandhi NR, Brust JCM, Moodley P, Weissman D, Heo M, Ning Y, et al. Minimal Diversity of Drug-Resistant Mycobacterium tuberculosis Strains, South Africa. Emerg Infect Dis. 2014;20 : 394–401.
11. Chihota VN, Müller B, Mlambo CK, Pillay M, Tait M, Streicher EM, et al. Population structure of multi - and extensively drug-resistant Mycobacterium tuberculosis strains in South Africa. J Clin Microbiol. 2012;50 : 995–1002. doi: 10.1128/JCM.05832-11 22170931
12. Moodley P, Shah NS, Tayob N, Connolly C, Zetola N, Gandhi N, et al. Spread of extensively drug-resistant tuberculosis in KwaZulu-Natal province, South Africa. PLoS One. 2011;6: e17513. doi: 10.1371/journal.pone.0017513 21655324
13. Tuberculosis MDR/XDR: The Msinga Experience 2005–2009. 2009. http://www.kznhealth.gov.za/tbmsinga.pdf
14. Müller B, Chihota VN, Pillay M, Klopper M, Streicher EM, Coetzee G, et al. Programmatically selected multidrug-resistant strains drive the emergence of extensively drug-resistant tuberculosis in South Africa. PLoS One. 2013;8: e70919. doi: 10.1371/journal.pone.0070919 24058399
15. Cohan FM, King EC, Zawadzki P. Amelioration of the Deleterious Pleiotropic Effects of an Adaptive Mutation in Bacillus subtilis. Evolution (N Y). 1994;48 : 81–95.
16. Schrag SJ, Perrot V. Reducing antibiotic resistance. Nature. 1996. pp. 120–121. 8700201
17. Reynolds MG. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics. 2000;156 : 1471–1481. 11102350
18. Sherman DR, Mdluli K, Hickey MJ, Arain TM, Morris SL, Barry CE, et al. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science. 1996;272 : 1641–3. 8658136
19. Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, et al. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. Nature Publishing Group; 2012;44 : 106–10.
20. Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. Nature Publishing Group; 2014;46 : 279–86.
21. Farhat MR, Shapiro BJ, Kieser KJ, Sultana R, Jacobson KR, Victor TC, et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat Genet. Nature Publishing Group; 2013;45 : 1183–9.
22. Zhang H, Li D, Zhao L, Fleming J, Lin N, Wang T, et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat Genet. Nature Publishing Group; 2013;45 : 1255–60.
23. Merker M, Blin C, Mona S, Duforet-frebourg N, Lecher S, Willery E, et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. 2015;47(3):242–249. doi: 10.1038/ng.3195 25599400
24. Ioerger TR, Koo S, No E-G, Chen X, Larsen MH, Jacobs WR, et al. Genome analysis of multi - and extensively-drug-resistant tuberculosis from KwaZulu-Natal, South Africa. PLoS One. 2009;4: e7778. doi: 10.1371/journal.pone.0007778 19890396
25. Koenig R. Tuberculosis. Few mutations divide some drug-resistant TB strains. Science. 2007;318 : 901–2. 17991835
26. Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29 : 1969–73. doi: 10.1093/molbev/mss075 22367748
27. WHO Updated interim critical concentration for first-line and second-line DST [Internet]. http://www.stoptb.org/wg/gli/assets/documents/Updatedcriticalconcentrationtable_1stand2ndlinedrugspdf
28. Bantubani N, Kabera G, Connolly C, Rustomjee R, Reddy T, Cohen T, et al. High rates of potentially infectious tuberculosis and multidrug-resistant tuberculosis (MDR-TB) among hospital inpatients in KwaZulu Natal, South Africa indicate risk of nosocomial transmission. PLoS One. 2014;9: e90868. doi: 10.1371/journal.pone.0090868 24625669
29. O’Donnell MR, Wolf A, Werner L, Horsburgh CR, Padayatchi N. Adherence in the treatment of patients with extensively drug-resistant tuberculosis and HIV in South Africa: A prospective cohort study. J Acquir Immune Defic Syndr. 2014;67(1):22–29. doi: 10.1097/QAI.0000000000000221 24872138
30. O’Donnell MR, Pym A, Jain P, Munsamy V, Wolf A, Karim F, et al. A novel reporter phage to detect tuberculosis and rifampicin resistance in an HIV endemic population. J Clin Microbiol. 2015;53 : 2188–2194. doi: 10.1128/JCM.03530-14 25926493
31. Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR. Genetic Manipulation of Mycobacterium tuberculosis. Curr Protoc Microbiol. 2007;Chapter 10: Unit 10A.2. doi: 10.1002/9780471729259.mc10a02s6 18770603
32. Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon : An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS One. 2014;9: e112963. doi: 10.1371/journal.pone.0112963 25409509
33. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25 : 1754–60. doi: 10.1093/bioinformatics/btp324 19451168
34. Picard [Internet]. http://broadinstitute.github.io/picard
35. Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22 : 2688–90. 16928733
36. Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103 : 2869–73. 16477032
37. Demay C, Liens B, Burguière T, Hill V, Couvin D, Millet J, et al. SITVITWEB—a publicly available international multimarker database for studying Mycobacterium tuberculosis genetic diversity and molecular epidemiology. Infect Genet Evol. 2012;12 : 755–66. doi: 10.1016/j.meegid.2012.02.004 22365971
38. Ester M, Kriegel H-P, Jörg S, Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. In: Evangelos Simoudis; Jiawei Han; UM Fayyad, editor. Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD-96). AAAI Press; 1996. pp. 226–231.
39. Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. Elsevier Ltd; 2013;13 : 137–46.
40. Walker TM, Lalor MK, Broda A, Saldana Ortega L, Morgan M, Parker L, et al. Assessment of Mycobacterium tuberculosis transmission in Oxfordshire, UK, 2007–12, with whole pathogen genome sequences: an observational study. Lancet Respir Med. 2014;2 : 285–92. doi: 10.1016/S2213-2600(14)70027-X 24717625
41. Guerra-Assunção J, Crampin A, Houben R, Mzembe T, Mallard K, Coll F, et al. Large scale population-based whole genome sequencing of Mycobacterium tuberculosis provides insights into transmission in a high prevalence area. Elife. 2015;4 : 1–17.
42. Bonnet E, Van de Peer Y. zt: a software tool for simple and partial Mantel tests. J Stat Softw. 2002;7 : 1–12.
43. Inman JW. Navigation and nautical astronomy for use of British seamen. 3rd ed. London: C. and J. Rivington; 1835. 20895631
44. Swofford DL. PAUP: Phylogenetic Analysis Using Parsimony, Version 3.1 Computer program distributed by the Illinois Natural History Survey, Champaign, Illinois. 1991.
45. Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45 : 784–90. doi: 10.1038/ng.2656 23749189
46. Bryant JM, Schürch AC, van Deutekom H, Harris SR, de Beer JL, de Jager V, et al. Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect Dis. 2013;13 : 110. doi: 10.1186/1471-2334-13-110 23446317
47. Roetzer A, Diel R, Kohl T a., Rückert C, Nübel U, Blom J, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. Neyrolles O, editor. PLoS Med. 2013;10: e1001387. doi: 10.1371/journal.pmed.1001387 23424287
48. Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet. 2011;43 : 482–6. doi: 10.1038/ng.811 21516081
49. Gagneux S, Small PM. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. 2007;7 : 328–37. 17448936
50. Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 2008;6: e311. doi: 10.1371/journal.pbio.0060311 19090620
51. Coll F, McNerney R, Guerra-Assunção JA, Glynn JR, Perdigão J, Viveiros M, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5 : 4812. doi: 10.1038/ncomms5812 25176035
52. South Africa KwaZulu-Natal location map [Internet]. https://commons.wikimedia.org/wiki/File:South_Africa_KwaZulu-Natal_location_map.svg
53. Ma Z, Lienhardt C, McIlleron H, Nunn AJ, Wang X. Global tuberculosis drug development pipeline: the need and the reality. Lancet. Elsevier Ltd; 2010;375 : 2100–9.
54. Song T, Park Y, Shamputa IC, Seo S, Lee SY, Jeon HS, et al. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β′ subunit of RNA polymerase. Mol Microbiol. 2014;91 : 1106–1119. doi: 10.1111/mmi.12520 24417450
55. De Vos M, Müller B, Borrell S, Black P a., Van Helden PD, Warren RM, et al. Putative compensatory mutations in the rpoc gene of rifampin-resistant mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother. 2013;57 : 827–832. doi: 10.1128/AAC.01541-12 23208709
56. Safi H, Lingaraju S, Amin A, Kim S, Jones M, Holmes M, et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-β-D-arabinose biosynthetic and utilization pathway genes. Nat Genet. Nature Publishing Group; 2013;45 : 1190–7.
57. Iseman MD. Tuberculosis therapy: past, present and future. Eur Respir J. 2002;20 : 87S–94s.
58. McEvoy CRE, Cloete R, Müller B, Schürch AC, van Helden PD, Gagneux S, et al. Comparative analysis of Mycobacterium tuberculosis pe and ppe genes reveals high sequence variation and an apparent absence of selective constraints. PLoS One. 2012;7: e30593. doi: 10.1371/journal.pone.0030593 22496726
59. Coscolla M, Barry PM, Oeltmann JE, Koshinsky H, Shaw T, Cilnis M, et al. Genomic Epidemiology of Multidrug-resistant Mycobacterium tuberculosis During Transcontinental Spread. J Infect Dis. 2015; 1–9.
60. Boehme CC, Nabeta P, Hillemann D, Nicol MP, Shenai S, Krapp F, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363 : 1005–15. doi: 10.1056/NEJMoa0907847 20825313
61. Cattamanchi A, Dantes RB, Metcalfe JZ, Jarlsberg LG, Grinsdale J, Kawamura LM, et al. Clinical characteristics and treatment outcomes of patients with isoniazid-monoresistant tuberculosis. Clin Infect Dis. 2009;48 : 179–85. doi: 10.1086/595689 19086909
62. Denkinger CM, Pai M, Dowdy DW. Do we need to detect isoniazid resistance in addition to rifampicin resistance in diagnostic tests for tuberculosis? PLoS One. 2014;9: e84197. doi: 10.1371/journal.pone.0084197 24404155
63. Dlamini-Mvelase NR, Werner L, Phili R, Cele LP, Mlisana KP. Effects of introducing Xpert MTB/RIF test on multi-drug resistant tuberculosis diagnosis in KwaZulu-Natal South Africa. BMC Infect Dis. 2014;14 : 442. doi: 10.1186/1471-2334-14-442 25129689
64. Mills HL, Cohen T, Colijn C. Community-wide isoniazid preventive therapy drives drug-resistant tuberculosis: a model-based analysis. Sci Transl Med. 2013;5 : 180ra49. doi: 10.1126/scitranslmed.3005260 23576815
65. Cohen T, Lipsitch M, Walensky RP, Murray M. Beneficial and perverse effects of isoniazid preventive therapy for latent tuberculosis infection in HIV-tuberculosis coinfected populations. Proc Natl Acad Sci U S A. 2006;103 : 7042–7. 16632605
66. Churchyard GJ, Fielding KL, Lewis JJ, Coetzee L, Corbett EL, Godfrey-Faussett P, et al. A Trial of Mass Isoniazid Preventive Therapy for Tuberculosis Control. N Engl J Med. 2014;370 : 301–310. doi: 10.1056/NEJMoa1214289 24450889
67. Eldholm V, Monteserin J, Rieux A, Lopez B, Sobkowiak B, Ritacco V, et al. Four decades of transmission of a multidrug-resistant Mycobacterium tuberculosis outbreak strain. Nat Commun. Nature Publishing Group; 2015;6 : 7119.
68. Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. Nature Publishing Group; 2015;47 : 242–249.
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2015 Číslo 9
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Současné postavení a přínos sartanů v klinické praxi
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
- Hypertrofická obstrukční kardiomyopatie ve světle moderní farmakoterapie – kazuistika
Nejčtenější v tomto čísle
- Asporin Is a Fibroblast-Derived TGF-β1 Inhibitor and a Tumor Suppressor Associated with Good Prognosis in Breast Cancer
- Simplified HIV Testing and Treatment in China: Analysis of Mortality Rates Before and After a Structural Intervention
- Effectiveness of Electronic Reminders to Improve Medication Adherence in Tuberculosis Patients: A Cluster-Randomised Trial
- Changes in Intake of Fruits and Vegetables and Weight Change in United States Men and Women Followed for Up to 24 Years: Analysis from Three Prospective Cohort Studies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy