
Publication Bias in Antipsychotic Trials: An Analysis of Efficacy Comparing the Published Literature to the US Food and Drug Administration Database
Background:
Publication bias compromises the validity of evidence-based medicine, yet a growing body of research shows that this problem is widespread. Efficacy data from drug regulatory agencies, e.g., the US Food and Drug Administration (FDA), can serve as a benchmark or control against which data in journal articles can be checked. Thus one may determine whether publication bias is present and quantify the extent to which it inflates apparent drug efficacy.
Methods and Findings:
FDA Drug Approval Packages for eight second-generation antipsychotics—aripiprazole, iloperidone, olanzapine, paliperidone, quetiapine, risperidone, risperidone long-acting injection (risperidone LAI), and ziprasidone—were used to identify a cohort of 24 FDA-registered premarketing trials. The results of these trials according to the FDA were compared with the results conveyed in corresponding journal articles. The relationship between study outcome and publication status was examined, and effect sizes derived from the two data sources were compared. Among the 24 FDA-registered trials, four (17%) were unpublished. Of these, three failed to show that the study drug had a statistical advantage over placebo, and one showed the study drug was statistically inferior to the active comparator. Among the 20 published trials, the five that were not positive, according to the FDA, showed some evidence of outcome reporting bias. However, the association between trial outcome and publication status did not reach statistical significance. Further, the apparent increase in the effect size point estimate due to publication bias was modest (8%) and not statistically significant. On the other hand, the effect size for unpublished trials (0.23, 95% confidence interval 0.07 to 0.39) was less than half that for the published trials (0.47, 95% confidence interval 0.40 to 0.54), a difference that was significant.
Conclusions:
The magnitude of publication bias found for antipsychotics was less than that found previously for antidepressants, possibly because antipsychotics demonstrate superiority to placebo more consistently. Without increased access to regulatory agency data, publication bias will continue to blur distinctions between effective and ineffective drugs.
: Please see later in the article for the Editors' Summary
Published in the journal:
. PLoS Med 9(3): e32767. doi:10.1371/journal.pmed.1001189
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1001189
Summary
Background:
Publication bias compromises the validity of evidence-based medicine, yet a growing body of research shows that this problem is widespread. Efficacy data from drug regulatory agencies, e.g., the US Food and Drug Administration (FDA), can serve as a benchmark or control against which data in journal articles can be checked. Thus one may determine whether publication bias is present and quantify the extent to which it inflates apparent drug efficacy.
Methods and Findings:
FDA Drug Approval Packages for eight second-generation antipsychotics—aripiprazole, iloperidone, olanzapine, paliperidone, quetiapine, risperidone, risperidone long-acting injection (risperidone LAI), and ziprasidone—were used to identify a cohort of 24 FDA-registered premarketing trials. The results of these trials according to the FDA were compared with the results conveyed in corresponding journal articles. The relationship between study outcome and publication status was examined, and effect sizes derived from the two data sources were compared. Among the 24 FDA-registered trials, four (17%) were unpublished. Of these, three failed to show that the study drug had a statistical advantage over placebo, and one showed the study drug was statistically inferior to the active comparator. Among the 20 published trials, the five that were not positive, according to the FDA, showed some evidence of outcome reporting bias. However, the association between trial outcome and publication status did not reach statistical significance. Further, the apparent increase in the effect size point estimate due to publication bias was modest (8%) and not statistically significant. On the other hand, the effect size for unpublished trials (0.23, 95% confidence interval 0.07 to 0.39) was less than half that for the published trials (0.47, 95% confidence interval 0.40 to 0.54), a difference that was significant.
Conclusions:
The magnitude of publication bias found for antipsychotics was less than that found previously for antidepressants, possibly because antipsychotics demonstrate superiority to placebo more consistently. Without increased access to regulatory agency data, publication bias will continue to blur distinctions between effective and ineffective drugs.
: Please see later in the article for the Editors' Summary
Introduction
Evidence-based medicine is valuable only to the extent that the available evidence is complete and unbiased. Unfortunately, whether research results are published, and how they are published, often depends on their statistical significance [1],[2], which alters the apparent risk–benefit ratio of drugs. Within medicine, a recent review found evidence for various forms of publication bias within 40 different indications [3].
Despite ample evidence for the existence of publication bias, there is little evidence of its quantitative impact on the apparent efficacy of most drugs. Most methods for studying publication bias provide indirect evidence for nonpublication or outcome reporting bias. A frequently used method is to examine a cohort of published studies for “small study effects” (smaller studies showing larger treatment effects) in the form of funnel plot asymmetry [4]. Despite its wide use, this approach has limitations. First, although a funnel plot may suggest that studies with smaller effect sizes have not been published, it cannot prove that such studies in fact ever existed. Second, the effect sizes plotted are based on results that are published, and one cannot be sure whether and to what extent such results have been affected by outcome reporting bias. The true underlying results—the results according to the prespecified outcomes—usually remain unknown.
By contrast, such data are often available from drug regulators, such as the US Food and Drug Administration (FDA). Because the FDA gathers data from premarketing trials both before inception and after completion, it functions as both a registry and a results database [5]. For any given cohort of trials, results according to FDA reviews can be compared to corresponding results according to the published literature. Any discrepancies between the two sources provide direct evidence of publication bias.
FDA reviews have been used to document publication bias across various medical indications in at least two studies [6],[7]. An advantage of looking broadly across medical indications is that it documents the wide scope of publication bias, while focusing on a single indication may lead some readers to assume that the phenomenon is specific to that indication. On the other hand, FDA data from a single indication may be more useful to meta-analysts, since data measuring the same construct allow for the calculation of an overall effect size. Such data should also be useful to clinicians, who are interested in the true efficacy of a specific drug class that they prescribe.
In a previous study of antidepressants, our group found that publication bias nearly doubled the apparent proportion of positive trials and increased the apparent effect size by one-third [8]. This raises the question as to whether publication bias similarly affects the apparent efficacy of other drug classes.
Schizophrenia has a lifetime prevalence of 0.55% [9], and its core symptom, psychosis, is the third most disabling condition worldwide [10]. The objective of this study is to use FDA data as an independent benchmark or control to determine whether, and to what extent, the apparent efficacy of second-generation antipsychotics has been influenced by publication bias.
Methods
Ethics Statement
This study was approved by the Research and Development Committee of the Portland Veterans Affairs Medical Center. It was reported according to the guidelines of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) group (Text S1).
Data Procurement
Procurement of data from FDA reviews
We identified the phase 2/3 clinical trial programs leading to the FDA's marketing approval of eight second-generation antipsychotic drugs for the treatment of schizophrenia. This retrospective cohort consisted of 24 FDA-registered short-term double-blind placebo-controlled trials conducted between December 1993 and May 2005. The FDA Drug Approval Packages were publicly available at, and downloaded from, the FDA's web site [5],[11] for all drugs except risperidone (Risperdal), for which we obtained the Drug Approval Package via request to the FDA's Freedom of Information Office. (Because risperidone was approved in 1993, it was not subject to the Electronic Freedom of Information Act of 1996 [12].) Within the Drug Approval Packages, we examined data relevant to the agency's determination of drug efficacy in medical reviews, statistical reviews, and administrative correspondence.
In order to make these documents more accessible to readers and researchers, the FDA Drug Approval Package documents were processed using Adobe Acrobat as follows: (1) reviews of the same type but presented as multiple PDF files (e.g., Medical Review Parts 1, 2, 3, and 4) were combined into single PDFs; (2) page numbers were added as footers; (3) because the FDA had presented the documents in an unsearchable format [5], they were rendered searchable using optical character recognition; and (4) text directly quoted in the present article was highlighted. These documents have been placed in a digital repository of Oregon Health & Science University. The reader may find them by navigating to (1) http://www.ohsu.edu/library/, (2) “Digital Resources Library,” then (3) “FDA Drug Approval Documents.”
Procurement of data from journal articles (literature search)
The published literature was searched for journal articles matching each FDA-reviewed trial, with a cutoff date of May 5, 2010. The best match for each trial was identified using the following parameters: drug name, name of active comparator (when used), dosage groups, their sample sizes, trial duration, and name of principal investigator.
The initial searches employed two databases, PubMed and Cochrane Central Register of Controlled Trials. The search strategy was for the title field to include the name of the drug and either “schizophrenia” or “schizoaffective,” and for the word “placebo” to appear in any field (e.g., title, keywords, abstract). As an example, when searching Cochrane Central Register of Controlled Trials for relevant aripiprazole trials, the search syntax was “(aripiprazole and schizo$).ti. and placebo.af.”
From the search output, titles and abstracts were screened so as to exclude journal articles focused on topics other than the overall efficacy of the drug for schizophrenia or schizoaffective disorder. Thus, articles focused on the following topics were excluded: other indications (e.g., bipolar disorder, treatment-resistant schizophrenia), subsets with specific comorbid conditions, particular symptom clusters (e.g., agitation, weight change), safety (as opposed to efficacy), specific demographic samples, trials lacking a parallel design (add-on, open-label, crossover), trials that were not placebo-controlled, trials not involving acute treatment (long-term trials, including maintenance trials), and trials involving other routes of administration.
Trials were counted as published according to the method of our earlier study [8]. The intent was to include journal articles that provided data sufficient for meta-analysis yet were reasonably discoverable by, and accessible to, the average clinician. Articles in languages other than English were excluded. Meeting abstracts were excluded—trials were required to be fully published [13]. An FDA-registered trial was considered published if it could be matched with a primary publication [14]. The preferred type of primary publication was a stand-alone publication (i.e., an article devoted to reporting the results of a single trial). If no stand-alone publication could be found, aggregate publications, in which multiple trials were covered in a single article, were sought. However, aggregate publications were accepted only if all trials included in the article presented primary data; aggregate publications that were heterogeneous, i.e., those reporting a mix of primary and secondary trial data, were excluded.
If the original searches using PubMed and the Cochrane Central Register of Controlled Trials suggested that an FDA-registered trial was unpublished, Ovid Medline was searched for the three most recent review articles focused on the efficacy of the drug in question. These review articles were then examined for trial bibliographic information. The Ovid Medline search strategy was similar to that given above for the Cochrane Central Register of Controlled Trials, except that the search was restricted to review articles in English and that “placebo” was omitted as a search term. For example, when the original search yielded no journal articles matching two FDA-registered trials of aripiprazole, the following search strategy was used: (1) “(aripiprazole and schizo$).ti. and review.pt.”; (2) limit (1) to English language.
Finally, because pharmaceutical companies pledged in 2004 to increase transparency by posting trial results publicly, the PhRMA Clinical Study Results Database (decommissioned December 2011) and the drug sponsors' own web sites were searched for information as to whether these trials were published.
If all the above steps yielded no evidence of publication, it was concluded that that the trial in question was not reasonably accessible to the average clinician, and it was considered unpublished.
Data Extraction and Entry
Double data extraction and entry was employed in this study. The primary outcome was identified for each trial by two authors (D. K. and L. S.) working independently, with the stipulation that it be either the Brief Psychiatric Rating Scale (BPRS) [15] or the Positive and Negative Syndrome Scale (PANSS) [16]. The two raters' sets of entries were compared, and any discrepancies were resolved through consensus. The primary rating scale according to the FDA matched that in the journal article for all trials but one.
Throughout the FDA reviews and journal articles, the primary analyses involved modified intention-to-treat methods [17] for handling dropouts: mixed-effects model repeated measures [18] was used for some of the iloperidone trials (in both FDA reviews and journal articles); last observation carried forward (LOCF) [19] was used for all other trials. The numerical results according to the primary outcome (p-values, means, standard deviations, standard errors, and/or confidence intervals) were extracted and entered independently by D. K. and L. S., for the FDA data followed by the journal article data. Boolean formulas in Excel were used to compare and flag any mismatches between the two sets of entries. For each mismatch, the original data source (FDA review or journal article) was re-examined by E. H. T. to determine which of the two entries was correct.
In addition to the numerical or continuous data mentioned above, categorical data were extracted. The FDA's regulatory decision on each trial was rated independently by D. K. and L. S. at one of three levels: positive (supportive of efficacy), questionable (neither clearly positive nor clearly negative), or negative (not supportive of efficacy). Any discrepancies between the two sets of ratings were discussed among the three authors while consulting the FDA review materials, and consensus was reached. When the judgment in the FDA's review was unclear, the clinical trials section of the original product labeling was downloaded from the FDA web site and referred to for clarification. For each journal publication, the presentation of the result on the primary outcome was rated, again independently by D. K. and L. S., as positive, negative, or questionable. Again, any differences between the two sets of ratings were resolved by consensus.
For trials where the FDA ratings and journal ratings disagreed, the authors shared and discussed their observations as to how they disagreed. In contrast to our previously published study [8], the differences found were, we felt, too varied and nuanced to be meaningfully and reliably captured using a categorical rating system and then subjected to statistical analysis. Instead, the differences between the FDA and journal presentations of the trial results are described in narrative format. This text was drafted by E. H. T., critically revised by D. K. and L. S., and includes several direct quotes from the data sources.
Data Analysis
Trial outcome versus publication status
As noted above, the FDA's regulatory decisions regarding the trials were classified in this study as (1) positive (clearly supportive of efficacy), (2) questionable (marginal or borderline support for efficacy), or (3) negative (clearly not supportive of efficacy). Categories 2 and 3 were combined into a not-positive grouping. The strength of the association between the FDA regulatory decision and publication status (published versus unpublished) was calculated as Fisher's exact p (two-tailed) using the csi command with the exact option in Stata 11 [20].
Meta-analysis
As described previously [8], two meta-analyses were conducted: a conventional meta-analysis using published data, and a control meta-analysis using FDA data. Data from active comparators were excluded, so that each drug's effect size was derived solely from data collected by that drug's sponsor. The measure of effect size used was Hedges's g [21], calculated using the following equation [22]:(1)The values for g were adjusted using Hedges's correction for small sample size [23]. To calculate t, as previously described [8], precise p-values were used together with the degrees of freedom as arguments in Excel's TINV function. If the p-value was instead reported as a range (e.g., “p<0.05”), collateral data to calculate effect size were used according to the following hierarchy: standard deviations, standard errors, and 95% confidence interval around the mean difference. In the few cases where none of these data were available in the journal article, data were imputed from the FDA database. Conversely, when the FDA database lacked the necessary data, data were imputed from the corresponding journal article. The purpose of this imputation was to err in the direction of the null hypothesis of no difference in effect size between the FDA and the published literature. By convention, positive and negative effect size values were used to signify superiority and inferiority to placebo, respectively.
The typical trial compared multiple doses of study drug to placebo (fixed-dose design). In such cases, a single trial-level effect size and standard error was calculated using a fixed effects model [24] to pool the values from that trial's multiple treatment arms. To avoid a spuriously low standard error, each trial's shared placebo n was counted once rather than redundantly for each dose group. A limitation of this method is that it only partially addresses error due to correlation between the comparisons [25].
Using the trial-level effect size values, the random effects pooling method [26] in Stata [20],[27] was used to calculate mean weighted values for each drug and for the entire drug class. Heterogeneity was assessed using the I2 statistic [28]. As has been recommended [29], 95% confidence intervals around I2 were calculated using the non-central chi-squared-based approach within the heterogi module [30] for Stata.
As an error check, author E. H. T. returned to the dataset from the original double data extraction and entry, repeated the calculations, and electronically compared the resulting values with those calculated earlier.
Meta-regression was conducted using the Metareg module for Stata 11 [27] to contrast the FDA-based effect sizes of the published trials with those of the unpublished trials. The method used for estimating the between-study variance was residual maximum likelihood, the default method in Stata 11. A similar analysis was used to contrast the effect sizes derived from the FDA data, from both published and unpublished trials, with the effect sizes derived from the published literature.
Results
Number of Trials
According to the FDA reviews, the eight drugs examined were approved based on the efficacy results from 24 short-term double-blind placebo-controlled trials. Thus, three trials were required, on average, to approve one second-generation antipsychotic. Table 1 shows the characteristics of these trials according to the FDA. Table 2 shows bibliographic information on these trials. As seen, 20 of the trials (83%) were published and four (17%) were unpublished. The 20 published trials were published in 19 journal articles. The lack of one-to-one correspondence occurred because (1) one article [31] reported the results of three iloperidone trials (included because their results were not reported elsewhere in stand-alone form—see Methods), and (2) one FDA-registered trial of risperidone was published in two separate articles, one for the Canadian sites and another for the US sites, as if they were separate trials (details below).
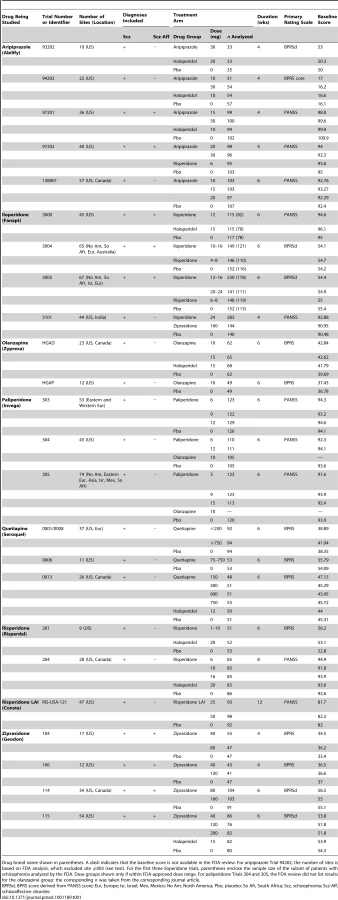

Trial Outcome versus Publication Status
While most of the published trials (15/20 = 75%) were FDA-positive, most of the unpublished trials (3/4 = 75%) were FDA-negative or -questionable. The relationship between trial outcome and publication status, shown in Figure 1, did not reach statistical significance (Fisher's exact test p = 0.09). Handling the FDA-questionable category in alternate ways yielded similar p-values (see Table S1). In a post hoc analysis conducted after reclassifying unpublished ziprasidone Trial 115 from positive to questionable (in part because of its statistical inferiority to the active comparator haloperidol—see narrative trial details below), this relationship did reach statistical significance (p = 0.012). When this trial was excluded from the analysis, it remained significant (p = 0.03).

Overall, according to the FDA reviews, two-thirds of the trials (16/24 = 67%, binomial 95% confidence interval [CI95%] 45% to 84%) were positive, with the remaining one-third either questionable or negative. By contrast, according to the journal articles, there were 21 trials, all of them positive (CI95% 84% to 100%).
Meta-Analysis
The dose groups reported on in the journal articles matched those in the FDA reviews. The statistical output from the two meta-analyses, i.e., that of the FDA and that of the journal data, is reproduced in Table S2 and Table S3, respectively. Figure 2 is a forest plot of the effect size and confidence intervals based on the data from the FDA reviews. For the published trials, the effect size was 0.47 (CI95% 0.40 to 0.54; I2 38%, CI95% 0% to 62%). For the unpublished trials, the effect size was less than half that, 0.23 (CI95% 0.07 to 0.39; I2 0%, CI95% 0% to 68%). By meta-regression, the difference between the effect sizes for the published versus unpublished trials was statistically significant (β = −0.25, CI95%−0.47 to −0.03, t = −2.36, p = 0.027). Because the unpublished trials were confined to two of the eight drugs, this latter analysis was repeated while adding drug as an explanatory variable. Here the effect of drug was not statistically significant (β = 0.02, CI95% −0.01 to 0.05, t = 1.35, p = 0.19), and the difference between the published and unpublished trials remained significant (β = −0.28, CI95% −0.49 to −0.06, t = −2.62, p = 0.016).
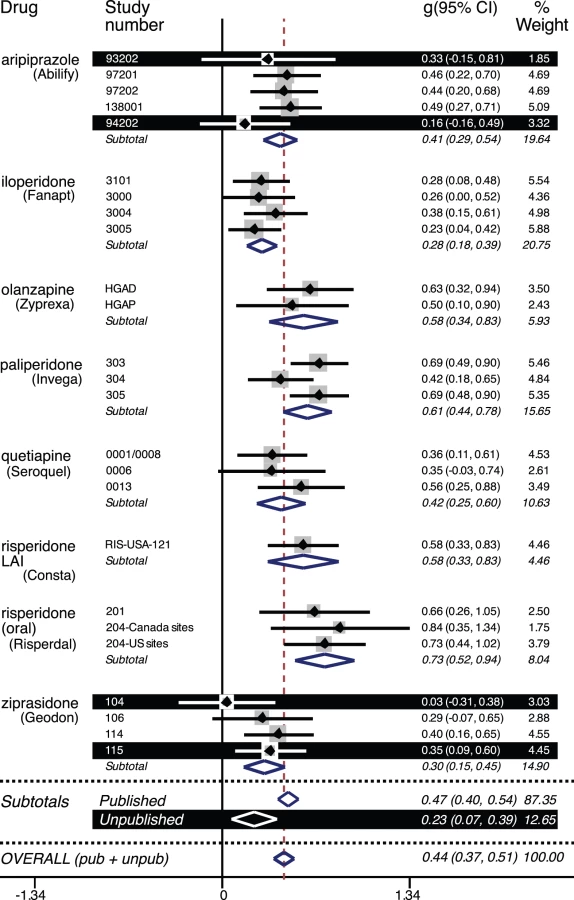
Within the published trials, there was essentially no difference between the effect sizes derived from the FDA reviews (0.47 from above) and those derived from the journal articles (0.48, CI95% 0.40 to 0.56; I2 46%, CI95% 0% to 66%). By meta-regression, this difference was not significant (β = 0.002, CI95% −0.11 to 0.11, t = 0.03, p = 0.98).
The FDA data from the published and unpublished trials were combined into overall FDA-based effect sizes for each of eight second-generation antipsychotics. These effect sizes, along with their confidence intervals, are shown alongside their corresponding journal-based effect sizes in Figure 3. For individual drugs, the difference in effect size ranged from a 4% decrease (risperidone LAI) to a 20% increase (ziprasidone). With all drugs combined, the overall FDA-based effect size was 0.44 (CI95% 0.37 to 0.51; I2 43%, CI95% 0% to 63%). Compared to this, the overall journal-based effect size (0.48 from above) represented a slight (8%) increase in effect size due to publication bias, which was nonsignificant by meta-regression (β = 0.03, CI95% −0.08 to 0.14, t = 0.62, p = 0.54). For each of the four effect sizes reported above, the lower confidence limit for I2 was zero, and the upper confidence limit ranged from 62% to 68%, between proposed landmarks for moderate and high levels of heterogeneity [32].

Unpublished Trials (n = 4 Trials)
There were four trials for which we were unable to find any evidence of publication. The literature search efforts are detailed for each trial in Text S2. Following are details regarding the conduct and results of these trials.
Unpublished aripiprazole trials (n = 2)
Aripiprazole Trial 94202 was one of two unpublished aripiprazole trials (Table 2). As shown in Table 1, it involved over 200 patient-participants at 22 sites in the US. According to the FDA medical officer review (pages 43 and 168 of 238), the data from one of the sites (site 003) were excluded because its investigator (Richard Borison, M.D., Ph.D.) was “disqualified due to allegations of research misconduct and conviction on criminal charges.” Neither the 10-mg dose nor the 30-mg dose separated from placebo (p = 0.89 and p = 0.12, respectively). The active comparator haloperidol separated from placebo on one of the primary rating scales (BPRS core, p = 0.0495) but not the other (Clinical Global Impression Scale–Improvement, p = 0.08). Consistent with the former, the statistical reviewer deemed the trial negative; consistent with the latter, the medical reviewer deemed the trial “failed.” For our purposes, we adopted the more conservative judgment of the study as failed, and thus classified it as questionable (Figure 1).
Aripiprazole Trial 93202 involved over 100 patients at ten US sites (Table 1). Like aripiprazole Trial 94202, aripiprazole did not separate statistically from placebo (p = 0.173), but unlike Trial 94202, the active comparator haloperidol clearly separated from placebo (p = 0.010). Thus, the FDA deemed Trial 93202 a negative, rather than a failed, trial (Figure 1). This trial was not published.
Unpublished ziprasidone trials (n = 2)
Ziprasidone Trial 115 was one of two unpublished ziprasidone trials (see Text S2 for results of literature search). As seen in Table 1, ziprasidone Trial 115 involved 53 US sites and over 400 patients and five treatment arms: three doses of ziprasidone (20 mg bid, 60 mg bid, and 100 mg bid), placebo, and haloperidol 10 mg as an active comparator. All active treatment arms demonstrated statistical superiority to the placebo arm, and there was evidence of a positive dose–response relationship. Because the FDA judges efficacy based on whether the study drug demonstrates superiority to placebo, the trial was ultimately considered positive.
Earlier, however, efficacy results from this trial, together with a safety concern, proved to be a temporary obstacle to marketing approval. When ziprasidone was first being reviewed, the FDA was concerned about the drug's tendency to prolong the QT interval and how that might affect the overall risk–benefit ratio and, consequently, its approvability. The cardiology consultant is quoted within the medical review (page 2 of 223):
Dr. Ganley recommended that, “unless efficacy data suggests superior benefit over currently available drugs, ziprasidone should be considered for second line therapy with adequate warnings of risk associated with drugs that prolong the QT interval.”
Ziprasidone Trial 115 was the only premarketing trial in which ziprasidone was compared to a marketed antipsychotic as well as to placebo. In this trial, ziprasidone failed to show the above-mentioned “superior benefit over currently available drugs.” In fact, it showed the opposite, that ziprasidone was statistically inferior to the active comparator haloperidol. According to the statistical review (page 7 of 61):
The haloperidol (active control) seemed to have a larger decrease in changes from baseline for most of the primary efficacy endpoints.
This statement was followed in the statistical review by Table 1R, which documented the statistical superiority of haloperidol over ziprasidone in this trial. In the column comparing the combination of all three ziprasidone dose groups to haloperidol, the p-values achieved on the scales BPRS total, BPRS core, Clinical Global Impression Scale–Severity, PANSS total, and PANSS negative symptoms were 0.037, 0.002, 0.002, 0.017 and 0.335, respectively. Thus, ziprasidone was statistically inferior to haloperidol on four of the five scales. The statistical reviewer also noted (page 7 of 61):
The secondary objective comparing haloperidol with ziprasidone was changed to that of comparing haloperidol with placebo after the completion of the trial [emphasis in original].
Thus, with respect to the original secondary objective, Trial 115 was negative. (The fact that this trial could be considered positive is because secondary outcomes are trumped by primary outcomes, which involved comparisons of ziprasidone to placebo.) Weighing the overall risk–benefit ratio of ziprasidone during the earlier review cycle, the medical review (page 3 of 223) quoted the Division Director:
“[Z]iprasidone's comparative performance (Study 115) supports a conclusion that it is less efficacious than haloperidol, a long marketed antipsychotic drug.” Dr. Leber [the Division Director] recommended a nonapprovable action….The nonapprovable letter of June 17th, 1998 asserted that a sufficient advantage over currently marketed antipsychotics had not been demonstrated that could outweigh the risk of potentially fatal arrhythmias because of the demonstrated QTc prolongation.
Later, a July 2000 Advisory Committee recommended that the FDA approve ziprasidone in spite of the QT issues. Following this, the inferior performance of ziprasidone relative to haloperidol ceased to be a concern, so Trial 115 could be considered, from a regulatory standpoint, supportive of efficacy. Based on this final judgment of the FDA, Trial 115 is classified as positive in Figure 1 and in our primary statistical analysis and as questionable in a post hoc analysis.
The results of another ziprasidone trial, Trial 104, were not published. Table 1 shows that, in Trial 104, two doses of ziprasidone were tested against placebo. There was also a third dose group of 10 mg/d, but it is not shown in the table or included in our analyses because that dose was not approved by the FDA. This trial involved nearly 200 patients (including the patients assigned to the low-dose group) at 17 US sites. According to the FDA statistical review (pages 16 and 12 of 61):
None of the three primary efficacy endpoints reached statistical significance at two-sided 0.05 level either based on the ITT LOCF or the Completer analyses in Study 104….There was no dose-response trend either including or excluding placebo with respect to any of the primary endpoints.
Outcome Reporting Bias (n = 5 Trials)
Among the eight trials classified as having FDA-negative or -questionable outcomes (Figure 1), five were published. The FDA reviews of these five trials presented data that raised concerns about drug efficacy. Below we note whether and how these concerns were conveyed in the corresponding journal articles.
Iloperidone trials (n = 3)
One efficacy issue that was apparent from the FDA review of iloperidone, but not from the corresponding journal articles, was that the drug frequently proved to be statistically inferior to active comparators (Table 3 and detailed below).

Another issue apparent from the FDA review was that iloperidone's efficacy relative to placebo varied according to the patient diagnostic population studied. Consequently, as has been reported previously [33], iloperidone's path to FDA approval was somewhat convoluted. To elaborate, of the four iloperidone premarketing trials, the first three called for the recruitment of patients diagnosed with either schizophrenia or schizoaffective disorder: Trials 3000, 3004, and 3005. The protocols for these three trials were submitted to and approved by the FDA, and these trials were conducted between 1998 and 2001. The results of these three trials were presented together in a single 2008 journal article [31].
In contrast to the way these trials were reported in the journal article (details below), the FDA review initially judged only one of the three trials to be positive (Trial 3004; Table 3). Because the FDA requires two positive studies in order to approve a drug for marketing, the agency informed the drug's sponsor in 2001 that another positive trial would be required for approval. The agency added that such a trial should be restricted to patients with schizophrenia, i.e., excluding patients with schizoaffective disorder.
A fourth premarketing trial, restricted to patients with schizophrenia, was undertaken in 2005. The results from this trial were positive, and they were submitted to the FDA in a New Drug Application (NDA) in November 2007. On July 25, 2008, the agency issued a not-approvable letter, stating that the sponsor had failed to demonstrate efficacy (through two or more positive trials) in patients with schizophrenia.
After the sponsor appealed the decision, the FDA conducted a post hoc reanalysis of the first three trials, examining efficacy within the subset of patients diagnosed with schizophrenia, i.e., excluding data from the patients in those trials diagnosed with schizoaffective disorder. This reanalysis yielded a positive result for one of the initial three trials, Trial 3005 (Table 3). This positive result from Trial 3005, together with the positive result from Trial 3101, yielded a total of two positive trials in patients with schizophrenia. The FDA's criterion for approval, i.e., two or more positive trials in a defined patient population, was now achieved, thus allowing the agency to approve iloperidone in May 2009.
Iloperidone Trial 3000
In iloperidone Trial 3000, according to the FDA, the dose group prespecified in the protocol as primary was the combination of patients taking either 8 mg or 12 mg of iloperidone per day. For this combined dose group, using the full schizophrenia-plus-schizoaffective sample of patients, the drug–placebo difference was nonsignificant (p = 0.065). The corresponding journal article [31] reported this nonsignificant p-value in the text of the results section but preceded this with the report of a significant finding (p = 0.047) obtained with the 12-mg dose alone, a secondary outcome. This significant result was also reported in the abstract, while the nonsignificant result on the primary dose group was not. The FDA's findings that iloperidone was statistically inferior to haloperidol for this all-patients sample (p = 0.027; Table 3) was not reported in the corresponding journal article. Additionally, the FDA's finding of a lack of statistical superiority to placebo (p = 0.148) with the schizophrenia-only subset was not reported in the journal article [31]. However, the sponsor did not learn of the FDA's intent to focus on the schizophrenia-only subset until the not-approvable letter was issued (July 2008), some months after the journal article was published (April 2008).
Iloperidone Trial 3004
Among the three iloperidone trials that recruited patients diagnosed with either schizophrenia or schizoaffective disorder, Trial 3004 was the only one for which the FDA found significant results for the all-patients sample (Table 3). We have classified it as questionable for two reasons. First, although the FDA initially judged this trial to be positive based on the all-patients sample, it later conducted a post hoc analysis on the subset of patients with schizophrenia, which yielded a nonsignificant result (p = 0.306). Commenting on this finding, the FDA medical reviewer stated (page 110 of 247):
The results of Study 3004 do not provide evidence of efficacy of iloperidone…in the treatment of schizophrenia versus placebo over 42 days of treatment.
Second, the FDA found that iloperidone was significantly inferior to the active comparator risperidone (Table 3), both for the subset of patients with schizophrenia (p = 0.021) and for the all-patients group (p = 0.034). According to the summary review by Division Director Thomas Laughren (page 6 of 21):
Thus, either approach to defining the sample for this study yields a result that favors a standard control agent over iloperidone.
This statistical inferiority to risperidone was not reported in published version of this trial [31].
Iloperidone Trial 3005
Trial 3005 also yielded a mix of positive and not-positive efficacy results. As with the other iloperidone trials, the FDA review revealed evidence of iloperidone's statistical inferiority to the active comparator (Table 3). Additionally, for the all-patients (schizophrenia-plus-schizoaffective) sample, the FDA found that the 0.05 threshold for statistical significance was not achieved for either dose group, while the journal publication [31] reported a significant result (p = 0.01) for one of the two dose groups.
In the FDA's post hoc analysis of the subset of patients with schizophrenia, the results were significant for both dose groups (p = 0.033 and p = 0.005, respectively). Thus, as a result of the FDA's reanalysis using this patient subset, the results of Trial 3005 changed from nonsignificant to significant, the opposite of what occurred with the reanalysis of Trial 3004. As was the case for the other two trials covered in this journal article [31], the results based on this patient subset were not reported. As can be seen in Table 3, these results were favorable for Trial 3005 (though unfavorable for Trials 3000 and 3004). As stated above, the fact that Trial 3005 was positive in the schizophrenia-only subset allowed the FDA to approve iloperidone: by focusing on patients with schizophrenia and combining this result with that of Trial 3101, the FDA's requirement of two positive trials was achieved.
Iloperidone Trial 3101
Trial 3101 was the only iloperidone premarketing trial to include only patients with schizophrenia and exclude those with schizoaffective disorder. As shown in Table 3, iloperidone demonstrated superiority to placebo (p = 0.007). However, the FDA also reported that iloperidone was significantly inferior to the active comparator (ziprasidone in this trial) in most of the comparisons. (These p-values were not reported in the FDA review, so they do not appear in Table 3.) As with the above-mentioned trials, iloperidone's statistical inferiority to the active comparator was not reported in the corresponding journal article [34].
Quetiapine Trial 0006
We also classified quetiapine Trial 0006 as questionable, rather than clearly positive or negative. According to the FDA medical officer's conclusion (page 82 of 245):
On balance, this study provides marginal support for antipsychotic efficacy of quetiapine, when titrated to a wide dose range. Strictly speaking, however, the data fall short of meeting the customary level of statistical proof, particularly for the observed cases [completers] analyses.
Regarding this same trial, the statistical reviewer stated (page 2 of 40):
The borderline statistical result of the LOCF [the protocol-prespecified primary method for handling dropouts; p = 0.07] is likely due to the less than anticipated treatment difference at 6 weeks (−8.1 versus −2.1). Note that there was no difference at all for the completers. [For example, using the BPRS rating scale, p = 0.95.] Thus, Figure 5 indicates that the entire treatment difference in the LOCF analysis is due to dropouts in the first 4 weeks of the trial.
The corresponding journal article [35] communicated the results of this trial with more favorable language. The abstract opened its presentation of the results with the following:
Significant differences (p equal to or less than 0.05) between treatment groups, which favored ICI 204,636 [quetiapine], were identified throughout the trial.
The results section opened its efficacy section with the following:
On days 14, 28, and 35, when statistically significant differences between treatment groups were detected, mean changes in BPRS total scores were….
In the sentence that followed, the above-mentioned p-value of 0.07 was reported and described as “marginally significant and favored ICH 204,636 [quetiapine].” Following that, four p-values above the 0.05 significance threshold were listed and described as “marginally significant.” The results of the completers analyses, described by the FDA as showing “no difference at all” (see above), were not reported in the journal article.
Ziprasidone Trial 106
We have classified ziprasidone Trial 106 as neither clearly positive nor negative, but rather questionable, for the following reasons. First, the FDA medical review stated:
Because this study showed statistical significance in only two of the three primary efficacy variables in week 4 only, it merely provides fair evidence for the antipsychotic properties of ziprasidone at a dose of 60 mg bid.
The FDA's point—that the efficacy of the 120-mg dose was in question because it failed to demonstrate superiority to placebo on all primary outcomes—was not apparent in the journal article [36], which stated:
In the intent-to-treat analysis of mean changes from baseline at 4 weeks, ziprasidone 120 mg/day was significantly more effective than placebo in improving mean BPRS total and CGI-S [Clinical Global Impression Scale–Severity] scores (P<0.05).
Second, a 40-mg/d (20 mg twice daily) dose group was included in this trial, and though it demonstrated efficacy in other trials and thus became an FDA-approved dose, it did not demonstrate statistical superiority to placebo in this trial on any of the three primary variables (e.g., p = 0.657 on the BPRS).
In the published version of this trial [36] the nonsignificance of the results at this dose were acknowledged, but without p-values and, compared to the 120-mg findings, less prominently in terms of placement. In the results section, while the 120-mg/d results were mentioned in the first line of the efficacy subsection, the 40-mg results were mentioned in the ninth line. In the discussion section, while the significant 120-mg results were mentioned in the first line, the nonsignificant 40-mg results were mentioned in the middle of the third paragraph. The abstract reported several significant results for the 120-mg dose but no results for the 40-mg dose.
Selective Reporting of Sites within Trials (n = 2 Trials)
Risperidone Trial 201
According to the FDA review, risperidone Trial 201 involved nine US sites. The total baseline sample consisted of 160 patients, of whom 156 were included in the LOCF analysis, this trial's primary method of handling dropouts. The corresponding journal article [37] reported a total sample size of only 36. The number of sites was not reported. No additional publications arising from this trial could be identified in our literature search or within a meta-analysis on risperidone [38] published four years later.
Because the FDA review did not break down Trial 201's results by site, it is unknown whether the published results were any more favorable to risperidone than those that were not published. Nevertheless, the overall results were positive according to both the FDA and the journal article. According to the FDA:
Study 201 provides unequivocal support for the effectiveness of risperidone as an antipsychotic agent.
Thus, there was evidence that the data from this trial were selectively reported in terms of the patients reported on, but this situation does not appear to meet criteria for publication bias in that it did not affect the strength and direction of the results [39], at least with respect to efficacy.
Risperidone Trial 204
According to the FDA review, risperidone Trial 204 was a single multicenter trial conducted at 26 sites, 20 in the US and six in Canada. Being a multicenter trial, the data from all sites were pooled in the FDA analysis. Rather than publish the results of this trial in a single journal article, the sponsor published reported the data results from the six Canadian sites in a 1993 journal article [40] as one positive trial and the data from the 20 US sites in a 1994 article [41] as a separate positive trial. The first article did not mention the US sites. The second article stated:
In the present article we report the results of a multicenter study of schizophrenic patients recruited at 20 centers in the United States. The study is part of a U.S.-Canadian collaborative investigation of risperidone in schizophrenia. The results of the Canadian arm of the investigation have been published [citation referencing [42]].
Three years later, the drug's sponsor reported the US and Canadian sites' results in a single article [42], but the article's first sentence stated:
Two pivotal controlled trials of risperidone have been conducted in North America, the Canadian study of Chouinard et al [citation referencing [40]] and the United States study of Marder and Meibach [citation referencing [41]]. In this report, we present the results of an analysis of the combined data from the two trials….
Therefore, while the FDA was aware that this was a single trial and analyzed it as such, readers, editors, and reviewers of journal articles would have thought they were reading two different trials. This “split” is shown with a horizontal dotted line in Figure 1, and it contributed to the lack of one-to-one correspondence between the FDA studies and journal articles shown in Table 1.
Discussion
Recapitulation of Findings
These data provide mixed evidence for publication bias for antipsychotic drug trials. One-sixth (17%) of the trials were unpublished. Among the four unpublished trials, three failed to show that the drug was superior to placebo, and one showed that the drug was statistically inferior to the active comparator. Among the 20 published trials, 15 (two-thirds) were FDA-positive; the five that were not positive showed some evidence of outcome reporting bias. However, the association between trial outcome and publication status was not statistically significant. The mean effect size derived from the published literature was only slightly higher than that derived from the FDA reviews, and the difference between them did not reach statistical significance. On the other hand, within the FDA dataset, the mean effect size of the published trials was approximately double that of the unpublished trials.
Publication Bias and Antipsychotics
Previous work on publication bias among antipsychotic trials has focused on alleged advantages of specific antipsychotics over one another [43],[44]. And within the context of a meta-analysis of second-generation antipsychotics, small study bias was suggested by an asymmetric funnel plot [45]. In the present study, FDA data provided a relatively unbiased control dataset with which to measure the influence of publication bias on apparent drug efficacy. Our approach is perhaps best compared to two previous studies that employed drug regulatory data to examine publication bias for antidepressants: one by our group, which used FDA data and similar methodology [8], and an earlier study, which used data from the Swedish drug regulatory authority [46].
Publication Bias for Antipsychotics versus Antidepressants
Compared to the findings for antidepressants, these findings for antipsychotics are less striking. (The overall results for the two drug classes are compared at the bottom of Figure 3.) We believe this is fundamentally related to the larger FDA-based effect size for antipsychotics compared to antidepressants [8], which increases the probability that, in any given trial, the drug–placebo difference will reach statistical significance. Indeed, two-thirds of the antipsychotic trials were positive, compared to one-half of the antidepressant trials [8]. With a larger proportion of positive trials, it should not be surprising that a larger proportion of the antipsychotic trials were published. We do not know whether this is due to a higher manuscript submission rate by drug companies and their investigators, a higher acceptance rate by journals, or both.
As a further consequence of the increased proportion of positive trials, fewer trials were needed, on a per-drug basis, to attain the two positive trials required for FDA approval (three here versus approximately six for antidepressants). This, combined with the fact that we were working with fewer drugs (eight here versus 12 for antidepressants), led to a smaller total number of trials (24 versus 74) and thereby less statistical power. In the meta-analyses, the smaller n associated with the unpublished negative and questionable trials carried little meta-analytic weight relative to the larger n associated with the positive and published trials. This diluted the impact of the unpublished trials when the overall effect size was calculated, leading to a smaller gap between the FDA - and journal-based overall effect size values (8% here versus 32% for antidepressants).
Comparing drug classes, the published literature suggests that the effect size for antipsychotics is only slightly greater than that for antidepressants, but FDA data reveal that the effect size gap between these two drug classes is much larger (Figure 3). A similar discrepancy between published and FDA data can be seen when one compares the proportion of positive trials for the two drug classes. Examined either way, publication bias can blur distinctions between effective and ineffective drugs.
Effect Size in Context
The overall effect size we found, 0.44, was somewhat lower than those from two previous meta-analyses that also made use of FDA data from placebo-controlled trials, probably because of methodological differences. One of these meta-analyses [45] reported an overall effect size of 0.51. It covered somewhat different drugs and comingled (limited) FDA data with published data, while we deliberately kept these two data sources separate in order to contrast them. The other meta-analysis [47] reported an effect size of 0.53. It was based on FDA data exclusively, but it covered only three of the eight drugs in the present study. That meta-analysis was published before FDA data became available on the two drugs found in this study to have the lowest effect sizes, ziprasidone (0.30) and iloperidone (0.28).
Because the effect size point estimate of 0.44 is less than 0.5, there is the risk that some parties will declare that antipsychotics have failed a critical litmus test for clinical significance, as has been declared for antidepressants [48],[49]. Space does not permit a full discussion as to why this reasoning is problematic [50], but we will touch briefly on some of the issues. (1) This 0.5 cutoff seems to derive from Cohen's [51] suggestion that 0.5 be used as a landmark for a medium effect, but Cohen never mentioned clinical significance nor, for that matter, clinical trials. (2) In proposing these landmarks, Cohen cautioned, “The values chosen had no more reliable a basis than my own intuition.” Put another way, they are not evidence-based. (3) It seems simplistic to think that one cutoff should apply uniformly across all fields of scientific inquiry, including all classes of drugs. Rather, as has been stated, “The most challenging and urgent task remains unsolved: developing the principles that underlie the thresholds of clinical significance in different clinical contexts” [52]. (4) Because of publication bias, effect sizes derived from the published literature should be interpreted with some caution. Some effect sizes that appear to exceed 0.5 might need to be revised downward once they are recalculated using data that are less vulnerable to publication bias, such as FDA data. Until such time, considering the evidence that publication bias is pervasive throughout medicine [3], we believe that any decisions on thresholds for clinical significance are premature.
Limitations
Lack of statistical power, due to the small number of trials analyzed (see above), was a major limitation of this study. Because of this, some of the statistically nonsignificant results could represent type II error (false negatives). Further, the statistical test for trial outcome versus publication status examined whether trials were published but not how they were published (outcome reporting bias).
Although the forest plots might seem to suggest significant between-drug differences, this study was not designed to address comparative effectiveness. The degree of heterogeneity is unclear: the 95% confidence intervals around I2 were wide, as appears to be the case in the majority of meta-analyses of medical interventions [29].
While this study addresses the efficacy of antipsychotics, it does not address their safety, an integral component of the clinician's risk–benefit analysis, nor their “real world” effectiveness [53]. In the trials we studied, efficacy was measured using scales that give little weight to disabling cognitive and negative symptoms [54]. These data apply only to adult, not pediatric or geriatric, patients. And they apply only to schizophrenia and schizoaffective disorder, not to other conditions for which these drugs are used.
This study assumes that the FDA database serves as a gold standard that is complete and unbiased, but caveats must be acknowledged. Drug companies must register trials with the FDA before they can begin them, but this applies only if the drug company is already pursuing marketing authorization in the US. Sometimes drug companies conduct clinical trials programs and obtain marketing approval outside the US and only approach the FDA at a later point in time. In such cases it is conceivable that the FDA, having not had the benefit of a priori trial registration, might not learn about the existence of certain non-US trials. A second caveat arises from the fact that what the FDA routinely makes publicly available, the drug approval packages [5],[11], contain data primarily from premarketing trials. Postmarketing trials are generally omitted, and these may represent the majority of the trials conducted on a given drug.
Implications
Selective reporting of research results undermines the integrity of the evidence base, which ultimately deprives clinicians of accurate data for prescribing decisions. With further studies investigating publication bias in other drug classes, a more accurate evidence base can emerge. To that end, increased access to FDA reviews has been advocated [5],[55]. At the present time, the FDA is not as transparent with its clinical trial data as it could be. For example, we pointed out in 2004 that the reviews for several antipsychotic drugs were posted on the FDA web site, but only for the original indication of schizophrenia and not for bipolar mania [5]. More than seven years later, the mania reviews remain inaccessible. On the other hand, it is encouraging that the FDA has convened a Transparency Task Force [56]. If the agency fulfills its mission to increase transparency, the public health will surely benefit.
Supporting Information
Zdroje
1. DwanKAltmanDGArnaizJABloomJChanAW 2008 Systematic review of the empirical evidence of study publication bias and outcome reporting bias. PLoS ONE 3 e3081 doi:10.1371/journal.pone.0003081
2. HopewellSLoudonKClarkeMJOxmanADDickersinK 2009 Publication bias in clinical trials due to statistical significance or direction of trial results. Cochrane Database Syst Rev 2009 MR000006
3. McGauranNWieselerBKreisJSchülerY-BKölschH 2010 Reporting bias in medical research—a narrative review. Trials 11 37
4. SterneJAGavaghanDEggerM 2000 Publication and related bias in meta-analysis: power of statistical tests and prevalence in the literature. J Clin Epidemiol 53 1119 1129
5. TurnerEH 2004 A taxpayer-funded clinical trials registry and results database. PLoS Med 1 e60 doi:10.1371/journal.pmed.0010060
6. RisingKBacchettiPBeroL 2008 Reporting bias in drug trials submitted to the Food and Drug Administration: review of publication and presentation. PLoS Med 5 e217 doi:10.1371/journal.pmed.0050217
7. LeeKBacchettiPSimI 2008 Publication of clinical trials supporting successful new drug applications: a literature analysis. PLoS Med 5 e191 doi:10.1371/journal.pmed.0050191
8. TurnerEHMatthewsAMLinardatosETellRARosenthalR 2008 Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med 358 252 260
9. GoldnerEMHsuLWaraichPSomersJM 2002 Prevalence and incidence studies of schizophrenic disorders: a systematic review of the literature. Can J Psychiatry 47 833 843
10. UstünTBRehmJChatterjiSSaxenaSTrotterR 1999 Multiple-informant ranking of the disabling effects of different health conditions in 14 countries. WHO/NIH Joint Project CAR Study Group. Lancet 354 111 115
11. US Food and Drug Administration 2012 Drugs@FDA: FDA approved drug products [database]. Available: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. Accessed 12 February 2012
12. US Department of Justice 1996 The Freedom of Information Act 5 U.S.C. § 552, as amended by Public Law No. 104-231, 110 Stat. 3048. FOIA Update, Vol. XVII, No. 4 Washington (District of Columbia) US Department of Justice Available: http://www.justice.gov/oip/foia_updates/Vol_XVII_4/page2.htm. Accessed 3 February 2012
13. SchererRWLangenbergPvon ElmE 2007 Full publication of results initially presented in abstracts. Cochrane Database Syst Rev 2007 MR000005
14. Council of Biology 1969 Proposed definition of a primary publication. CBE Newsletter November 1968 1 2
15. WoernerMGMannuzzaSKaneJM 1988 Anchoring the BPRS: an aid to improved reliability. Psychopharmacol Bull 24 112 117
16. KaySROplerLALindenmayerJP 1989 The Positive and Negative Syndrome Scale (PANSS): rationale and standardisation. Br J Psychiatry Suppl 59 67
17. AbrahaIMontedoriA 2010 Modified intention to treat reporting in randomised controlled trials: systematic review. BMJ 340 c2697
18. MallinckrodtCHClarkSWSCarrollRJMolenberghG 2003 Assessing response profiles from incomplete longitudinal clinical trial data under regulatory considerations. J Biopharm Stat 13 179 190
19. WoolleySBCardoniAAGoetheJW 2009 Last-observation-carried-forward imputation method in clinical efficacy trials: review of 352 antidepressant studies. Pharmacotherapy 29 1408 1416
20. StataCorp 2009 Stata statistical software: release 11 [computer program] College Station (Texas) StataCorp
21. HedgesLV 1982 Estimation of effect size from a series of independent experiments. Psychol Bull 92 490 499
22. RosenthalR 1991 Meta-analytic procedures for social research Newbury Park (California) Sage
23. HedgesLVOlkinI 1985 Statistical methods for meta-analysis New York Academic Press
24. SuttonAJAbramsKRJonesDRSheldonTASongF 2000 Fixed effects methods for combining study estimates. Methods for meta-analysis in medical research Chichester John Wiley & Sons 57 72
25. HigginsJGreenS 2011 Cochrane handbook for systematic reviews of interventions, version 5.1.0. Section 16.15.14. The Cochrane Collaboration
26. DerSimonianRLairdN 1986 Meta-analysis in clinical trials. Control Clin Trials 7 177 188
27. SterneJACBradburnMJEggerM 2001 Meta–analysis in Stata. EggerMSmithGDAltmanD Systematic reviews in health care: meta-analysis in context, 2nd edition 347 369
28. HigginsJPThompsonSG 2002 Quantifying heterogeneity in a meta-analysis. Stat Med 21 1539 1558
29. IoannidisJPPatsopoulosNAEvangelouE 2007 Uncertainty in heterogeneity estimates in meta-analyses. BMJ 335 914 916
30. OrsiniNHigginsJBottaiMBuchanI 2005 Heterogi: Stata module to quantify heterogeneity in a meta-analysis, revised 2006-01-25 [computer program]. Statistical Software Components S449201 Boston Boston College Department of Economics Available: http://EconPapers.repec.org/RePEc:boc:bocode:s449201. Accessed 12 February 2012
31. PotkinSGLitmanRETorresRWolfgangCD 2008 Efficacy of iloperidone in the treatment of schizophrenia: initial phase 3 studies. J Clin Psychopharmacol 28 S4 S11
32. HigginsJPTThompsonSGDeeksJJAltmanDG 2003 Measuring inconsistency in meta-analyses. BMJ 327 557 560
33. CitromeL 2010 Iloperidone redux: a dissection of the Drug Approval Package for this newly commercialised second-generation antipsychotic. Int J Clin Pract 64 707 718
34. CutlerAJKalaliAHWeidenPJHamiltonJWolfgangCD 2008 Four-week, double-blind, placebo - and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol 28 S20 S28
35. BorisonRLArvanitisLAMillerBG 1996 ICI 204,636, an atypical antipsychotic: efficacy and safety in a multicenter, placebo-controlled trial in patients with schizophrenia. U.S. SEROQUEL Study Group. J Clin Psychopharmacol 16 158 169
36. KeckPBuffensteinAFergusonJFeighnerJJaffeW 1998 Ziprasidone 40 and 120 mg/day in the acute exacerbation of schizophrenia and schizoaffective disorder: a 4-week placebo-controlled trial. Psychopharmacology (Berl) 140 173 184
37. BorisonRLPathirajaAPDiamondBIMeibachRC 1992 Risperidone: clinical safety and efficacy in schizophrenia. Psychopharmacol Bull 28 213 218
38. de OliveiraIRMiranda-ScippaAMde SenaEPPereiraELRibeiroMG 1996 Risperidone versus haloperidol in the treatment of schizophrenia: a meta-analysis comparing their efficacy and safety. J Clin Pharm Ther 21 349 358
39. DickersinKRennieD 2003 Registering clinical trials. JAMA 290 516 523
40. ChouinardGJonesBRemingtonGBloomDAddingtonD 1993 A Canadian multicenter placebo-controlled study of fixed doses of risperidone and haloperidol in the treatment of chronic schizophrenic patients. J Clin Psychopharmacol 13 25 40
41. MarderSRMeibachRC 1994 Risperidone in the treatment of schizophrenia. Am J Psychiatry 151 825 835
42. MarderSRDavisJMChouinardG 1997 The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry 58 538 546
43. MakhinsonM 2010 Biases in medication prescribing: the case of second-generation antipsychotics. J Psychiatr Pract 16 15 21
44. HeresSDavisJMainoKJetzingerEKisslingW 2006 Why olanzapine beats risperidone, risperidone beats quetiapine, and quetiapine beats olanzapine: an exploratory analysis of head-to-head comparison studies of second-generation antipsychotics. Am J Psychiatry 163 185 194
45. LeuchtSArbterDEngelRRKisslingWDavisJM 2009 How effective are second-generation antipsychotic drugs? A meta-analysis of placebo-controlled trials. Mol Psychiatry 14 429 447
46. MelanderHAhlqvist-RastadJMeijerGBeermannB 2003 Evidence b(i)ased medicine—selective reporting from studies sponsored by pharmaceutical industry: review of studies in new drug applications. BMJ 326 1171 1173
47. WoodsSWStolarMSernyakMJCharneyDS 2001 Consistency of atypical antipsychotic superiority to placebo in recent clinical trials. Biol Psychiatry 49 64 70
48. KirschIDeaconBJHuedo-MedinaTBScoboriaAMooreTJ 2008 Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med 5 e45 doi:10.1371/journal.pmed.0050045
49. FournierJCDeRubeisRJHollonSDDimidjianSAmsterdamJD 2010 Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA 303 47 53
50. TurnerEHRosenthalR 2008 Efficacy of antidepressants. BMJ 336 516 517
51. CohenJ 1988 Statistical power analysis for the behavioral sciences New York Lawrence Erlbaum Associates
52. KraemerHCKupferDJ 2006 Size of treatment effects and their importance to clinical research and practice. Biol Psychiatry 59 990 996
53. MöllerH-J 2008 Do effectiveness (“real world”) studies on antipsychotics tell us the real truth? Eur Arch Psychiatry Clin Neurosci 258 257 270
54. Villalta-GilVVilaplanaMOchoaSHaroJMDolzM 2006 Neurocognitive performance and negative symptoms: are they equal in explaining disability in schizophrenia outpatients? Schizophr Res 87 246 253
55. O'ConnorAB 2009 The need for improved access to FDA reviews. JAMA 302 191 193
56. AsamoahAKSharfsteinJM 2010 Transparency at the Food and Drug Administration. N Engl J Med 362 2341 2343
57. SmallJGHirschSRArvanitisLAMillerBGLinkCG 1997 Quetiapine in patients with schizophrenia. A high - and low-dose double-blind comparison with placebo. Seroquel Study Group. Arch Gen Psychiatry 54 549 557
58. ArvanitisLAMillerBG 1997 Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry 42 233 246
59. KaneJMCarsonWHSahaARMcQuadeRDIngenitoGG 2002 Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 63 763 771
60. PotkinSGSahaARKujawaMJCarsonWHAliM 2003 Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry 60 681 690
61. McEvoyJPDanielDGCarsonWHMcQuadeRDMarcusRN 2007 A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res 41 895 905
62. KaneJCanasFKramerMFordLGassmann-MayerC 2007 Treatment of schizophrenia with paliperidone extended-release tablets: a 6-week placebo-controlled trial. Schizophr Res 90 147 161
63. MarderSRKramerMFordLEerdekensELimP 2007 Efficacy and safety of paliperidone extended-release tablets: results of a 6-week, randomized, placebo-controlled study. Biol Psychiatry 62 1363 1370
64. DavidsonMEmsleyRKramerMFordLGuohuaP 2007 Efficacy, safety and early response of paliperidone extended-release tablets (paliperidone ER): results of a 6-week, randomized, placebo-controlled study. Schizophr Res 93 117 130
65. KaneJMEerdekensMLindenmayerJ-PKeithSJLesemM 2003 Long-acting injectable risperidone: efficacy and safety of the first long-acting atypical antipsychotic. Am J Psychiatry 160 1125 1132
66. BeasleyCMTollefsonGTranPSatterleeWSangerT 1996 Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology 14 111 123
67. BeasleyCMSangerTSatterleeWTollefsonGTranP 1996 Olanzapine versus placebo: results of a double-blind, fixed-dose olanzapine trial. Psychopharmacology (Berl) 124 159 167
68. DanielDGZimbroffDLPotkinSGReevesKRHarriganEP 1999 Ziprasidone 80 mg/day and 160 mg/day in the acute exacerbation of schizophrenia and schizoaffective disorder: a 6-week placebo-controlled trial. Ziprasidone Study Group. Neuropsychopharmacology 20 491 505
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2012 Číslo 3
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Rána vizitkou (nejen) chirurga
Nejčtenější v tomto čísle
- Guidance for Evidence-Informed Policies about Health Systems: Assessing How Much Confidence to Place in the Research Evidence
- Uterine Rupture by Intended Mode of Delivery in the UK: A National Case-Control Study
- Guidance for Evidence-Informed Policies about Health Systems: Linking Guidance Development to Policy Development
- Improving Ethical Review of Research Involving Incentives for Health Promotion
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy