
Dissecting Inflammatory Complications in Critically Injured Patients by Within-Patient Gene Expression Changes: A Longitudinal Clinical Genomics Study
Background:
Trauma is the number one killer of individuals 1–44 y of age in the United States. The prognosis and treatment of inflammatory complications in critically injured patients continue to be challenging, with a history of failed clinical trials and poorly understood biology. New approaches are therefore needed to improve our ability to diagnose and treat this clinical condition.
Methods and Findings:
We conducted a large-scale study on 168 blunt-force trauma patients over 28 d, measuring ∼400 clinical variables and longitudinally profiling leukocyte gene expression with ∼800 microarrays. Marshall MOF (multiple organ failure) clinical score trajectories were first utilized to organize the patients into five categories of increasingly poor outcomes. We then developed an analysis framework modeling early within-patient expression changes to produce a robust characterization of the genomic response to trauma. A quarter of the genome shows early expression changes associated with longer-term post-injury complications, captured by at least five dynamic co-expression modules of functionally related genes. In particular, early down-regulation of MHC-class II genes and up-regulation of p38 MAPK signaling pathway were found to strongly associate with longer-term post-injury complications, providing discrimination among patient outcomes from expression changes during the 40–80 h window post-injury.
Conclusions:
The genomic characterization provided here substantially expands the scope by which the molecular response to trauma may be characterized and understood. These results may be instrumental in furthering our understanding of the disease process and identifying potential targets for therapeutic intervention. Additionally, the quantitative approach we have introduced is potentially applicable to future genomics studies of rapidly progressing clinical conditions.
Trial Registration: ClinicalTrials.gov NCT00257231
: Please see later in the article for the Editors' Summary
Published in the journal:
. PLoS Med 8(9): e32767. doi:10.1371/journal.pmed.1001093
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1001093
Summary
Background:
Trauma is the number one killer of individuals 1–44 y of age in the United States. The prognosis and treatment of inflammatory complications in critically injured patients continue to be challenging, with a history of failed clinical trials and poorly understood biology. New approaches are therefore needed to improve our ability to diagnose and treat this clinical condition.
Methods and Findings:
We conducted a large-scale study on 168 blunt-force trauma patients over 28 d, measuring ∼400 clinical variables and longitudinally profiling leukocyte gene expression with ∼800 microarrays. Marshall MOF (multiple organ failure) clinical score trajectories were first utilized to organize the patients into five categories of increasingly poor outcomes. We then developed an analysis framework modeling early within-patient expression changes to produce a robust characterization of the genomic response to trauma. A quarter of the genome shows early expression changes associated with longer-term post-injury complications, captured by at least five dynamic co-expression modules of functionally related genes. In particular, early down-regulation of MHC-class II genes and up-regulation of p38 MAPK signaling pathway were found to strongly associate with longer-term post-injury complications, providing discrimination among patient outcomes from expression changes during the 40–80 h window post-injury.
Conclusions:
The genomic characterization provided here substantially expands the scope by which the molecular response to trauma may be characterized and understood. These results may be instrumental in furthering our understanding of the disease process and identifying potential targets for therapeutic intervention. Additionally, the quantitative approach we have introduced is potentially applicable to future genomics studies of rapidly progressing clinical conditions.
Trial Registration: ClinicalTrials.gov NCT00257231
: Please see later in the article for the Editors' Summary
Introduction
Trauma is the number one killer of individuals aged 1–44 y [1], a source of some of the top health care costs in the United States [2], and a major global health priority [3],[4]. Trauma injuries frequently lead to infections, sepsis, and multiple organ failure (MOF) [5],[6], which contribute to 51%–61% of late trauma mortality [7]. A number of clinical trials for treating late trauma complications have failed, believed partly due to the inability to identify a proper patient population as well as the limited understanding of the interplay of biological processes underlying post-injury inflammatory complications [8],[9]. A more comprehensive characterization of the genomic response to trauma is therefore required in order to increase our understanding of the molecular basis of clinical outcomes, leading to improvements in diagnosis and treatment.
Despite the public health implications of improved trauma care, relatively few studies have been carried out to understand the molecular basis of trauma recovery, particularly from a genome-wide perspective [10]. An endotoxin experiment on healthy volunteers [11] and a retrospective sepsis study [12] have shown a strong genomic response to trauma-related phenotypes. However, to date there has been no in-depth, prospective longitudinal characterization of the genome-wide expression response to blunt-force trauma that (a) identifies which pathways are fundamental determinants of the patient's recovery trajectory, and (b) elucidates the time period post-injury when these molecular signatures are most informative. Uncovering these factors can reveal new therapeutic strategies and the dynamic regimens for their administration.
To this end, the “Inflammation and the Host Response to Injury” (IHRI) research program conducted a large-scale, 28-d prospective clinical genomics study involving 168 patients, 797 microarrays, and 393 clinical variables. The key statistical challenge we faced was how to accurately associate early longitudinal gene expression measured at multiple time points with 28 d clinical trajectories captured by a constellation of clinical variables. We developed and applied a tractable and robust quantitative framework to analyze this complex clinical genomics study. Specifically, we sought to capitalize on the longitudinal structure within an individual, combining bioinformatics and statistical tools to elucidate pathway dynamics from the gene expression data.
We found that approximately one quarter of the genome changes during early stages of treatment in concordance with the observed variation in 28-d clinical outcomes. These expression changes are coordinated into five distinct modules, which together provide a fine-scale separation of patient outcomes. We pinpointed several pathways that appear to be key drivers of these modules and may be instrumental in furthering our understanding of the disease process and identifying potential targets for therapeutic intervention [13],[14]. We investigated the dynamics of these pathways and found that several discriminate among 28-d post-injury patients trajectories. Specifically, we identified p38 MAPK signaling pathway and MHC-class II genes as having the strongest discrimination in the first 40–80 h. Such information is potentially useful in determining the exact timing and effective dosage of drugs targeting these pathways in trauma patients.
A lack of reproducibility of clinical genomics results [15] has been shown to be largely due to patient heterogeneity, latent sources of confounding, and platform-dependent non-biological variation [16], all difficult to deal with when associating clinical outcome with a single snapshot of gene expression. Taking advantage of the longitudinal design of our study, we developed and applied an approach modeling “within-patient” gene expression dynamics for extracting robust signatures, thereby accounting for patient-specific effects and being more likely to reproduce in future patients. Our framework is likely applicable to other complex clinical genomics studies, especially in rapidly progressing clinical conditions.
Methods
Study Design and Patient Samples
In the IHRI prospective clinical genomics study, we studied a cohort of 168 patients (ages 16–55 y; 107 males) from a larger epidemiological study, involving 1977 severe blunt-force trauma patients, conducted from 2003 to 2011 through 7 U.S. Level I trauma centers across the United States. Figure 1 provides the flow chart leading to the 168 patients analyzed in this paper (epidemiological study: ClinicalTrials.gov identifier: NCT00257231). These 168 patients were followed for up to 28 hospital days post-injury, and their longitudinal genome-wide gene expression was measured. To ensure patients were at risk of developing MOF, infectious complications, and death (thereby satisfying the study requirements), the consortium employed a set of inclusion/exclusion enrollment criteria (Text S1). Patients with isolated traumatic brain injury were excluded. Samples were taken at fixed time points following injury according to study design and independent of physician influence. Thus there was no physician or severity of illness bias in the sample collection process.

The institutional review board of each center approved the study, and written informed consent was obtained from all patients or their legal next of kin. The same standardized patient care protocol was used to minimize the impact of variability in clinical care across centers. Patient clinical information, typically consisting of >300 variables (some longitudinal), was collected by trained nurses and entered into a central database to maintain conformity and consistency across all participating centers. For each patient, genome-wide longitudinal gene expression was measured for total blood leukocytes isolated from peripheral blood samples (collection, processing, and normalization described in Text S1 and Figure S1). The data (de-identified as defined by the Health Insurance Portability and Accountability Act of 1996; see http://www.gluegrant.org/trdb.htm) are freely available at http://www.gluegrant.org; see http://www.gluegrant.org/glueadmin/register_consortium.jsp for details.
Statistical Analysis
Our proposed statistical framework (Figure 2), derived from two a priori statistical hypotheses (Results), consists of three key steps:

Step 1
We used a time-dependent modified Marshall score [17] (excluding the neurological component) as the measure of developing MOF, providing an up-to-28-d Marshall score time course trajectory for each patient. We applied hierarchical clustering to the 168 patient Marshall score trajectories, yielding five clusters (Figure 2, Step 1; Text S2). We used relevant patient information, such as the 28-d mortality and morbidity rates, to order these five subgroups (Figure 3; Table 1). This yielded a clinically interpretable, ordered categorical MOF score (ocMOF) ranging from i (good outcome) to v (bad outcome).


Step 2
For the gene expression analysis, we considered 126 patients with three or more arrays meeting the RNA quality requirements among hours 12–250 (Text S2 and Figure S2). We sought to characterize within-patient expression changes (WPEC) by quantifying per hour log-fold change. To compute WPEC for each probeset, we regressed log gene expression on time (in hours) and extracted the linear slope (Figure 2, Step 2).
Step 3
We tested each probeset's WPEC for an association with ocMOF using an adjusted Spearman rank-based correlation test and obtained a p-value for each probeset (Figure 2, Step 3; Text S2). Associating WPEC with ocMOF through a rank-based test enhances robustness, as WPEC values are used to establish an ordering of directional changes across patients, but do not rely on the actual magnitudes. Statistical significance of the 54,675 resulting p-values (Figure 4a) was assessed using the false discovery rate (FDR) [18] yielding an estimate of the total percentage of probesets associated with ocMOF as well as specific probesets identified as significant at various FDR thresholds (Figure 4b).
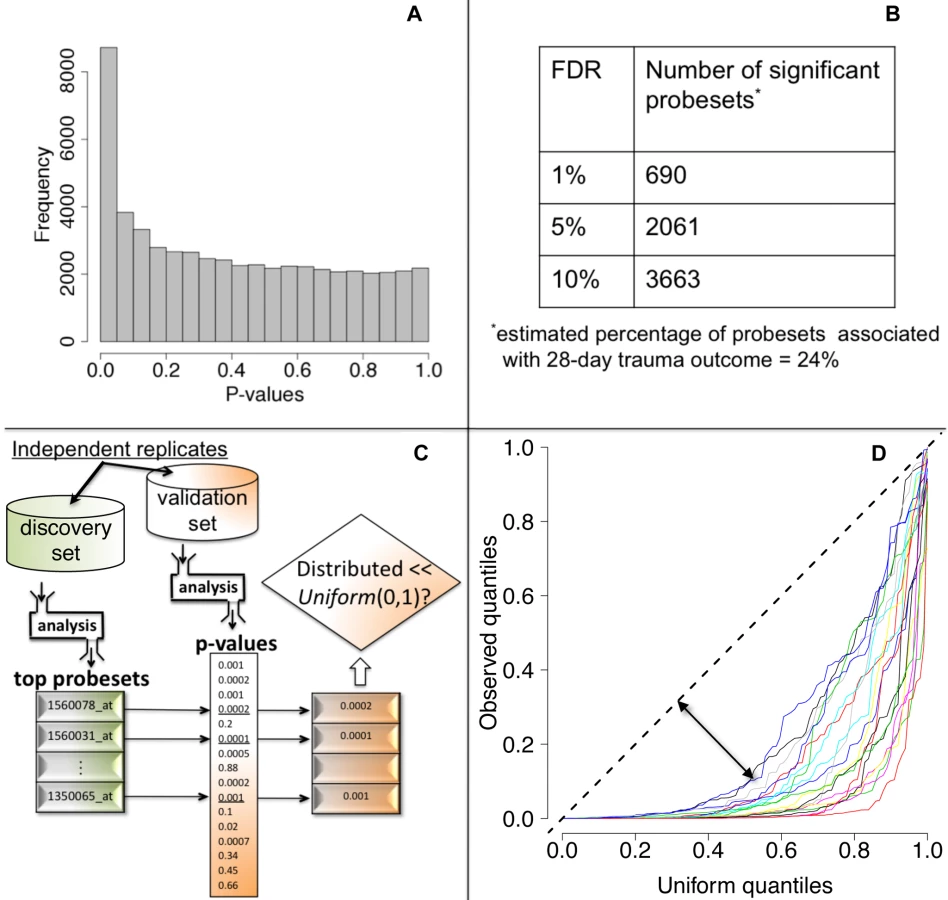
All the methodological, algorithmic, and data-filtering decisions in Step 1 (clinical data) and in Step 2 (genomic data) were made completely independently by two different analysts to avoid any potential over-fitting.
Additional mathematical and algorithmic details about the statistical methods can be found in Text S2. The R statistical software environment (http://www.r-project.org/) was utilized to perform all data analyses. Computer code that reproduces all results is available as Dataset S1 with detailed documentation provided in Text S2.
PCA-Based Analysis of WPEC Matrix
To assess the overall effectiveness of WPEC in explaining trauma response, we performed principal component analysis (PCA) [19] on the WPEC matrix and determined the number of principal components (PCs) via the scree plot. We used these PCs as explanatory variables to model each of the 393 measured clinical variables and identified the top ten most significant clinical variables (Text S3).
Assessment of Reproducibility
We took a principled strategy to assess reproducibility (Figure 4c and 4d). Our strategy (a) obtains the most significant probesets using a “discovery set,” (b) computes their corresponding p-values in a “validation set,” and (c) assesses whether the validation set p-values show systematic, reproducible significance. We complete the last step by comparing the validation set p-values to the Uniform (0,1) distribution [18], which corresponds to the distribution of p-values when there is no significance. We performed 20 cross-validations by randomly splitting the study into discovery and validation datasets (Text S3).
Dominant Expression Trajectories and Dynamic Co-expression Modules
We utilized the DAVID software package to classify probesets with FDR <10% into 54 functionally related gene sets. We further aggregated these gene sets into five modules according to the similarity of their dominant trajectories across the ocMOF subgroups (Text S4). We used the average Pearson correlation of dominant trajectories across ocMOF subgroups as the similarity metric. If the signs of these Pearson correlations were inconsistent across ocMOF subgroups, then the similarity metric was set to zero (no similarity). Next, we used Ward as the agglomeration method and performed hierarchical clustering to obtain five modules.
Specifically, to obtain the five dominant expression trajectories for a gene set, we first removed patient–probeset-specific effects by standardizing the gene expression values (scaling each patient using the sample standard deviation of all of its probesets and separately mean-centering each scaled probeset), and subsequently fitted a loess curve to each ocMOF subgroup. These five dominant trajectories were aligned to a common initial reference.
For each module, the five dominant trajectories were obtained by averaging the corresponding gene sets trajectories (Text S4). We then applied Ingenuity Pathways Analysis (IPA) to identify significant (p-value<0.002, after Bonferroni correction) canonical pathways among the probesets making up each module. The exact settings employed for DAVID and IPA are discussed in Text S4.
Results
We conducted a large-scale 28-d prospective clinical genomics study involving 168 patients, 797 microarrays, and 393 clinical variables (Methods) in order to understand the molecular basis of clinical responses to trauma from a genome-wide perspective. To this end, the central data-analytic challenge we faced was to associate longitudinal gene expression and 28-d Marshall score time course trajectories without over-fitting the data and while maintaining clinical interpretability. To address this challenge, we developed a within-patient longitudinal gene expression framework (Figure 2), derived from the following a priori statistical hypotheses: (a) there are several distinct trauma recovery trajectories (or physiological responses to trauma), reflected in the time varying clinical measures of interest, and (b) by using each patient as her/his own internal control while modeling the gene expression, inter-patient heterogeneity and confounding are reduced. The framework collapses Marshall score trajectories into clinically interpretable, ocMOF scores and longitudinal gene expression into within-patient expression changes (WPEC). We associated ocMOF and WPEC for each probeset using a rank-based correlation test (Methods).
Composite Longitudinal ocMOF Score Captures Relevant Clinical Variation
The ocMOF score is designed to capture the clinical variation among patients across the 28-d treatment window (Table 1; Figure 3; Text S5). Note that ocMOF is not introduced here to replace standard clinical measures, but instead serves as a 28-d longitudinal composite of overall patient variation and outcomes in which the higher the score, the worse the patient experience.
The ocMOF i subgroup captures uncomplicated recovery with minor or no inflammatory and infectious complications, whereas ocMOF v group captures complications leading to MOF and death. Figure 2, Step 1 shows the average Marshall score trajectories for the five ordinal patient subgroups. Note, for example, that the patients with ocMOF iii (0% mortality rate) and v (100% mortality rate) have very similar Marshall score trajectories during the first 7-d (Figures S3 and S4), and hence are difficult to separate using just Marshall scores (e.g., for the first 7-d mean Marshall scores, the two-sample t-test p-value is 0.506).
WPEC Measure Robustly Captures Relevant Clinical Variation
Instead of associating absolute expression values with ocMOF, which is the de facto analysis strategy, we instead sought to associate the within-patient change in gene expression with clinical outcome. For each probeset and patient, a within-patient expression change (WPEC) was formed by quantifying per-hour log-fold change over hours 12–250 post injury (i.e., regressing log gene expression on time and estimating the linear slope), which was adequate (Text S6 and Figures S5 and S6).
We first performed a PCA-based analysis to assess the overall effectiveness of WPEC in explaining trauma response (Text S6). Eight PCs were obtained from the WPEC matrix (54,675 probesets by 126 patients), capturing 31% of total variation (Figure S7a). Among the 393 clinical variables, those related with Marshall and Denver scores are among the top ten most significant clinical variables associated with these eight PCs collectively, with ocMOF being the most significant (Table S1). We repeated the same analysis on the mean expression matrix (taking the mean across the time course, which effectively combines baseline expression and WPEC) for hours 12–250 (eight PCs, 62% variation, Figure S7b) and found sampling phase and trauma center among the ten most significant variables (Table S2), implying patient-specific baseline expression is susceptible to confounders.
Significance Analysis of WPEC Associations with 28-d Trauma Outcome
We then performed a test of association between each probeset's WPEC measure and ocMOF to identify probesets that show a statistically significant association with clinical outcome (Methods). Both the resulting p-values (Figure 4a) and the FDR calculations (Figure 4b) indicate strong statistical significance. This information and mean WPEC for each ocMOF subgroup for all 54,675 probesets are provided in Table S3. The estimated percentage of probesets associated [18] with ocMOF was ≥24%, indicating that at least one-quarter of the genome undergoes early within-patient expression changes associated with 28-d trauma outcome.
A lack of reproducible results has been a major hurdle for translational research in clinical genomics [20],[21]. With thousands of genes tested for association and many being involved in the disease, it is important to assess the reproducibility of our association analysis. Therefore, we developed and applied a principled cross-validation strategy to assess reproducibility of significance (Figure 4c). We consistently observed small p-values for the top 100 probesets (Figure 4d), suggesting strong reproducibility of significant associations.
Dynamic Co-expression Modules Discriminating Trauma Outcome
We took a functional genomics approach to characterize the dynamic expression variation driving the WPEC and trauma outcome associations (Methods). We first applied DAVID [22],[23], a state-of-the-art classification algorithm that groups genes based on their co-occurrences in annotation terms, to functionally cluster 1,256 of the 3,663 probesets with FDR <10% (Table S4). We further aggregated these gene sets into five modules according to the similarity of their dominant trajectories across the ocMOF subgroups (Figure S8). These modules were then organized in descending order of their average pairwise similarity metric, Module A (largest) to Module E (smallest); their dominant trajectories are provided in Figures S9, S10, S11, S12, S13. The average dominant trajectory of each ocMOF subgroup in each module shows highly coordinated dynamic co-expression patterns that discriminate the ocMOF groups in an ordered manner (Figure 5), especially in the early time window post-injury.

We then applied IPA, a hand-curated database of biological interactions and functional annotations, to identify statistically significant canonical pathways among the probesets making up each module (Methods). In general, the significant canonical pathways within each module were relevant and related to inflammation and immunity (see Figure 5 caption and Table S5). Module A's dominant trajectory for ocMOF v is notably different from the other ocMOF subgroups, and its significant canonical pathway is oxidative phosphorylation. Metabolic dysfunction from trauma and infections has adverse effects on organ systems, and it generally originates from the mitochondrion, which is involved in the metabolism processes through oxidative phosphorylation [24],[25]. For Module E, the most significant canonical pathway is protein ubiquitination; targeting ubiquitin-mediated signaling is thought to regulate nuclear factor-κB (NF-κB), currently of interest as a therapeutic target in inflammatory diseases [26].
Key Pathways Associated with Trauma Outcome
To identify the key drivers of the genomic response to trauma, we performed ontological analyses on the top 500 most significant probesets using DAVID and IPA. For these 500 probesets, the heatmap of all the gene expression data collected throughout the study (stratified by day and ocMOF outcome) is provided in Figure S14. We sought to identify gene sets that are enriched for biological processes leading to poor trauma outcomes, show tightly coordinated dynamic expression trajectories, and have strong discriminatory power for post-injury MOF.
The top six canonical pathways (p-values<2.2×10−5) identified by IPA are dendritic cell maturation, Toll-like receptor (TLR) signaling, p38 mitogen-activated protein kinase (p38 MAPK) signaling, interleukin(IL)-6 signaling, production of nitric oxide and reactive oxygen species in macrophages, and antigen presentation (see Table S6 for the top 20). All six are involved in cellular immune responses, implying a common theme. Two of the pathways, TLR and p38 MAPK signaling, were recently identified in a genome-wide expression study on early sepsis [12]. We applied DAVID to functionally cluster the top 500 probesets (Table S7), yielding five gene groups that strongly support the IPA results. Both analyses identified several genes that have been individually targeted in previous model system studies [27],[28]. We discuss below two gene sets identified from the analyses, with analogous results being shown for three others.
Antigen presentation pathway
The top gene group from DAVID is enriched with the major histocompatibility complex class II (MHC-II) genes, in which 16 of the 17 probesets in that group are MHC-II, with four being in top 50. One of the top six canonical pathways from IPA is antigen presentation (p-value = 2.2×10−5), which also consists of MHC-II genes (Text S7 and Figure S15). The MHC-II molecules are relevant to MOF because they present foreign antigens on the cell surface, which is essential for adaptive or innate immunity [29],[30]. We used the 16 MHC-II probesets (representing HLA-DMB, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DRA, HLA-DRB1, HLA-DRB3, HLA-DRB4, LOC100294318, and LOC100133678 genes) identified by DAVID to comprise the MHC-II gene set for subsequent analysis (Figure 6a–6c).

The boxplots of WPEC suggest that, in moving from ocMOF i to v, the WPEC decreases for all 16 MHC-II probesets, exhibiting a dosage effect (Figure S16). The dominant expression trajectories also exhibited a similar trend (Figure 6a). The difference among dominant trajectories is particularly pronounced during the 40–80 h window, suggesting that early expression changes can be used to discriminate among the patient ocMOF scores (Figure 6b). Using the two time points closest to 40–80 h post-trauma, we counted the number of up-regulated MHC-II probesets within each patient, and observed consistently high counts for ocMOF i, ii, and iii and low counts for ocMOF iv and v (Figure 6c).
p38 MAPK signaling pathway
All of the top five canonical pathways from IPA contain the mitogen-activated protein kinase 14 (MAPK14) gene, and all four probesets representing it are among the top 500 with three being in top 50. The MAPK14 gene is an isoform of the p38 MAPK gene, the signaling pathway of which (one of the top five canonical pathways, Figure S17) is known to play an important role in driving the inflammatory response to either microbial products (via PAMPs), endogenous danger signals (via DAMPs or alarmins), and pro-inflammatory cytokines by phosphorylating transcription factors, resulting in the further expression of inflammatory mediators [31],[32],[33]. This suggests that the p38 MAPK signaling pathway is an integral signaling mechanism for the other top five canonical pathways and hence the presence of MAPK14 in those pathways. Using IPA, we obtained an additional eight (out of the top 500) probesets representing the genes involved in the p38 MAPK signaling pathway (Text S7), giving us the p38 MAPK gene set (12 probesets, representing MAPK14, CREB5, IL1R1, IL1RN, IRAK2, IRAK3, MAP2K6, and TIFA) for further analysis (Figure 6d–6f).
For the p38 MAPK probesets, we observed a trend opposite that of the MHC-II probesets. Generally, WPEC increased in moving from ocMOF i to v (Figure S18), and the early dominant trajectories also discriminated the ocMOF groups (Figure 6e–6f). A similar analysis was performed on three other gene sets, representative of the remaining top six canonical pathways, suggesting trends similar to those of p38 MAPK (Text S7 and Figures S19, S20, S21, S22, S23, S24).
Using a controlled endotoxin experiment dataset [11], which served as a corroborative experiment, we obtained dominant trajectories for MHC-II and p38 MAPK among healthy individuals administered with endotoxin (Text S7). The trends of these trajectories from ≥5 h were similar to those seen with ocMOF i and ii (Figure S25).
Discussion
In this paper we have provided a comprehensive analysis of the longitudinal IHRIP study, taking into account all major sources of collected data. Despite inherent complexities in clinical genomic data, we showed that robust and relevant genomic signatures can be obtained with our framework, aimed at facilitating straightforward translation into a clinical setting. Our results have implications for the design and analysis of future large-scale clinical genomics studies.
We showed that clinical association using WPEC is straightforward to calculate and appears to be robust to confounders. Our framework collapses Marshall score trajectories and longitudinal gene expression into clinically interpretable quantities, permitting reliable statistical modeling without over-fitting the data. Using our framework we identified genes whose WPEC discriminate among the ocMOF outcomes. One of the main advantages of utilizing WPEC is that it leads more directly than do other measures to a clinical translation of the results, because it captures the change in expression within a patient, regardless of the patient's baseline value, which is susceptible to patient heterogeneity, confounders, and technical effects. On the other hand, any snapshot, baseline, or average expression profile will be susceptible to these effects. We repeated the above analyses using both an estimate of the hour 12 expression value and the average over the entire time course. Both measures showed evidence of being influenced by confounders (particularly batch and trauma center effects), and neither produced biological significance greater than WPEC.
We performed a global functional genomics analysis of the top 3,663 statistically significant WPEC associations with trauma outcome (FDR = 10%), identifying five dynamic co-expression modules highly enriched for immune pathways. We also performed a more focused pathway analysis of the top 500 associations (FDR = 0.6%) and identified a number of relevant gene sets. We pinpointed the MHC-II and p38 MAPK gene sets, showing that their expression dynamics suggest their potential as biomarkers. Moreover, our analysis suggests that the strongest discrimination occurs in the first 40–80 h post-injury.
From the dynamic co-expression modules results, one can consider the configuration of ocMOF-specific trajectories within and among these modules along with the biological significance of the modules to construct a spectrum of biologically relevant gene expression variation discriminating the clinical outcomes (ocMOF i to v). For example, our module-based analysis pointed to the NF-κB pathway. Previous studies have indicated that this pathway, which is downstream of the TLR, is critical in the context of post-traumatic immune dysfunction-induced poor outcomes [34],[35]. Taken as a whole, this systems analysis revealed a large and coordinated gene expression response to trauma, characterized by the modules and gene sets, indicating that something is to be gained from a systems-level understanding [36] of the molecular biology of post-injury MOF in forming therapeutic targets and prognostic procedures.
Our findings on the down-regulation of MHC-II genes among patients from ocMOF iv to v are consistent with persistently low HLA-DR expression that has been associated with septic complications [37],[38], because a marked depression of cell-mediated immune function (i.e., immunosuppression) is believed to play a role in sepsis after severe trauma. HLA-DR is a promising molecular surrogate marker for treating post-injury inflammatory complications [39], and monitoring HLA-DR expression to treat trauma patients with immunomodulatory drugs such as interferon-γ has been studied. Importantly, our genome-wide approach suggests the association of the entire MHC-II gene set (represented by 16 probesets, Kruskal-Wallis p-value = 0.00004, Figure 6c), besides HLA-DR (represented by five of these 16 probesets, Kruskal-Wallis p-value = 0.00029), may be more informative. Persistent systemic inflammatory response syndrome (SIRS) is shown to be predictive of nosocomial infection in trauma patients [40],[41], which is consistent with the up-regulation of genes in the p38 MAPK signaling pathway among patients from ocMOF iii to v.
It has been posited that MOF is an outcome of an inappropriate generalized inflammatory response, involving the interplay between mediators (e.g., cytokines and chemokines) and effector cells (e.g., neutrophils and macrophages) [42]. The SIRS and compensatory response syndrome (CARS) are proposed to be involved in the etiology of MOF [7]. It may well be that the increase in p38 MAPK expression seen here after severe trauma reflects the development of SIRS while a persistent decrease in MHC-II expression reflects the development of CARS (Text S8), which is supported from our agnostic, genome-wide analysis. Beyond the definitions of CARS and SIRS, the modern concept of post-traumatic immune suppression is also believed to be a major cause of secondary infections or organ dysfunction [43],[44].
Although it was to date the largest clinical genomic study on trauma response of which we are aware, the current study has certain limitations. Since gene expression was measured for total blood leukocytes isolated from peripheral blood samples, some of the differential expression changes identified could be confounded by changes in individual leukocyte subpopulations. Arguably, the predictive utility of the identified biomarkers still exists.
In this study, we identified relevant pathways and gene sets showing a coordinated pattern of expression variation associated with response to trauma at a genome-wide scale. These findings potentially provide the most comprehensive picture of the gene expression response to trauma to date, thereby demonstrating the power of moving beyond candidate gene studies [45] of this clinical condition. The expression variation at the genomic level that we have characterized among patients may help to provide a more comprehensive set of drug targets and a means to identify relevant subsets of patients for which these may be effective.
Supporting Information
Zdroje
1. SasserSMHuntRCSulliventEEWaldMMMitchkoJ 2009 Guidelines for field triage of injured patients. Recommendations of the National Expert Panel on Field Triage. MMWR Recomm Rep 58 1 35
2. SoniA 2009 The five most costly conditions, 1996 and 2006: Estimates for the U.S. civilian noninstitutionalized population. Statistical Brief Rockville Agency for Healthcare Research and Quality 1 5
3. PedenMMcGeeKKrugE 2002 Injury: A leading cause of the global burden of disease, 2000. Geneva World Health Organization
4. HofmanKPrimackAKeuschGHrynkowS 2005 Addressing the growing burden of trauma and injury in low - and middle-income countries. Am J Public Health 95 13 17
5. DeCampMMDemlingRH 1988 Posttraumatic multisystem organ failure. J Am Med Assoc 260 530 534
6. MarshallJCVincentJLSibbaldWJ 1995 Clinical Trials for the Treatment of Sepsis. VincentJLSibbaldWJ Berlin Springer-Verlag 122 138
7. DewarDMooreFAMooreEEBaloghZ 2009 Postinjury multiple organ failure. Injury 40 912 918
8. OpalSM 2003 Clinical trial design and outcomes in patients with severe sepsis. Shock 20 295 302
9. BaueAE 1997 Multiple organ failure, multiple organ dysfunction syndrome, and systemic inflammatory response syndrome. Why no magic bullets? Arch Surg 132 703 707
10. CobbJPMindrinosMNMiller-GrazianoCCalvanoSEBakerHV 2005 Application of genome-wide expression analysis to human health and disease. Proc Natl Acad Sci U S A 102 4801 4806
11. CalvanoSEXiaoWRichardsDRFelcianoRMBakerHV 2005 A network-based analysis of systemic inflammation in humans. Nature 437 1032 1037
12. JohnsonSBLissauerMBochicchioGVMooreRCrossAS 2007 Gene expression profiles differentiate between sterile SIRS and early sepsis. Ann Surg 245 611 621
13. CollinsF 2010 Has the revolution arrived? Nature 464 674 675
14. FeeroWGGuttmacherAECollinsFS 2010 Genomic Medicine: Genomic Medicine – An Updated Primer. N Engl J Med 362 2001 2011
15. FrantzS 2005 An array of problems. Nat Rev Drug Discov 4 362 363
16. PotterJD 2001 At the interfaces of epidemiology, genetics and genomics. Nat Rev Genet 2 142 147
17. MarshallJCCookDJChristouNVBernardGRSprungCL 1995 Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med 23 1638 1652
18. StoreyJDTibshiraniR 2003 Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100 9440 9445
19. JolliffeIT 2002 Principal Component Analysis NY Springer
20. IoannidisJPA 2005 Microarrays and molecular research: Noise discovery? Lancet 365 454 455
21. MichielsSKoscielnySHillC 2005 Prediction of cancer outcome with microarrays: A multiple random validation strategy. Lancet 365 488 492
22. DennisGShermanBTHosackDAYangJGaoW 2003 DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4 P3
23. HuangDWShermanBTTanQCollinsJRAlvordWG 2007 The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 8 R183
24. HasselgrenPOHubbardWJChaudryIH 2007 Metabolic and inflammatory responses to trauma and infection. FischerJE Mastery of Surgery. Fifth ed Philadelphia Lippincott Williams & Wilkins 2 23
25. DareAJPhillipsARHickeyAJMittalALovedayB 2009 A systematic review of experimental treatments for mitochondrial dysfunction in sepsis and multiple organ dysfunction syndrome. Free Radic Biol Med 47 1517 1525
26. WullaertAHeyninckKJanssensSBeyaertR 2006 Ubiquitin: tool and target for intracellular NF-kappa B inhibitors. Trends Immunol 27 533 540
27. HanJLeeJDBibbsLUlevitchRJ 1994 A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265 808 811
28. RosengartMRNathensABArbabiSNeffMJGarciaI 2003 Mitogen-activated protein kinases in the intensive care unit: Prognostic potential. Ann Surg 237 94 100
29. ChainBMKayePMShawMA 1988 The biochemistry and cell biology of antigen processing. Immunol Rev 106 33 58
30. CresswellP 1987 Antigen recognition by T lymphocytes. Immunol Today 8 67 69
31. KyriakisJMAvruchJ 2001 Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81 807 869
32. DongCDavisRJFlavellRA 2002 MAP kinases in the immune response. Annu Rev Immunol 20 55 72
33. ZhangQRaoofMChenYSumiYSursalT 2010 Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464 104-U115
34. LiuSFMalikAB 2006 NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 290 L622 L645
35. BohrerHQiuFZimmermanTZhangYMJllmerT 1997 Role of NF kappa B in the mortality of sepsis. J Clin Invest 100 972 985
36. AhnACTewariMPoonCSPhillipsRS 2006 The limits of reductionism in medicine: could systems biology offer an alternative? PLoS Med 3 e208 doi:10.1371/journal.pmed.0030208
37. DitschkowskiMKreuzfelderERebmannVFerencikSMajetschakM 1999 HLA-DR expression and soluble HLA-DR levels in septic patients after trauma. Ann Surg 229 246 254
38. MonneretGLepapeAVoirinNBohéJVenetF 2006 Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med 32 1175 1183
39. SpruijtNEVisserTLeenenLPH 2010 A systematic review of randomized controlled trials exploring the effect of immunomodulative interventions on infection, organ failure, and mortality in trauma patients. Crit Care 14 R150
40. BochicchioGVNapolitanoLMJoshiMKnorrKTracyJK 2002 Persistent systemic inflammatory response syndrome is predictive of nosocomial infection in trauma. J Trauma 53 245 251
41. HooverLBochicchioGVNapolitanoLMJoshiMBochicchioK 2006 Systemic inflammatory response syndrome and nosocomial infection in trauma. J Trauma 61 310 316
42. TsukamotoTChanthaphavongRSPapeH-C 2010 Current theories on the pathophysiology of multiple organ failure after trauma. Injury 41 21 26
43. MunfordRSPuginJ 2001 Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Resp Crit Care 163 316 321
44. ReddyRCChenGHTekchandaniPKStandifordTJ 2001 Sepsis-induced immunosuppression - From bad to worse. Immunol Res 24 273 287
45. MockCNDriesDJJurkovichGJMaierRV 1996 Assessment of two clinical trials: interferon-gamma therapy in severe injury. Shock 5 235 240
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2011 Číslo 9
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Současné postavení a přínos sartanů v klinické praxi
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
- Hypertrofická obstrukční kardiomyopatie ve světle moderní farmakoterapie – kazuistika
Nejčtenější v tomto čísle
- Living Alone and Alcohol-Related Mortality: A Population-Based Cohort Study from Finland
- Cardiovascular Risk with Non-Steroidal Anti-Inflammatory Drugs: Systematic Review of Population-Based Controlled Observational Studies
- , , and Variants Additively Predict Response to Therapy in Chronic Hepatitis C Virus Infection in a European Cohort: A Cross-Sectional Study
- Towards Improved Measurement of Financial Protection in Health
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy