
Genome-Wide Assessment of AU-Rich Elements by the ARE Algorithm
In mammalian cells, AU-rich elements (AREs) are well known regulatory sequences located in the 3′ untranslated region (UTR) of many short-lived mRNAs. AREs cause mRNAs to be degraded rapidly and thereby suppress gene expression at the posttranscriptional level. Based on the number of AUUUA pentamers, their proximity, and surrounding AU-rich regions, we generated an algorithm termed AREScore that identifies AREs and provides a numerical assessment of their strength. By analyzing the AREScore distribution in the transcriptomes of 14 metazoan species, we provide evidence that AREs were selected for in several vertebrates and Drosophila melanogaster. We then measured mRNA expression levels genome-wide to address the importance of AREs in SL2 cells derived from D. melanogaster hemocytes. Tis11, a zinc finger RNA–binding protein homologous to mammalian tristetraprolin, was found to target ARE–containing reporter mRNAs for rapid degradation in SL2 cells. Drosophila mRNAs whose expression is elevated upon knock down of Tis11 were found to have higher AREScores. Moreover high AREScores correlate with reduced mRNA expression levels on a genome-wide scale. The precise measurement of degradation rates for 26 Drosophila mRNAs revealed that the AREScore is a very good predictor of short-lived mRNAs. Taken together, this study introduces AREScore as a simple tool to identify ARE–containing mRNAs and provides compelling evidence that AREs are widespread regulatory elements in Drosophila.
Published in the journal:
. PLoS Genet 8(1): e32767. doi:10.1371/journal.pgen.1002433
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002433
Summary
In mammalian cells, AU-rich elements (AREs) are well known regulatory sequences located in the 3′ untranslated region (UTR) of many short-lived mRNAs. AREs cause mRNAs to be degraded rapidly and thereby suppress gene expression at the posttranscriptional level. Based on the number of AUUUA pentamers, their proximity, and surrounding AU-rich regions, we generated an algorithm termed AREScore that identifies AREs and provides a numerical assessment of their strength. By analyzing the AREScore distribution in the transcriptomes of 14 metazoan species, we provide evidence that AREs were selected for in several vertebrates and Drosophila melanogaster. We then measured mRNA expression levels genome-wide to address the importance of AREs in SL2 cells derived from D. melanogaster hemocytes. Tis11, a zinc finger RNA–binding protein homologous to mammalian tristetraprolin, was found to target ARE–containing reporter mRNAs for rapid degradation in SL2 cells. Drosophila mRNAs whose expression is elevated upon knock down of Tis11 were found to have higher AREScores. Moreover high AREScores correlate with reduced mRNA expression levels on a genome-wide scale. The precise measurement of degradation rates for 26 Drosophila mRNAs revealed that the AREScore is a very good predictor of short-lived mRNAs. Taken together, this study introduces AREScore as a simple tool to identify ARE–containing mRNAs and provides compelling evidence that AREs are widespread regulatory elements in Drosophila.
Introduction
Gene expression is extensively regulated at both transcriptional and posttranscriptional levels. In the cytoplasm, numerous mechanisms act on mRNAs to ensure their proper localization, translation and stability [1]. Together with the rate of transcription, the lifespan of an mRNA is a key determinant of the level at which any given gene is expressed. Half-lives differ widely between transcripts, ranging in human cells from 5 minutes to >10 hours [2], [3]. In yeast, mRNAs are degraded more rapidly and their half-lives range from 3 to >90 minutes [4].
AU-rich elements (AREs) are well-characterized cis-acting regulatory sequences that strongly accelerate the degradation of mammalian mRNAs. AREs were initially discovered in 3′ untranslated regions (UTRs) of short-lived transcripts encoding cytokines [5], [6], and since have been proposed to reside in 5–8% of all transcripts [7]. However, the frequency of functional AREs in a given cell type is certainly lower because genome-wide measurements of mRNA decay rates showed that the presence of AU-rich sequences correlates only to a limited extent with rapid mRNA decay: In primary human T-cells, only about 25% of mRNAs with AU-rich sequences were found to decay rapidly [2], and in the hepatocellular carcinoma cell line HepG2 this proportion was only 10–15% [8].
Although there is no strict consensus sequence for AREs, the following key motifs have been identified: AUUUA pentamers that frequently occur in multiple copies, which may overlap or localize in close proximity [6], [9]; a related nonameric motif UUAUUUAUU or UUAUUUA(U/A)(U/A), which is strongly linked to rapid mRNA decay [10], [11], [12], [13]; and a generally U-rich or AU-rich context required for maximum efficiency of either pentamers or nonamers [9]. AREs can be distinguished according to different deadenylation kinetics, which gave rise to a widely used classification published by Shyu et al. [14]. Class I AREs (e.g. c-myc, c-fos) contain a few scattered pentamers within a larger U - or AU-rich context, and mediate synchronous deadenylation indicative of a distributive exoribonuclease. Class II AREs (e.g. GM-CSF, IL-3 and TNFα) have a cluster of 4–7 partially overlapping pentamers within a U-rich context, and mediate asynchronous deadenylation indicative of a processive exoribonuclease. Class III AREs (e.g., c-jun) lack pentamers and have been less well characterized. Khabar et al. proposed an alternative classification of AREs into five groups based on the number of overlapping AUUUA pantamers [15]. This classification has been used to mine databases for the occurrence of ARE-regulated genes [7], yet the functional implication of this classification has not been thoroughly tested.
ARE-mediated mRNA decay (AMD) depends on specific RNA-binding proteins (BPs) that recognize AREs and target the mRNA for rapid degradation [16]. The Tis11 zinc finger proteins are ARE-BPs with a major role in AMD. The mammalian Tis11 family comprises TTP [17], BRF1 [18] and BRF2 [19], all of which are potent inducers of mRNA degradation. These proteins share a highly conserved tandem C3H zinc finger domain required for RNA binding. TTP is the best characterized member of this family and acts as a suppressor of inflammation in mice by controlling the expression of tumor necrosis factor-α (TNFα) [17]. Further studies showed that TTP causes the degradation of many additional mRNAs related to the immune response (reviewed in [20]). TTP induces the degradation of its target mRNAs by recruiting the components of the general RNA degradation machinery such as the exosome [21], the decapping complex [22] and the Ccr4-Caf1-Not deadenylation complex [23]. Moreover, TTP is regulated through phosphorylation by the p38-MAPK – MK2 kinase cascade. Direct phosphorylation by MK2 causes binding of 14-3-3 adaptor proteins and decreases the activity of TTP [24], [25] by interfering with the ability of TTP to recruit the Ccr4-Caf1-Not deadenylation complex [26], [27]. In turn, the protein phosphatase 2A dephosphorylates TTP and thereby activates AMD [28].
Very little is known about AMD in Drosophila melanogaster. So far, only the mRNAs encoding CecA1 and bnl were shown to contain a functional ARE [29], [30], [31]. The CecA1 ARE binds to Tis11, the homologue of TTP in D. melanogaster, which in turn promotes rapid degradation of CecA1 mRNA by enhancing deadenylation [29]. Interestingly, expression of mammalian TTP could compensate for the knock down of Tis11 in Drosophila cells [30], suggesting evolutionary conservation. While the regulation of CecA1 mRNA degradation has been well characterized, there is no experimental study addressing more generally the role of AMD in D. melanogaster.
Here we report the development of AREScore, a software application by which mRNAs can be assessed for the presence of AREs. After validating the AREScore using half-life measurements of human and mouse mRNAs, the transcriptome-wide AREScore distribution was analyzed across 14 metazoan species. The AREScore was then applied to the analysis of AMD in Drosophila SL2 cells. By combining biochemical and bioinformatic approaches, we provide evidence for a specific set of mRNAs regulated by Tis11, and for the broader role of AREs in controlling mRNA degradation in D. melanogaster.
Results
AREScore provides a numerical assessment for AREs
With the aim to identify genes containing AREs in any given set of sequences, we developed an algorithm termed AREScore, schematically depicted in Figure 1A. Its purpose is to provide a numerical measure of the potential strength of an ARE, and assess the occurrence of AREs on a transcriptome-wide level. The AREScore is based on quantifying three typical features of AREs: the number of AUUUA pentamers, the proximity between pentamers, and the presence of a region with high AU content surrounding AUUUA pentamers. The UUAUUUA(U/A)(U/A) nonamer was not counted as a separate parameter because it largely corresponds to two overlapping AUUUA pentamers or a pentamer within a region of high AU content. The algorithm first counts AUUUA pentamers and attributes a fixed value of 1 for each pentamer to generate a basal score. It then calculates the distance between neighboring pentamers, and adds a value to the basal score if pentamers are close to each other. Likewise, a value is added if pentamers are located within a region of high AU content, herein termed an AU-block. To increase the flexibility of AREScore, users can change the values that are added to the basal score, and alter the settings that define an AU-block. Thereby users can adapt the algorithm to their needs and particular questions. A web-based version of AREScore is available at http://arescore.dkfz.de/arescore.pl.

To validate the algorithm, we calculated the AREScore for every human mRNA in the RefSeq database with a 3′UTR length ≥10 nucleotides (nt), whereby many falsely annotated 3′UTRs could be excluded from the analysis. In Figure 1B, the AREScore was then compared to previously measured mRNA half-lives in human DG75 B-cells [32]. The AREScore shows a slight, but statistically highly significant, negative correlation with mRNA half-life (Spearman rank correlation coefficient RS = −0.155, p<0.0001). The correlation was more apparent when mRNAs were classified into groups with similar AREScores and the average half-life was plotted for each group (Figure 1C). We then used Receiver Operating Characteristic (ROC) analysis to assess the predictive power of the AREScore in this dataset (Figure 1D). Every possible AREScore value was tested for its ability to discriminate the 10% most short-lived mRNAs from the 10% most long-lived ones. For instance, mRNAs with an AREScore ≥3.9 make up 53% of the short-lived mRNAs (true positive rate), but only 14% of the long-lived mRNAs (false positive rate). By plotting true positive rate against false positive rate for every possible AREScore, the ROC curve is obtained. The area under this curve (AUC) corresponds to the probability that a random short-lived mRNA has a higher AREScore than a random long-lived mRNA. With a value of 0.75, the AUC is well above that of a random predictor (AUC = 0.5).
In a similar manner, we compared the AREScore of mouse mRNAs with half-lives measured previously in mouse NIH3T3 fibroblasts [33]. This analysis again showed a weak but highly significant negative correlation between AREScore and mRNA half-life (Figure 1E, RS = −0.147, p<0.0001, and Figure 1F). With an AUC of 0.73, the ROC curve of the mouse dataset (Figure 1G) is very similar to the curve of the human dataset (Figure 1D). Taken together, the comparison of AREScores with genome-wide measurements of mRNA half-lives showed that mRNAs with a high AREScore are more likely to be short-lived, both in human and mouse cell lines.
To further validate the AREScore, we analyzed a set of transcripts that we had previously identified as TTP-associated mRNAs in mouse macrophages using RNA-immunoprecipitation [13]. Figure 1H shows that the AREScore is very high among the 135 TTP-associated mRNAs (median 7.8) compared to the entire mouse transcriptome (median 1.3) or a more stringent control set of randomly chosen, concatenated 3′UTR sequences (median 4.65) whose lengths were matched to the lengths of the TTP-associated 3′UTRs. To test whether the AREScore distribution of the TTP-associated mRNAs was statistically different from the controls, we compared the frequency of mRNAs with an AREScore <4 and ≥4 in 2×2 contingency tables (Tables S1 and S2). P-values were calculated either by χ2-test or Fisher's exact test, and found to be <0.0001 for both comparisons. Thus, the AREScores of the TTP-associated mRNAs were significantly higher than the AREScores of both control groups. This confirmed that the AREScore is a useful tool to identify ARE-containing mRNAs.
AREs are more abundant in vertebrate and arthropod genomes
Having the AREScore at hand as a numerical tool to estimate the abundance and strength of AREs in any given genome, we calculated the AREScore of all annotated transcripts with a 3′UTR length ≥10 nt for Homo sapiens and four important metazoan model organisms, Caenorhabditis elegans, D. melanogaster, Danio rerio, and Mus musculus. The analysis shows that in all five species, the vast majority of mRNAs have a score below 4 (Figure 2A). Differences became apparent when frequencies were plotted on a logarithmic scale (Figure 2B). The highest AREScore is 17.4 in C. elegans and 34.3 in D. melanogaster, whereas in the two mammalian species AREScores go beyond 60. These differences are also visible in the plot of cumulative frequencies (Figure 2C), which shows the highest prevalence of low AREScores in C. elegans and of high AREScores in H. sapiens. It was interesting to note that the 3′UTR length follows a similar pattern (Figure 2D), with C. elegans having the by far shortest 3′UTRs (median: 140 nt), followed by D. melanogaster (207 nt) and D. rerio (402 nt), and considerably longer 3′UTRs in the two mammalian species (704 nt in mouse, 804 in man). This analysis shows that mRNAs with high AREScores as well as long 3′UTRs are more abundant in the two mammalian species, which likely reflects the need for additional elements regulating gene expression.

Evolutionary selection for AREs
To test whether AREs are truly enriched in any of the transcriptomes we analyzed, we compared the AREScore distribution in different species with sets of randomized sequences that have identical A/T/G/C contents and length distributions (Figure 3 and Figure S1). This comparison revealed that mRNAs with high AREScores (≥10) are overrepresented in the transcriptome of H. sapiens (Figure 3A). In D. melanogaster, the enrichment already starts at an AREScore of 4 (Figure 3B), whereas there is no enrichment of mRNAs with higher AREScores in C. elegans (Figure 3C). We then expanded this analysis to the transcriptomes of 11 additional species, covering most of the major branches of metazoan evolution (Figure S1). Only for Annelida and Crustacea, no properly annotated transcriptomes were available. In the 14 species analyzed, the frequency of mRNAs with an AREScore ≥10 was compared to the frequency of AREScores ≥10 in sets of randomized control sequences (Figure 3D). mRNAs with an AREScore ≥10 were found to be overrepresented in the transcriptomes of H. sapiens, M musculus, Gallus gallus (chicken), Danio rerio (zebrafish) and D. melanogaster. This is reflected by a positive Φ coefficient, a measure for how strongly AREScores ≥10 are associated with the actual transcriptome as compared to the randomized control. In all these cases, the difference was significant as determined by χ2-test. mRNAs with an AREScore ≥10 were also more abundant in Ixodes scapularis (deer tick), although in this case the difference was statistically not significant. In the 8 other species analyzed, mRNAs with an AREScore ≥10 were less abundant than in the randomized control sequences. Thus, our analysis suggested that AREs were selected for during the evolution of several vertebrate species (Xenopus laevis being the exception) as well as D. melanogaster.

AREScore of Tis11-sensitive mRNAs in Drosophila cells
Given that we found AREs to be overrepresented in the D. melanogaster transcriptome (Figure 3B) and that little is known about the general importance of AMD in this organism, we decided to experimentally address the functional relevance of the AREScore in Drosophila. We first established an assay to measure AMD in D. melanogaster SL2 cells by generating firefly luciferase (FL) reporter genes containing the ARE of mouse interleukin (IL)-3 in the 3′UTR (Figure S2A, IL3 ARE sequence depicted in Figure S3). Expression of the FL reporter gene was found to be strongly suppressed by the IL3 ARE in SL2 cells, both at the protein (luciferase activity) and mRNA level, and suppression was due to accelerated degradation of the reporter mRNA (Figure S2B–S2D). We then tested several factors for their involvement in Drosophila AMD by knocking down their expression using dsRNAs. Whereas knock down (kd) of Tis11 and Not1, a core protein of the cytoplasmic Ccr4-Caf1-Not deadenylation complex, caused elevated expression of the ARE-reporter, other proteins such as Rox8, AGO1, AGO2, LSm1 and pcm did not affect reporter gene expression (Figure 4A).

Since Drosphila Not1 is important for mRNA deadenylation in general [34], [35], we focused on Tis11 as an ARE-specific RNA binding protein. Our goal was to examine the AREScore of mRNAs regulated by Tis11. We first confirmed that the dsRNA against Tis11 potently suppressed the expression of Tis11 mRNA (Figure 4B), and that Tis11 kd stabilizes the FL-mIL3-ARE reporter mRNA (Figure 4C). To identify Drosophila mRNAs regulated by Tis11, we determined the mRNA expression profile in SL2 cells after knocking down Tis11 or, as a control, GFP. Since direct targets of Tis11 are expected to show higher expression levels after Tis11 kd, we concentrated on the 53 mRNAs that we found to be upregulated by a factor of at least 1.41 (0.5 log2-transformed) after Tis11 kd in the microarray analysis (Figure 5A). 20 out of these mRNAs were chosen for confirmation by qPCR, and for 18 of them we could verify that Tis11 kd causes a an increase in expression of minimally 1.41-fold (Figure S4), indicating that our microarray dataset was reliable. The Vir1 mRNA, which was strongly upregulated by Tis11 kd (Figure S4), has an AREScore of 5.6 and a readily detectable ARE (Figure S3). Indeed, an FL reporter mRNA containing the ARE of Drosophila Vir1 was stabilized by kd of Tis11 (Figure 4C).
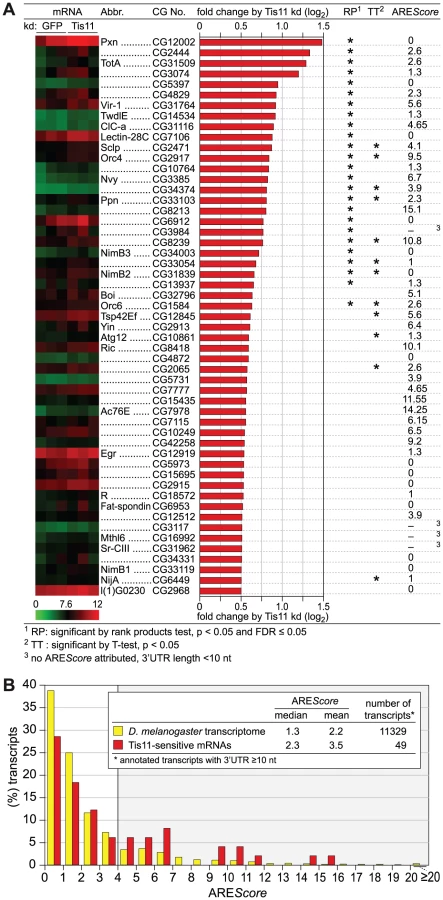
Out of the 53 mRNAs sensitive to Tis11 kd, we then determined the AREScore for those 49 transcripts whose annotated 3′UTR length is ≥10 nt. In comparison to the AREScore distribution of the entire D. melanogaster transcriptome, the Tis11-sensitive mRNAs showed an increased abundance of AREScores ≥4 (Figure 5B). By χ2-test, this increase was statistically significant with a p-value of 0.0011 (Table S3), suggesting that target mRNAs of Drosophila Tis11 share characteristics with mammalian AREs.
AREScore correlates with Drosophila mRNA half-life and expression level
After applying the AREScore to the subgroup of Tis11-sensitive mRNAs, we wanted to assess the importance of AREs in regulating Drosophila gene expression more generally. If AREs are wide-spread elements that promote mRNA degradation but do not affect transcription, a first prediction is that, on average, mRNAs with a high AREScore should be expressed at lower levels. A second prediction is that these mRNAs should have shorter half-lives. We tested the first prediction by comparing the expression levels of 6657 mRNAs, derived from our microarray analysis in SL2 cells, with their AREScores (Figure 6A). Indeed, we observed a tendency for mRNAs with high AREScores to be expressed at lower levels. We further grouped the mRNAs into 9 categories according to their AREScore, and compared the average expression levels of each group to the overall average (Figure 6B). The two groups with very high AREScores ≥12 showed average expression levels that were more than 3-fold (1.6 log2-transformed) below the overall average, and the reduction in expression was already significant above an AREScore of 8.

We then compared the 3′UTR lengths with the AREScores of all 6657 mRNAs (Figure 6C). As expected, we found a very strong correlation between these two parameters (RS = 0.57, p<0.0001). Thus, it was important to assess whether the 3′UTR length on its own had an influence on mRNA expression levels (Figure 6D and 6E). Two opposing correlations were apparent: mRNAs with very short 3′UTRs <100 nt, and mRNA with long 3′UTRs ≥1000 nt were expressed at significantly reduced levels, whereas mRNAs with 3′UTRs of intermediate length (100–999 nt) showed the highest expression levels. In fact, the 3′UTR length appeared to have a stronger influence on the expression level than the AREScore, as mRNAs with 3′UTRs ≥2000 nt were expressed more than 5-fold (2.4 log2-transformed) below the overall average. To examine whether the predictive power of the AREScore is independent of 3′UTR length, we chose to analyze a subgroup of 1781 mRNAs with 3′UTRs between 200 and 499 nt (pink bars in Figure 6E). In this group, the length of the 3′UTR per se does not negatively correlate with mRNA levels (Figure 6F), whereas mRNAs with higher AREScores do show a trend towards reduced expression levels (Figure 6G). Indeed, the 35 mRNAs that have an AREScore ≥8 within this group had a more than 3-fold (1.6 log2-transformed) reduced average expression level compared to the 1746 mRNAs that have an AREScore between 0 and 7.99 (Figure 6H), and this difference was highly significant (p<0.0005). In contrast, the average expression level of the 35 mRNAs with the longest 3′UTRs was only 1.4-fold (0.5 log2-transformed) below the expression level of the remaining 1749 mRNAs with the shorter 3′UTRs. From this comparison we concluded that a high AREScore correlates with lower mRNA expression levels independently of the 3′UTR length.
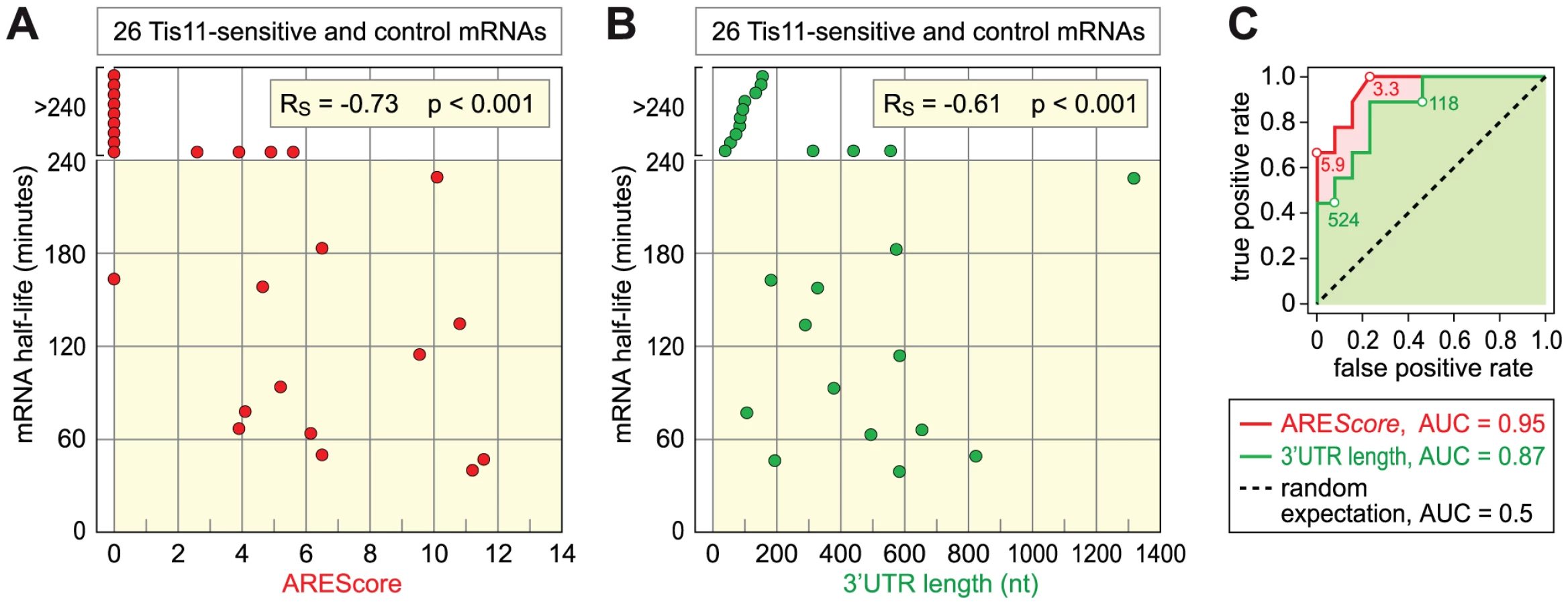
Finally, we tested the second prediction that mRNAs with higher AREScores should undergo more rapid decay. To this end, we measured the half-lives of 26 mRNAs with high accuracy by qPCR (Table 1, Figure S5). 12 mRNAs were chosen from the group of Tis-11 sensitive mRNAs, and 14 from the large pool of mRNAs that are not affected by Tis11 kd. To cover the entire range, 5 mRNAs had a high AREScore ≥8, 8 mRNAs had a medium AREScore between 4 and 7.99, and 13 had a low AREScore <4. In Figure 7A, we plotted the half-lives of these mRNAs against the AREScore. The most striking observation was that 9 out of 10 mRNAs with an AREScore of 0 degraded very slowly with half-lives >240 minutes. On the other side, the two mRNAs with the highest AREScore (CG115435 from the group of Tis11-sensitive mRNAs and Reck and from the control group) also had the shortest half-lives. In our analysis of 26 mRNAs, the Spearman's rank correlation coefficient RS between the two parameters equals −0.73, and this correlation was highly significant (p<0.001). We also compared the half-lives of these 26 mRNAs with their 3′UTR length (Figure 7B), and found a weaker correlation (RS = −0.61, p<0.001). ROC analysis was then applied to test the ability of both AREScore and 3′UTR length to discriminate labile mRNAs with half-lives <140 minutes from stable mRNAs with half-lives >240 minutes (Figure 7C). The AREScore performed extremely well in this test with an AUC of 0.95, better than 3′UTR length with an AUC of 0.87. Clearly, the AREScore identifies short-lived mRNAs in D. melanogaster, showing that AREs are general regulatory elements in this organism.

Discussion
In this report, we developed AREScore as an algorithm to identify AREs and provide a measure for their potential strength (Figure 1). The AREScore was validated using genome-wide mRNA half-life measurements in human DG75 B-cells [32] and mouse NIH3T3 fibroblast [33]. Although the correlation between AREScore and mRNA half-life was weak (RS = −0.155 and −0.147 in the two data sets, respectively), it was statistically highly significant. To our knowledge, this is the best correlation observed so far between any parameter and mRNA half-lives on a genome-wide scale.
The potential of the AREScore could be further demonstrated with a set of TTP-associated mRNAs that we had previously identified by RNA-IP in mouse macrophages [13]. AREScores were much higher in this set of mRNAs than in the two control groups (Figure 1H). Among the Tis11-sensitive mRNAs that we identified in Drosophila SL2 cells, we also observed an increased frequency of mRNAs with higher AREScores (Figure 5), suggesting that Drosophila Tis11 recognizes AREs with sequence features similar to mammalian AREs.
Khabar et al. used bioinformatic tools to generate the ARE-database (ARED), a comprehensive list of potential AREs in the human, mouse and Drosophila genome [7], [31], [36]. The principle behind ARED is that it classifies AREs according to the number and density of AUUUA pentamers and surrounding AU-rich sequences, which correlates, to some degree, with the potential strength of the ARE. In contrast to ARED, the purpose of AREScore is not to make categories, but rather generate a single score that provides a measure for the likelihood and potential strength of an ARE. It is important to emphasize that in the absence of experimental validation, neither ARED nor AREScore is able to predict with absolute certainty whether a given mRNA contains a functional ARE. For the AREScore, the false positive rate was visualized by ROC analysis, whereby the AREScore is tested for its ability to discriminate between the 10% most short-lived and the 10% most long-lived mRNAs (Figure 1D and 1G). For ARED, the false positive rate is not known.
An advantage of AREScore is that it can be applied easily to any genome or set of sequences. Thus, we were able to compute the AREScore distribution for the transcriptomes of 14 species representing all but two of the major branches of metazoan evolution (Figure 2 and Figure S1). The analysis showed that mRNAs with high AREScores are most abundant in man and mouse, the two mammalian species analyzed. Comparison to randomized control sequences revealed that mRNAs with high AREScores (≥10) are overrepesented in man, mouse, chicken, zebrafish and the fruit fly (Figure 3). This suggests that AREs were under positive selection pressure during the evolution of these organisms. On the other hand, high AREScore mRNAs are underepresented in the sponge A. queenslandica, the freshwater cnidarian H. magnipapillata, the mollusc A. californica and the nematode C. elegans, suggesting that AREs did not expand in the genomes of metazoans with simpler body plans. Alternatively, the element corresponding to the ARE might have different sequence features in these organisms.
Given that very little is known about AREs in D. melanogaster, we then made use of the AREScore to address the role of AMD in Drosophila SL2 cells. Using an FL-based reporter assay, we first tested several factors and found that knocking down Tis11 or Not1 caused inhibition of AMD, whereas the kd of Rox8, AGO1, AGO2, LSm1 or pcm had no effect (Figure 4). The requirement of Tis11 for AMD is in good agreement with the well documented role of TTP in mammalian AMD [37] as well as previous reports demonstrating that Tis11 participates in AMD in Drosophila cells [29], [30], [31], [38]. The requirement for Not1 may be linked to our recent finding that mammalian TTP recruits the Caf1 deadenylase through its association with Not1 [23]. Not1 is the scaffold protein of the Ccr4-Caf1-Not deadenylase complex that plays a key role in cytoplasmic mRNA turnover. In Drosophila, Not1 was shown to be important for bulk mRNA deadenylation and, more specifically, for the rapid deadenylation of Hsp70 mRNA [34], [35]. A previous report had suggested that AGO1 and AGO2 are required for the rapid degradation of a reporter mRNA containing the ARE of mammalian TNFα in Drosophila S2 cells [38]. In our assay, kd of the argonaute proteins AGO1 and AGO2 did not affect expression of the reporter gene containing the ARE of mouse IL-3 (Figure 4A), indicating that AGO proteins are not generally required for AMD.
As potential substrates of AMD, we then identified 53 mRNAs whose expression was elevated after kd of Tis11 (Figure 5A). The AREScore of these Tis11-sensitive mRNAs was found to be higher in comparison to the distribution in the entire D. melanogaster transcriptome (Figure 5B), and the difference was statistically significant for mRNAs with AREScores ≥4 (Table S3). CecA1 mRNA, previously identified as a target of Tis11 [29], [30], [31], did not come up as Tis11-sensitive simply because this mRNA is not represented on the Affymetrix Drosophila Genome 2.0 array that we used for our study.
We then compared the expression levels of 6657 mRNAs in SL2 cells with their AREScore (Figure 6A–6E), and observed that mRNAs with high AREScores have reduced expression levels. However, this effect may be indirect because the AREScore strongly correlates with 3′UTR length. Indeed, when grouping mRNAs according to their 3′UTR length, we again observed that mRNAs with long 3′UTRs have lower expression levels. The impact of 3′UTR length was in fact stronger than the impact of the AREScore. Long 3′UTRs are likely to correlate with low expression levels through the presence of different suppressive elements including AREs and miRNA-binding sites. Moreover, the distance between the stop codon and the poly(A) tail is a determinant of nonsense-mediated mRNA decay [39] and may thereby as well contribute to mRNA suppression. We also noted that mRNAs with very short 3′UTRs <100 nt are expressed below the overall average. A possible explanation is that very short 3′UTRs might lack stabilizing elements, although there is little experimental evidence that such elements are abundant.
To examine the impact of the AREScore independently of its correlation with 3′UTR length, we chose a group of mRNAs with intermediate 3′UTRs (Figure 6F–6H). Within this group we could observe that mRNAs with an AREScore ≥8 had a more than 3-fold reduced average expression level compared to the mRNAs with AREScores <8. Since 3′UTR length had a much smaller effect on mRNA levels in this group, we concluded that the AREScore is an independent parameter that correlates with suppressed mRNA levels. Given the multitude of factors that affect mRNA stability and transcription rates, it is remarkable that the AREScore alone has a detectable influence on mRNA expression levels.
Finally, we measured the decay rates of 26 mRNAs in Drosophila SL2 cells (Table 1). Indeed, we observed a very strong, negative correlation between mRNA half-life and the AREScore (RS = −0.73, Figure 7), which was higher than the correlation with 3′UTR length (RS = −0.61,). Since we measured mRNA half-lives both in control GFP and Tis11 kd cells, we could also identify three mRNAs that are significantly stabilized by the absence of Tis11. These mRNAs encode for peroxidasin (Pxn), CG15435, a C2H2 zinc finger protein of unknown function, and CG7115, a protein phosphatase of the PP2C family. Taken together, our analysis provides compelling evidence that AREs are functional regulatory elements in D. melanogaster cells whose suppressive effect can be detected on a transcriptome-wide level. Interestingly, we found two short-lived mRNAs with a high AREScore (Reck and CG32512) in our control group of mRNAs that are not sensitive to Tis11 kd. This indicates that in addition to Tis11, other proteins also participate in regulating AMD. It is clear that we have only begun to understand the posttranscriptional regulatory network that controls gene expression through mRNA turnover in D. melanogaster.
Methods
Plasmid construction
Plasmid pRp128-RL (p2933) [40] contains the Drosophila RNA polymerase III 128 kDa subunit promoter to drive RL expression, and was kindly provided by Michael Boutros (German Cancer Research Center, Heidelberg). For pRp128-FL (p2934), pRp128-RL was digested with SpeI/NheI and religated to remove part of the polylinker. In the resulting construct, the RL-containing HindIII/XbaI fragment was replaced with the FL-containing HindIII/XbaI insert from pGL3-Basic (Promega). For pRp128-FL-mIL3-ARE (p2935), the mouse IL-3 ARE sequence (NM_010556.4, nt 680–744) was amplified by PCR using primers G1090/G1091 (Table S4) and inserted into the XbaI site of pRp128-FL. The control vector pRp128-FL-mIL3-INV (p2936) was constructed in the same way with the IL-3 ARE inserted in the opposite orientation.
To generate pAc5-FL-mIL3-ARE (p2937), a 3.8 kb FL-containing HindIII (blunt)–BglII fragment was excised from plasmid pRp128-mIL3-ARE and ligated to KpnI (blunt) - BglII fragment (2.4 kb) with Ac5 promoter obtained by digestion of pAc5.1b-EGFP-dmDcp1 (p2450) (kindly provided by Elisa Izaurralde, Max Planck Institute for Developmental Biology, Tübingen, Germany). For pAc5-FL-Vir1-ARE (p2938), the D. melanogaster Vir1 3′UTR (NM_165011.2, nt 1521–1830) was first amplified by RT-PCR using primers G1673/G1674. An ARE-containing 191 nt long fragment (NM_165011.2, nt 1640–1830) was re-amplified by PCR using XbaI site-containing primers G1681/G1679 and inserted into the XbaI site of pRp128-FL to generate pRp128-FL-Vir1-ARE. Finally, the Ac5 promoter was excised as a SapI–BglI fragment from pAc5-FL-mIL3-ARE and cloned into the SapI/BglI sites of pRp128-FL-Vir1-ARE, thereby replacing the pRp128 promoter. pAc5-FL (p2939) was generated in a similar manner by cloning the SapI–BglI fragment from pAc5-FL-mIL3-ARE into the SapI/BglI sites of pRp128-FL.
Cell culture and transfection
Drosophila SL2 cells were cultivated at 26°C under atmospheric CO2 in Schneider's Drosophila Medium (Invitrogen-Gibco, Cat. No. 11720-034) supplemented with 10% foetal bovine serum (Biochrome Superior FBS, Cat. No. S0615), 50 U/ml penicillin and 0.05 mg/ml streptomycin (both Pan Biotech).
All DNA transfections were performed using Effectene reagent (Qiagen, Cat. No. 301425) according to the manufacturer's instructions. When combined with RNAi, cells were first treated with dsRNA for two days, followed by DNA transfection for two additional days. For luciferase assays, 10.000 cells were seeded per well of a 384-well plate (Greiner), treated with 250 ng of dsRNA and transfected with 7 ng of FL-encoding and 3 ng of RL-encoding plasmids. Where indicated, ActinomycinD (Applichem, Cat. No. A1489) was used at a concentration of 5 µg/ml. Unless noted otherwise, cell lysis and RNA extraction were performed with Genematrix universal DNA/RNA/Protein purification kit (Eurx), according to manufacturer's instructions.
dsRNA preparation and treatment
DNA templates for in vitro transcription were amplified by PCR using primers containing Sp6 promoter sequences, as specified in Table S5. DNA templates were gel-purified using a gel extraction kit (QIAGEN, Cat. No. 28706). In vitro transcription reactions were assembled in a total volume of 50 µl containing 50–75 ng/µl DNA template, 3 mM NTPs each (Promega), 40 U RNasin (Promega, N2111), 0.5 U yeast pyrophospatase (Sigma, Cat. No. I1891), 200 U Sp6 RNA polymerase (Fermentas, EP0133), 80 mM HEPES-KOH pH 7.5, 32 mM MgCl2, 2 mM spermidine and 40 mM DTT. Reactions were incubated for 4 hours at 37°C. DNA was then digested by the addition of 1 U/µl DNase RQ1 (Promega) for 15 minutes at 37°C. The synthesized RNA was purified by gel filtration using pre-packed Sephadex G-50 columns (Roche, Cat. No. 11274015001). Strands were annealed by heating the purified RNA to 65°C and allowing it to slowly cool to room temperature. For RNAi, Drosophila cells were grown in 6 cm-dishes and incubated with 50 µg of dsRNA per 4 ml of medium for a minimum of 4 days.
Nothern blot analysis
Total RNA was extracted using the Genematrix universal RNA purification kit (Eurx). 5–12 µg of RNA was resolved by 1.1% agarose/2% formaldehyde/MOPS (morpho-linepropanesulfonic acid) gel electrophoresis and blotted over night with 8× saline-sodium citrate (SSC, 1× contains 0.15 M NaCl and 0.015 M sodium citrate) buffer onto Hybond-N+ Nylon membranes (Amersham, GE Healthcare). Membranes were hybridized overnight at 55°C with digoxigenin-labelled RNA probes synthesized in vitro using Sp6 polymerase (Fermentas) and DIG RNA labelling mix (Roche). 500 ng RNA probe was diluted in 10 ml hybridization buffer containing 50% formamide, 5× SSC, 5× Denhard's solution, 5 mM EDTA, 10 mM PIPES pH 7.0, 4 mg torula yeast RNA (US Biological) and 1% SDS. Membranes were washed twice with 2× SSC/0.1% SDS for 5 minutes, and twice with 0.5× SSC/0.1% SDS for 20 minutes at 65°C. Alkaline phosphatase-coupled anti-digoxigenin Fab fragments and CDP-Star substrate (both Roche) were used for detection according to the manufacturer's instructions. Sequences of primers that were used to generate templates for digoxigenin-labelled RNA probes are provided in Table S6.
Quantitative PCR
For qPCR, total RNA was extracted by Genematrix RNA purification kit (Eurx) and subjected to DNase treatment using RQI DNase (Promega, 1.5 U/column). cDNA was synthesized from 5 µg of total RNA using oligo-dT18 (Invitrogen) and M-MLV H(-) reverse transcriptase (Promega). 1∶40 volume of a cDNA reaction was used for PCR. PCR reactions were assembled in 384-well plates, 15 µl/well final volume. DNA SYBR Green I Master kit (Roche Cat. No. 04707516001) was used according to manufacturer's instructions. Quantitative PCR was performed with the Lightcycler 480 system (Roche). Gene-specific primers sequences are given in Table S7.
Luciferase assay
FL and RL activities were measured using the Dual Luciferase Reporter Assay system (Promega) or reagents developed in the lab of Michael Boutros (German Cancer Research Centre, DKFZ, Heidelberg). Chemiluminescence was measured using a Mithras LB940 plate reader.
Genome-wide expression profiling and bioinformatic analysis
Drosophila SL2 were treated with either dsRNA-GFP or dsRNA-Tis11 for 4 days. Total RNA was prepared using RNEasy kit from Qiagen (Cat. No. 74106). Efficiency of Tis11 knockdown was confirmed separately by Northern blot and qPCR analyses. Expression profiling was carried out on GeneChip Drosophila Genome 2.0 Arrays (Affymetrix, Cat. No. 900531) at European Molecular Biology Lab Genecore facility (Heidelberg, Germany). Microarray data were deposited at NCBI GEO, accession GSE28147. The RMA algorithm was used for normalization of raw data (RMAExpress software, http://rmaexpress.bmbolstad.com/). Further statistical analyses were performed in the multi-experiment viewer of the TM4 microarray software suite [41] or with R software (http://www.r-project.org). Beside standard Student t-test, we also used microarray-oriented Rank products test [42] to identify significant changes in expression.
AREScore
The AREScore algorithm was written in Perl and is accessible online at http://arescore.dkfz.de/arescore.pl. AREScore uses either a set of sequences provided in FASTA format or retrieves the 3′UTR sequences of properly annotated transcripts if Refseq IDs are entered. The algorithm first generates a basal score by adding a fixed value of 1 for each AUUUA pentamer identified. It then calculates the distance between neighboring pentamers, and adds a value to the basal score if pentamers are in close proximity. A value is also added when pentamers are located within a region of high AU content, termed an AU-block in AREScore. In its standard setting, AREScore adds a value of 1.5 for overlapping pentamers, 0.75 if pentamers are 0–3 nt apart, 0.4 if pentamers are 4–6 nt apart, 0.2 if pentamers are 7–9 nt apart, and 0.3 if pentamers are within an AU-block. By default, an AU-block starts when a sequence of 20 nt (word size) has an AU content of ≥80%. The block ends when the AU content drops below 55% within the chosen word size. To increase the flexibility of AREScore, users can change the values that are added to the basal score, and alter the settings that define an AU-block.
For the transcriptome-wide AREScore analysis, transcripts with properly formated feature fields (Genbank GeneID and Accession identifiers) were downloaded from Refseq, and the AREScore was determined for every 3′UTR ≥10 nt in length. If several transcripts map to the same gene locus (identical GeneIDs), only the mRNA with the highest AREScore was taken into the analysis. To generate a fully matched set of randomized control sequences, nucleotides were randomly chosen from the pool of all 3′UTRs analyzed, and assembled into sequences identical in length to the original 3′UTRs. The AREScore was then determined for the randomized control sequences as well.
Supporting Information
Zdroje
1. MooreMJ 2005 From birth to death: the complex lives of eukaryotic mRNAs. Science 309 1514 1518
2. RaghavanAOgilvieRLReillyCAbelsonMLRaghavanS 2002 Genome-wide analysis of mRNA decay in resting and activated primary human T lymphocytes. Nucleic Acids Res 30 5529 5538
3. HaoSBaltimoreD 2009 The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol 10 281 288
4. WangYLiuCStoreyJTibshiraniRHerschlagD 2002 Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA 99 5860 5865
5. CaputDBeutlerBHartogKThayerRBrown-ShimerS 1986 Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci USA 83 1670 1674
6. ShawGKamenR 1986 A conserved AU sequence from the 3′untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46 659 667
7. BakheetTWilliamsBRKhabarKS 2006 ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res 34 D111 114
8. YangEvan NimwegenEZavolanMRajewskyNSchroederM 2003 Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res 13 1863 1872
9. XuNChenCYShyuAB 1997 Modulation of the fate of cytoplasmic mRNA by AU-rich elements: key sequence features controlling mRNA deadenylation and decay. Mol Cell Biol 17 4611 4621
10. ZubiagaAMBelascoJGGreenbergME 1995 The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol 15 2219 2230
11. LagnadoCABrownCYGoodallGJ 1994 AUUUA is not sufficient to promote poly(A) shortening and degradation of an mRNA: the functional sequence within AU-rich elements may be UUAUUUA(U/A)(U/A). Mol Cell Biol 14 7984 7995
12. LaiWSCarrickDMBlackshearPJ 2005 Influence of nonameric AU-rich tristetraprolin-binding sites on mRNA deadenylation and turnover. J Biol Chem 280 34365 34377
13. StoecklinGTenenbaumSAMayoTChitturSVGeorgeAD 2008 Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J Biol Chem 283 11689 11699
14. ChenC-YAShyuA-B 1995 AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20 465 470
15. BakheetTFrevelMWilliamsBRGreerWKhabarKS 2001 ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res 29 246 254
16. BarreauCPaillardLOsborneHB 2005 AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res 33 7138 7150
17. CarballoELaiWSBlackshearPJ 1998 Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281 1001 1005
18. StoecklinGColombiMRaineriILeuenbergerSMallaunM 2002 Functional cloning of BRF1, a regulator of ARE-dependent mRNA turnover. EMBO J 21 4709 4718
19. LaiWSCarballoEThornJMKenningtonEABlackshearPJ 2000 Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to AU-rich elements and destabilization of mRNA. J Biol Chem 275 17827 17837
20. SchottJStoecklinG 2010 Networks controlling mRNA decay in the immune system. Wiley Interdiscip Rev RNA 1 432 456
21. ChenCYGherziROngSEChanELRaijmakersR 2001 AU Binding Proteins Recruit the Exosome to Degrade ARE-Containing mRNAs. Cell 107 451 464
22. Lykke-AndersenJWagnerE 2005 Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev 19 351 361
23. SandlerHKrethJTimmersHTStoecklinG 2011 Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res 39 4373 4386
24. StoecklinGStubbsTKedershaNWaxSRigbyWF 2004 MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J 23 1313 1324
25. ChrestensenCASchroederMJShabanowitzJHuntDFPeloJW 2004 MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J Biol Chem 279 10176 10184
26. ClementSLScheckelCStoecklinGLykke-AndersenJ 2011 Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol Cell Biol 31 256 266
27. MarcheseFPAubaredaATudorCSaklatvalaJClarkAR 2010 MAPKAP kinase 2 blocks tristetraprolin-directed mRNA decay by inhibiting CAF1 deadenylase recruitment. J Biol Chem 285 27590 27600
28. SunLStoecklinGVan WaySHinkovska-GalchevaVGuoRF 2007 Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J Biol Chem 282 3766 3777
29. LauwersATwyffelsLSoinRWauquierCKruysV 2009 Post-transcriptional Regulation of Genes Encoding Anti-microbial Peptides in Drosophila. J Biol Chem 284 8973 8983
30. WeiYXiaoQZhangTMouZYouJ 2009 Differential regulation of mRNA stability controls the transient expression of genes encoding Drosophila antimicrobial peptide with distinct immune response characteristics. Nucleic Acids Res 37 6550 6561
31. CairraoFHaleesASKhabarKSMorelloDVanzoN 2009 AU-rich elements regulate Drosophila gene expression. Mol Cell Biol 29 2636 2643
32. DolkenLMaltererGErhardFKotheSFriedelCC 2010 Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 7 324 334
33. DolkenLRuzsicsZRadleBFriedelCCZimmerR 2008 High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14 1959 1972
34. TemmeCZaessingerSMeyerSSimoneligMWahleE 2004 A complex containing the CCR4 and CAF1 proteins is involved in mRNA deadenylation in Drosophila. EMBO J 23 2862 2871
35. TemmeCZhangLKremmerEIhlingCChartierA 2010 Subunits of the Drosophila CCR4-NOT complex and their roles in mRNA deadenylation. RNA 16 1356 1370
36. HaleesASEl-BadrawiRKhabarKS 2008 ARED Organism: expansion of ARED reveals AU-rich element cluster variations between human and mouse. Nucleic Acids Res 36 D137 140
37. SandlerHStoecklinG 2008 Control of mRNA decay by phosphorylation of tristetraprolin. Biochem Soc Trans 36 491 496
38. JingQHuangSGuthSZarubinTMotoyamaA 2005 Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 120 623 634
39. NicholsonPYepiskoposyanHMetzeSZamudio OrozcoRKleinschmidtN 2010 Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci 67 677 700
40. BartschererKPelteNIngelfingerDBoutrosM 2006 Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 125 523 533
41. SaeedAIBhagabatiNKBraistedJCLiangWSharovV 2006 TM4 microarray software suite. Methods Enzymol 411 134 193
42. BreitlingRArmengaudPAmtmannAHerzykP 2004 Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett 573 83 92
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Poly(ADP-Ribose) Polymerase 1 (PARP-1) Regulates Ribosomal Biogenesis in Nucleoli
- Microenvironmental Regulation by Fibrillin-1
- Parallel Mapping and Simultaneous Sequencing Reveals Deletions in and Associated with Discrete Inherited Disorders in a Domestic Dog Breed
- Two-Component Elements Mediate Interactions between Cytokinin and Salicylic Acid in Plant Immunity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy