
Genome Engineering in : A Feasible Approach to Address Biological Issues
Although bacteria with multipartite genomes are prevalent, our knowledge of the mechanisms maintaining their genome is very limited, and much remains to be learned about the structural and functional interrelationships of multiple chromosomes. Owing to its bi-chromosomal genome architecture and its importance in public health, Vibrio cholerae, the causative agent of cholera, has become a preferred model to study bacteria with multipartite genomes. However, most in vivo studies in V. cholerae have been hampered by its genome architecture, as it is difficult to give phenotypes to a specific chromosome. This difficulty was surmounted using a unique and powerful strategy based on massive rearrangement of prokaryotic genomes. We developed a site-specific recombination-based engineering tool, which allows targeted, oriented, and reciprocal DNA exchanges. Using this genetic tool, we obtained a panel of V. cholerae mutants with various genome configurations: one with a single chromosome, one with two chromosomes of equal size, and one with both chromosomes controlled by identical origins. We used these synthetic strains to address several biological questions—the specific case of the essentiality of Dam methylation in V. cholerae and the general question concerning bacteria carrying circular chromosomes—by looking at the effect of chromosome size on topological issues. In this article, we show that Dam, RctB, and ParA2/ParB2 are strictly essential for chrII origin maintenance, and we formally demonstrate that the formation of chromosome dimers increases exponentially with chromosome size.
Published in the journal:
. PLoS Genet 8(1): e32767. doi:10.1371/journal.pgen.1002472
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002472
Summary
Although bacteria with multipartite genomes are prevalent, our knowledge of the mechanisms maintaining their genome is very limited, and much remains to be learned about the structural and functional interrelationships of multiple chromosomes. Owing to its bi-chromosomal genome architecture and its importance in public health, Vibrio cholerae, the causative agent of cholera, has become a preferred model to study bacteria with multipartite genomes. However, most in vivo studies in V. cholerae have been hampered by its genome architecture, as it is difficult to give phenotypes to a specific chromosome. This difficulty was surmounted using a unique and powerful strategy based on massive rearrangement of prokaryotic genomes. We developed a site-specific recombination-based engineering tool, which allows targeted, oriented, and reciprocal DNA exchanges. Using this genetic tool, we obtained a panel of V. cholerae mutants with various genome configurations: one with a single chromosome, one with two chromosomes of equal size, and one with both chromosomes controlled by identical origins. We used these synthetic strains to address several biological questions—the specific case of the essentiality of Dam methylation in V. cholerae and the general question concerning bacteria carrying circular chromosomes—by looking at the effect of chromosome size on topological issues. In this article, we show that Dam, RctB, and ParA2/ParB2 are strictly essential for chrII origin maintenance, and we formally demonstrate that the formation of chromosome dimers increases exponentially with chromosome size.
Introduction
Bacteria were long thought to have a simple genome architecture based on a unique circular chromosome, and it is only in the late 1980s that the first prokaryote with multiple chromosomes, Rhodobacter sphaeroides, was characterized [1]. Since this seminal observation, many other species possessing multiple circular or linear chromosomes have been characterized across numerous bacterial lineages [2]. More than 80 multipartite bacterial genomes have been sequenced, propagating various hypotheses to explain their extant nature and posing fundamental questions about the selective benefit of such a genome architecture.
Numerous studies have established the cholera pathogen, Vibrio cholerae, as the model for bacteria with multipartite genomes [3]. The genome of V. cholerae N16961 consists of two circular chromosomes, a primary 2.96 Mbp chromosome (chrI) and a secondary 1.07 Mbp chromosome (chrII). V. cholerae's genes are asymmetrically distributed between the two chromosomes [4]. ChrI has low interspecies sequence variability and harbors many genes coding for essential biosynthetic pathways. ChrII contains many more species-specific genes, unknown ORFs and proportionally fewer essential genes [4]–[5]. Furthermore, V. cholerae's particular genomic organization and genetic disparity is consistent within the Vibrionaceae family [6]–[8]. The unusual genome structure of V. cholerae has inspired numerous studies to better understand the mechanisms and purposes of maintaining such a genomic organization, resulting in an impressive body of experimental data [9]–[20]. To date, however, and despite the impressive collective effort of the cited studies along with other research on chromosome and plasmid maintenance systems, the mechanisms coordinating the maintenance of multiple chromosomes are largely unknown. In tackling such pervasive yet fundamental questions, we decided to construct a unique genetic tool allowing targeted massive chromosomal rearrangements in proteobacteria. We applied this powerful technique to answer two outstanding questions. Firstly, we addressed the specific case of the essentiality of Dam methylation in V. cholerae. Secondly, we focused our genetic system on more general questions concerning bacteria with circular chromosomes by examining the effect of chromosome size and genetic distribution on topological issues.
Unlike eukaryotic organisms, where chromosomes are managed by common machineries which coordinate up to 90 chromosomes [21], V. cholerae has evolved a relatively complex and highly targeted strategy involving interplay of specific and common machineries for the maintenance of each chromosome. Replication of each V. cholerae chromosome is controlled by a unique initiator molecule [11]. ChrI replication is initiated at oriI by DnaA, the common initiator of chromosomal DNA replication in most bacteria [11], while chrII replication is regulated at a plasmid-like oriII by the Vibrio-specific factor, RctB [22]–[23]. ChrII is nonetheless replicated only once per cell generation, unlike plasmids, which are not generally linked to the cell cycle [24]. Both E. coli and V. cholerae are members of a mono-phyletic clade of the gamma-proteobacteria defined by the acquisition of the dam-seqA-mutH genes ensuring restriction of chromosome replication initiation to once per cell cycle and probe mismatch repair of replication errors [25]. Dam methylates the palindromic GATC sequence on both strands, which become transiently hemi-methylated after replication. The origin of replication and other regions with clusters of GATC sites become sequestered after replication by SeqA for up to one third of the cell cycle, which serves to preclude new initiations of replication [25]. Both V. cholerae chrI and chrII origins have GATC methylation sites [12] and their sequestration by SeqA contributes to limiting initiation of DNA replication to only once per cell cycle [9]. Replication of the larger chrI is initiated significantly before chrII so as to insure replication is terminated synchronously, suggesting a coordinating mechanism which has yet to be explained [16]. Whereas Dam is not an essential factor in E. coli, V. cholerae mutants lacking Dam methylation are not viable [26], implying the existence of differences in replication regulation between the two organisms. Methylation by CcrM, the counterpart of Dam in the α-proteobacteria, is also known to be essential for the viability of bacteria with multipartite genomes [27]–[29]. For these reasons, it was strongly suspected that the crucial role of Dam in V. cholerae could be related to its atypical genome arrangement, and Dam appeared to be a good candidate to investigate the coordinated replication initiation of the two chromosomes. In vitro studies showed that Dam methylation of RctB binding sites increases RctB binding and possibly serves a critical function in chrII replication [9]. The requirement for Dam in order to initiate replication at oriI was first studied in vivo using plasmids and monitoring the transformation efficiency of plasmids driven by oriI. These plasmids failed to transform E. coli dam mutants suggesting that Dam was essential for oriI replication initiation [12]. A reciprocal experiment involving oriC-plasmids and a mutant of E. coli where oriC was substituted by V. cholerae oriI (ΔoriC::oriI) showed that oriC-plasmids failed to transform E. coli ΔoriC::oriI Δdam [9]. Confronted with this last result and knowing that Dam is not essential in E. coli, it was hypothesized that the additional oriI-plasmid copies out-competed replication from chromosomal oriC, thus creating incompatibility conditions where Dam was required for viability of the transformants [9]. To prevent plasmid-mediated competition, the Dam requirement of V. cholerae oriI was directly assessed on the chromosome in E. coli ΔoriC::oriI [9], [30]. Two conflicting experiments, differing from the manner in which oriC was substituted by oriI, showed Dam methylation to be either required for the viability of E. coli ΔoriC::oriI [30] or not [9]. Therefore, the question of whether Dam was essential or dispensable for replication initiation of V. cholerae oriI remained unresolved. Reciprocal experiments consisting of testing oriII Dam requirement in a E. coli chromosomal context could not be tested because all attempts to replace oriC with oriII were unsuccessful [9].
After replication, partitioning of the resulting homologous chromosomes is fundamental to maintain genome stability [31]. In V. cholerae, the segregation of oriI and oriII are mediated by distinct partition factors, ParA1/B1 for chrI and ParA2/B2 for chrII [13]. ParA/B partitioning activity requires centromere-like, cis-acting sites called parS, which are bound by ParB to form a nucleoprotein complex that is a target for the ParA ATPase protein [32]. ParA1/B1 are chromosomal-like and mediate an asymmetric segregation of oriI [14]. On the other hand, ParA2/B2 are plasmid-like and carry out a symmetric segregation of oriII [20]. ParA1/B1 are not essential for chrI segregation, indicating that other factors contribute to the segregation of chrI [14] while ParA2/B2 are essential for chrII segregation and cell viability [20]. Many other bacteria with multipartite genomes have integrated distinct plasmid-like origins of replication and partitioning mechanisms to maintain their secondary chromosomes [3], which supports the hypothesis that secondary chromosomes were originally acquired as megaplasmids.
V. cholerae uses an interesting combination of mechanisms derived from both chromosomes and plasmids for the maintenance of chrII. In contrast to the above-mentioned plasmid-like mechanisms, terminal segregation of both chrI and chrII is controlled by a common bona fide chromosomal maintenance system involved in the generation of monomeric chromosome substrates for partitioning [19]. Circular chromosomes convey specific topological problems, such as the formation of dimeric chromosomes, which threatens the partition of genetic information to daughter cells (for a review, see [33]). Chromosome dimers are a side-product of homologous recombination associated with recombinational DNA repair between replicating or newly replicated circular chromosomes [33]. If an odd number of crossovers occur between sister strands, chromosome dimers are formed and must be resolved into monomers to allow chromosome segregation. This process is carried out by the combined action of the site specific tyrosine recombinases XerC and XerD that introduce an additional crossover at dif, a 28 bp site located opposite of the origin of replication [33]. V. cholerae carries two distinct recombination sites, dif1 and dif2, located in the terminus region of chrI and chrII, respectively [19]. Resolution of chromosome dimers of chrI and chrII links chromosome segregation to the late stages of cell division via the septal protein FtsK [19].
The presence of multiple chromosomes has posed challenges for in vivo studies of chromosome maintenance in bacteria as it is difficult to attribute observed phenotypes to a specific chromosome. To circumvent this issue, we designed a strategy based on specific genome rearrangements to directly study biological systems in their endogenous host. We developed a genetic tool based on two distinct site-specific recombination machineries, which allow targeted, oriented and reciprocal DNA exchanges throughout the genome. We used V. cholerae as a bi-chromosomal bacterial model to show the power of our genetic tool and how its use can help address important biological questions. Using this strategy, we examined the requirement of Vibrio-specific essential factors involved in chromosome maintenance for which functions could not be strictly attributed to a specific chromosome. We also investigated the correlation between chromosome size and the rate of formation of chromosome dimers that are the inevitable by-products of frequent recombination associated with recombinational DNA repair. To address all these questions, we created a mutant of V. cholerae with all its genetic content reorganized onto a single chromosome. We further refined our study by making additional chromosomal rearrangements to individually decipher each biological issue. In this article, we show that Dam, RctB and ParA2/ParB2 are only essential for chrII origin maintenance. We further demonstrate that the odds of forming chromosome dimers exponentially increases with chromosome size.
Results/Discussion
One from two: Reorganizing the genome of V. cholerae
We generated a mutant of V. cholerae with all its genetic content reorganized onto a single chromosome. To do so, we fused chrI with chrII in a calculated and conservative manner respecting known criteria for chromosome organization and maintenance. Prokaryotic genomes show intolerance towards various chromosome rearrangements such as inversions or relocations of DNA fragments [34]–[44]. Nevertheless, bacterial chromosomal structure can be drastically altered [45]–[48] provided that organizational features are respected (for reviews [49]–[51]). The fused chromosome was constructed to conserve the “ori-ter” axial symmetry, gene synteny, strand bias and the polarities of the original replichores. Replication of the fused chromosome initiates at oriI of chrI and finishes in the terminus of chrII near dif2. The single fused chromosome carries exclusively chromosomal-like attributes for replication and chromosome segregation (oriI, ParA1/B1, dif2), like other mono-chromosomal bacteria. By initiating replication at oriI, we conserve the replication-associated gene dosage on chrI [10]. Lastly, comparative genomics has shown that the ter region of chrI is flexible and would likely tolerate the integration of the 1 Mbp chrII [7], [52].
To perform the above-mentioned genome rearrangements, we developed a genetic tool which allows efficient and directional manipulations of any DNA segment. It involves two site-specific recombination systems which normally promote precise excision of the temperate phage genomes, λ and HK022, from their chromosomal location [53]. We used λ and HK022 integrases (Intλ and IntHK), their respective excision factors (Xisλ and XisHK) and their associated left and right excision sites (attRλ/attLλ and attRHK/attLHK). Unlike other site-specific recombination systems used for precise genome manipulation such as Cre/loxP [54] or Flp/FRT [55], the λ and HK022 recombination reactions have the calculated advantage of being directionally controlled, as the presence of the Xis excision factors orientates the catalytic reactions in one direction. This characteristic is very useful for two reasons: first, it insures that the mutant strain will not revert to the wild-type configuration after chromosomal rearrangement. Second, the newly formed sites (attB/P) react poorly with the substrate sites (attR/L) [56]. Therefore the same system can be reused in the mutant strain to perform new rearrangements at other positions by integrating new attR/L sites. In theory, this system could be used an infinite number of times in the same strain.
To fuse the two chromosomes of V. cholerae, each partner attL and attR sites specific to the same integrase were inserted on separate chromosomes: attRHK/attLλ were inserted at the junction between the two replichores in the terminus region of chrI and attLHK/attRλ were placed flanking [parA2/B2-oriII-rctA/B] in the origin region of chrII (Figure 1A). The consecutive recombination reactions between attRHK/attLHK and attRλ/attLλ sites, upon expression of Int and Xis, led to the fusion of chrI with chrII (Figure 1B, 1C). To visualize chromosomal rearrangement events, we used a colorimetric screen based on recombination-dependent reconstitution and expression of the lacZ gene (Figure 1E). We obtained a stable MonoCHromosomal V. cholerae mutant strain (MCH1) with a single chromosome of the expected 4 Mbp size (Figure 1F) observable by pulsed field gel electrophoresis (PFGE). MCH1 cells attain a generation time of 29 minutes when grown in fast-growing conditions (Table S1). Under the microscope, MCH1 fixed cells are indistinguishable from N16961 wild-type (WT) (Figure 1G) and the counting of viable cells forming microcolonies confirmed that MCH1 incurs no increase in the rate of mortality compared to the WT (data not shown). We measured the DNA distribution in exponentially growing cultures by flow cytometry and compared these distributions with modeled distributions (Figure S1). Whereas WT has a replication pattern which can be successfully modeled by assuming that chrII initiates late and terminates at approximately the same time as chrI as previously described [16], our analysis of MCH1's replication pattern was consistent with a single chromosome replicated at a constant rate (Figure S1).

RctB initiator and ParA2/B2 partitioning factors are essential for chrII maintenance only
We have taken a radical genetic approach by rearranging the genome of V. cholerae to investigate the specific biological functions of RctB and ParA2/B2. Since chrII is indispensable, these factors, essential for chrII initiation and partition, are ultimately essential for cell viability [11], [20], [22]. However, an additional role in the maintenance of chrI could never be formally tested due to the essentiality of their functions for chrII perpetuation. Recombinational fusion of the two chromosomes in MCH1 resulted in the excision of an 8 kb circular molecule carrying [parA2/B2-oriII-rctA/B-aph] (Figure 1C). The excised molecule encoded a functional aph gene conferring kanamycin resistance to the parental strain of MCH1, MV127. This circular molecule was readily lost in absence of selection observable by the absence of kanamycin resistance in MCH1 cells (Figure 1D). Loss of the 8 kb molecule was further confirmed by PCR, showing an absence of amplification of parB2 and rctB loci from MCH1 genomic DNA, while these loci could normally be amplified from MV127 genomic DNA (data not shown). Loss of the 8 kb molecule was surprising since it harbored the oriII origin of replication and a centromere-like parS2-B site (within rctA) [57] along with associated replication (rctA/B) and partitioning (parA2/B2) factors that should allow it to replicate autonomously in the cell. We have no experimental evidence that could explain this loss, but it could be the result of partition-mediated incompatibility [58] between parS2 sites located on separate entities, the fused chromosome and the 8 kb circular molecule. Yet, by physically linking chrII to chrI in MCH1, we placed replication and partitioning of chrII under the control of chrI machinery rendering chrII factors for replication initiation (RctB) and partitioning (ParA2/B2) non-essential.
Most of the centromere-like parS2 sites are located near oriII, ensuring its partition, but a functional parS2 site, parS2-1, was found located near the chrI terminus [57]. Therefore, ParA2/B2 could have an important function for the segregation of the terminus region of chrI. Under the microscope, MCH1 cells are indistinguishable from WT (Figure 1G). Nucleoid staining with DAPI shows no evident segregation or division problems that would be easily detectable by the presence of anucleoid cells, filaments and chromosomes trapped in the septum of division (Figure 1G). Our approach allowed us to readily demonstrate that the essential functions of RctB and ParA2/B2 in V. cholerae are strictly limited to chrII maintenance.
The essential activity of Dam is restricted to replication initiation at oriII
All previous in vivo Dam studies were undertaken in E. coli, where Dam is not essential. Here we investigate the essential function of Dam directly in V. cholerae to eliminate confusion arising from extrapolated results from E. coli. MCH1 enabled us to test the essentiality of Dam in replication initiation, since it only carries a single origin of replication, oriI. We deleted dam in MCH1 and the WT. Deletion of dam was done in the presence of pGD93, a complementing temperature sensitive replicating plasmid expressing V. cholerae dam under the control of an arabinose-inducible (permissive conditions) and glucose-repressible (restrictive conditions) promoter [9]. In the presence of Dam, both WTΔdam-[pGD93] and MCH1Δdam-[pGD93] grew normally (Figure 2A). Under restrictive conditions when Dam was depleted, WTΔdam colonies were hardly visible (Figure 2B) confirming the essentiality of Dam in V. cholerae. MCH1Δdam, on the other hand, grew and formed colonies under restrictive conditions (Figure 2B), indicating that Dam is no longer essential. This result demonstrates that initiation of replication at oriI doesn't require Dam. To more precisely characterize the role of Dam, we created a second mutant of V. cholerae where we maintained two distinct chromosomes but placed replication of chrII under the control of an additional copy of oriI, since Dam is not essential to initiate replication at oriI. To substitute oriII with oriI, we used the dual site-specific recombination tool previously described (Text S1). We generated a mutant of V. cholerae carrying two Identical Chromosomal-like oriI Origins (ICO1). The rctB deletion did not affect the viability of ICO1 confirming that its essential function was only required for replication initiation of oriII. We further tested the essentiality of Dam in ICO1, as described previously, and found that ICO1Δdam cells were viable under restrictive conditions (Figure 2B) demonstrating that Dam is no longer essential when chrII replication is initiated at oriI. Therefore, we can assert that Dam is required for replication initiation of chrII from oriII only.

It was previously thought that the critical function of Dam in V. cholerae could be related to its atypical genome arrangement. However, Dam is also essential in Yersinia pseudotuberculosis and Aeromonas hydrophila [59], bacteria with single chromosomes and members of the gamma subdivision of proteobacteria with V. cholerae. Therefore, the essential function of Dam could possibly be unrelated to the management of a multipartite genome. It is known that DNA methylation exerts an effect on diverse bacteria via its role as a global regulator of gene expression. In E. coli, many genes involved in DNA mismatch repair, SOS response, motility, host-pathogen interactions or cell cycle regulation are mis-regulated in the absence of Dam [59]. Thus, the role of DNA methylation in diverse cellular processes via gene expression regulation could also explain Dam's essential function in V. cholerae. The viability of MCH1Δdam mutants allowed us to rule out the potential role of Dam as an essential global regulator of gene expression since they have nearly the same genetic background as the WT where dam deletion is lethal. We further demonstrated in MCH1 that Dam was not required for initiation of replication at oriI. We tested the essentiality of Dam in the mutant ICO1, which carries two chromosomes both initiated at oriI. Since ICO1Δdam is viable, this precisely defined oriII as the region where Dam executes its essential function. This result substantiates earlier in vitro work showing that RctB preferentially binds methylated oriII [9]. We propose that in absence of Dam, GATC sites in oriII do not become methylated, preventing the binding of RctB to oriII and therefore precluding chrII replication initiation and maintenance which is fatal to the cell.
Chromosome dimer formation increases exponentially with the size of the chromosome
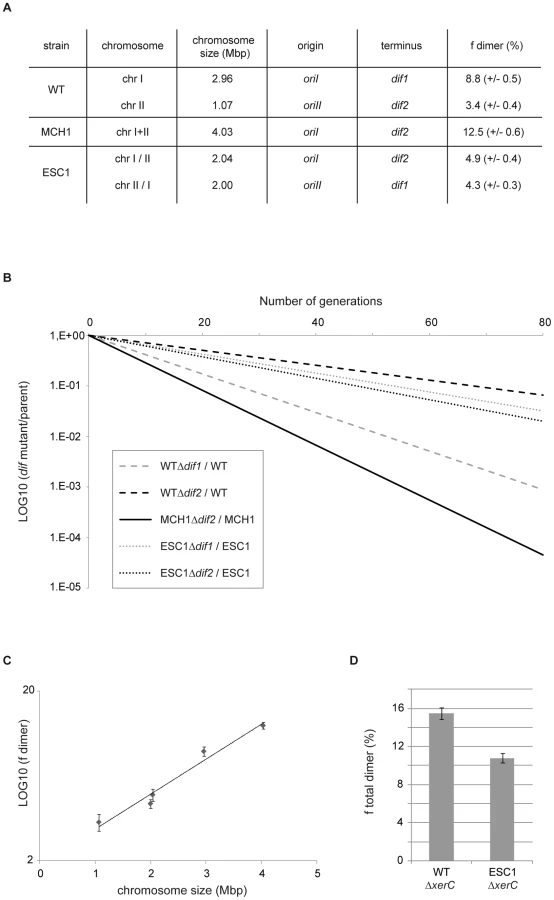
Formation of dimeric chromosomes is a particular problem associated with the circularity of bacterial chromosomes. We used V. cholerae as a bacterial model to determine how genome architecture affects the odds of topological difficulties during replication by assaying the effect of chromosome size on the rate of chromosome dimer formation. Very few cells carrying a dimer are expected to yield viable progeny in the absence of resolution. Inactivation of chromosome dimer resolution in E. coli results in ∼15% cell death per generation, which corresponds to the estimated rate of chromosome dimers formed at each cell generation [60]–[61]. We measured the fitness defect of a dif mutant by growth competition experiments, in which the growth of the mutant strain was directly compared to the growth of its parent (Figure 3B) to quantify the rate of dimers formed on a dif-carrying chromosome. In V. cholerae WT, 8.8% of dimers per cell per generation are formed on the 3 Mbp chrI (Δdif1) and 3.4% of dimers are formed on the 1 Mbp chrII (Δdif2) when grown in rich LB media (Figure 3A, 3B). In MCH1, 12.5% of dimers per cell per generation are formed on the 4 Mbp chromosome (Δdif2) under the same growth conditions (Figure 3A, 3B). These results suggested that dimer formation increases with replicon size. To strengthen our interpretation, we decided to construct an additional mutant of V. cholerae with two equally sized chromosomes of 2 Mbp and measure the rate of dimer formation on each chromosome. We transferred 1 Mbp from chrI to chrII by swapping the 1.05 Mbp DNA fragment evenly surrounding dif1 with the 0.12 Mbp DNA fragment evenly surrounding dif2, resulting in the exchange of dif1 and dif2 using the genetic tool described above (Text S1, Figure S2). We obtained a mutant of V. cholerae with Equally Sized Chromosomes (ESC1 with chrI/II and chrII/I) observable by PFGE (Figure S2D). A measure of the rate of chromosome dimers formed on the two 2 Mbp chromosomes was performed in ESC1. Our results show that 4.9% of dimers per cell per generation are formed on the 2 Mbp chrI/II (Δdif2) and 4.3% of dimers are formed on the 2 Mbp chrII/I (Δdif1) (Figure 3A, 3B). We plotted the rate of chromosome dimer formation as a function of chromosome size and observed a linear relationship between chromosome size and the logarithm of the frequency of dimer formation (r2 = 0.97) (Figure 3C, Methods). This result indicates that chromosome dimer formation increases exponentially with the size of the chromosome. In ESC1, the two equally sized chromosomes, chrI/II and chrII/I, have an asymmetric distribution of genes, specific machineries for their respective maintenance, distinct terminus regions and, very certainly, distinct chromosome structure, and yet the probability of dimer formation for each chromosome is essentially equivalent (Figure 3A). This implicated chromosome size as the primary influence on the rate of dimeric chromosome formation in an identical genetic background.
Homologous recombination involves a Holliday junction intermediate which is resolved by the RuvABC complex leading to either crossover or non-crossover potential products with only crossovers leading to the formation of chromosome dimers [62]. In E. coli, the RuvABC pathway is biased towards generating non-crossover products [63]–[64]. Since this bias can vary between species, it is not possible to infer the effects of genome architecture on the formation of chromosome dimers by direct comparison between bacteria with single and multiple chromosomes or between bacteria with multiple chromosomes of different sizes. V. cholerae allowed us to modify the size of the chromosomes by transferring DNA from one chromosome to the other, with minimal modifications of the genetic background.
Genetic information distribution between the two chromosomes impacts chromosome dimer formation
We tested the effect of DNA distribution between multiple chromosomes on the total rate of chromosome dimer formation. To do so, we measured the fitness defect of xerC mutants to obtain a quantification of the total rate of chromosome dimers formed in the cells (Figure 3D). As a consequence of more dimer formation in WT compared to ESC1, we observed that a xerC deletion had a greater effect on WT than on ESC1 (15.5% in WT , 10.8% in ESC1). The unequal (WT) or equal (ESC1) genetic distribution influences the chances for chromosome dimers to arise. Based on this result, it might be considered surprising that the extant WT genome configuration has been selected and all vibrios characterized to date have been shown to possess two unequally sized chromosomes [8]. This suggests that dimer formation has little impact on the selection of DNA distribution on multiple chromosomes.
One possible explanation for V. cholerae's unequally sized replicons and distinct replication initiation mechanisms may lie in adjusting the balance between genes found on separate chromosomes in response to drastic changes in growth conditions [10], [17], [65]. Gene dosage tends to shape chromosome organization of fast-growing bacteria, favoring placement of genes involved in translation and transcription near the origin of replication [65]. Differential gene dosage depends on replication rate, chromosome size and doubling time. This effect is particularly important for V. cholerae with its two chromosomes of uneven size and extremely short generation time. Indeed, when V. cholerae growth rate increases, origin-proximal loci of chrI are amplified by up to four copies per cell, yet origin-proximal loci of chrII never total more than two copies per cell [17]. Consistent with its larger size, gene dosage effects on chrI are greater than on chrII [10], [16]. Differently sized replicons may thus be selectively advantageous as a means to allow for a more nuanced gene dosage effect. This is certainly the case for the vibrios, where a higher abundance of growth-essential and growth-contributing genes are located near the origin of replication of chrI coupled with a dearth of such genes on chrII. This theory lends itself well to further investigation using our genetic engineering tools.
New insights into bacterial genome organization
We developed a site-specific recombination-based engineering tool, which provides us with a powerful means to massively reorganize in principle any prokaryotic genome provided that necessary host factors are present. This genetic tool consists in harnessing the λ and HK022 recombination systems to perform a large panel of genome reorganizations. By controlling the location and the orientation of each partner recombination site, we can obtain a large variety of genome rearrangements, such as chromosome fusion (e.g. MCH1), transfer and exchange of DNA fragments (e.g. ESC1), deletion, insertion, inversion or substitution of DNA (e.g. ICO1). Thanks to the construction and analysis of various synthetic mutants, we were able to tackle important biological issues on chromosome maintenance in V. cholerae. We showed that Dam, RctB and ParA2/ParB2 factors are essential for chrII maintenance. We further revealed that the odds of forming chromosome dimers exponentially increase with the size of a circular chromosome.
Our construction of mutants with massive genome rearrangements demonstrates the incredible plasticity of prokaryotic genomes. All of these genomic mutants conserved the rapid growth characteristic of vibrios, although with a slightly extended generation time (Table S1) that may be linked to their alternative genomic structure. This is currently under investigation. Recent advancements in the field of synthetic biology have demonstrated that the de novo creation of artificial genomes is now an attainable objective [66]. The recent assembly of the 580 kb genome of Mycoplasma genitalium starting from chemically synthesized oligonucleotides [67] and the successful demonstration that one can maintain and engineer a bacterial genome in a yeast and then transfer it to a bacterial recipient cell to generate an engineered bacterium [68] pave the way for many applications previously thought to be out of reach [69]. The current understanding of bacterial genomic organization and its connection with precise phenotypic properties is insufficient to propose an optimized genome arrangement to the field of synthetic biology. MCH1 is by far the closest isogenic mono-chromosomal model that can be used to make comparisons with the bi-chromosomal V. cholerae N16961 strain. A previous work has been done in Sinorhizobium meliloti, in which spontaneous fusions of the three natural replicons occurs at low frequency through recombination between repeated sequences in the genome [70]. In these experiments, the three different fused molecules all conserved their functional origins of replication, and the resulting fusion was reversible, rendering the results inconclusive in terms of the relationship between growth advantage and genome organization. On the contrary, the single chromosome of our engineered MCH1 is stable, contains only a single origin and terminus of replication and therefore provides us with a powerful new tool to investigate the selective advantage(s) of the characteristic multipartite genome organization of vibrios. New insights into bacterial genome organization and determination of how genomes are arranged can help us to design more optimized chromosomes, which will undoubtedly open novel developments in the field of synthetic biology.
Methods
Bacterial strains and growth conditions
Bacterial strains and plasmids used in this study are listed in Table S2. Cells were grown at 37°C in Luria broth. Antibiotics were used at the following concentrations: ampicillin, 75 µg/mL; chloramphenicol, 25 µg/mL for E. coli and 5 µg/mL for V. cholerae; kanamycin 25 µg/mL; spectinomycin 100 µg/mL; zeocin 25 µg/mL. Diaminopimelic acid was used at 0.3 mM, X-Gal (40 µg/mL); IPTG(1 mM); arabinose (0.2%) and glucose (1%).
General cloning procedures
DNA cassettes containing the att recombination sites were transferred from a plasmid vector to the chromosome by two homologous recombination steps. To provide homology for integration, two 500 bp regions spanning the point of insertion were amplified from N16961 chromosomal DNA by PCR. The amplified fragments were cloned into an R6K γ-ori-based suicide vector, pSW7848 that encodes the ccdB toxin gene under the control of an arabinose-inducible and glucose-repressible promoter, PBAD. The sequences containing the att recombination sites of interest were then cloned between the two chromosomal fragments. For cloning, Π3813 was used as a plasmid host [71]. For conjugal transfer of plasmids to V. cholerae strains, E. coli β3914 was used as the donor [71]. Selection of the plasmid-borne drug marker resulted in integration of the entire plasmid in the chromosome by a single crossover. Elimination of the plasmid backbone resulting from a second recombination step was selected for by arabinose induction of the ccdB toxin gene.
MCH1 construction
V. cholerae N16961 El Tor strain deleted for lacZ was used to create the mono-chromosomal MCH1 strain [4], [72]. Following the above-mentioned cloning and genome engineering procedures, four attR/L sites were inserted at precise chromosomal loci near dif1 on chrI and near oriCII on chrII using pSW7848-derivitave KO vectors pMP36 (attRλ), pMP42 (attLλ), pMP35 (attRHK), pMP49 (attLHK) (Table S2). First, [attRλ-3′lacZ-FRT-aph-FRT] was inserted downstream of rctB on chrII using pMP36 in N16961ΔlacZ generating strain MV122. The aph cassette was excised using pCP20 for expression of Flp recombinase that catalyses recombination between the two FRT sites [73]–[74]. After Flp-mediated recombination, a single FRT site remained near oriCII and the strain became sensitive to kanamycin, MV122Δaph. Second, [attLλ-5′lacZ-FRT-aph-FRT] was inserted upstream of dif1 on chrI using pMP42 in MV122Δaph generating strain MV124. The aph cassette was excised using pCP20, generating the mutant MV124Δaph. We checked MV124Δaph by PCR to make sure that undesirable recombination events between the remaining FRT site on chrII with FRT sites on chrI didn't occur. Third, dif1 was replaced by [attRHK-FRT-aph-FRT] using pMP35, yielding strain MV125. To insert attRHK close to dif1, it was necessary to delete dif1 to prevent site-specific integration of a dif1-carrying KO-vector mediated by the endogenous V. cholerae XerC/D recombinases [19]. The aph cassette was not excised, this antibiotic resistance cassette serving as a reporter to follow the subsequent loss of the excised 8 kb circular molecule resulting from the fusion of chr1 with chr2. Fourth, [attLHK] with no antibiotic resistance cassette was inserted downstream of parB2 using pMP49 generating mutant MV127 (Figure 1A).
A temperature-sensitive replicating vector pMP6 expressing [intλ-xisλ , intHK-xb isHK] was conjugated into MV127. Donor cells β2163 [pMP6] and recipient cells (MV127) were conjugated for one hour at 30°C and plated on LB-agar at 30°C supplemented with ampicillin, X-Gal and IPTG to select for pMP6 and monitor recombination events between attLλ and attRλ. Reconstitution and expression of the β-galactosidase encoding gene led to appearance of blue cells when grown in presence of X-Gal and IPTG. After 36 hours of growth at 30°C, blue quarters appeared within single white conjugant colonies (Figure 1E). From blue/white colonies of mixed population, cells were grown at 30°C in LB in presence of ampicillin to enrich for chromosome rearrangements. Cells were plated on LB supplemented with X-Gal, IPTG to monitor attLλ and attRλ recombination events and incubated at 42°C to cure pMP6. All selected colonies were completely blue. Ten clones were isolated and tested by PCR using primers flanking both recombined attBλ and attBHK sites to verify that recombination occurred between all four recombination sites. All tested blue clones also had recombined attRHK×attLHK. Fusion of the two chromosomes resulted in the excision of an 8 kb circular molecule. In absence of antibiotic pressure that selected for this 8 kb circular molecule (aph gene formerly located in the terminus region of chrI), the molecule was rapidly lost. All remaining and undesired FRT and attP sites were excised within the 8 kb molecule and subsequently lost. The resulting mutant carries a single circular chromosome, free of antibiotic resistance cassettes and containing only two short 50 bp attB sites that delimit chrI from chrII. Genomic stability of the mutant was established over 1000 generations carried out during a long-term evolution experiment.
Pulsed field gel electrophoresis
The preparation of genomic DNA embedded in agarose gels and the protocol for PFGE was performed as previously described [8], [75].
Dam depletion
WT, MCH1 and ICO1 strains were deleted for dam using pGD121 knock-out vector in the presence of pGD93 (Dam complementing vector) and then depleted for Dam as previously described [9].
Growth competition assay
The proportion of cells that a mutant strain deficient in dimer resolution fails to produce at each doubling time of its parent can be measured by growth competition experiments. Growth competitions of V. cholerae strains are described in [19]. V. cholerae cells were grown at 37°C with a 10−3 dilution in LB media every 8–12 h. Colony-forming units (CFUs) of mutant and parental cells in the cultures were determined by plating on appropriate antibiotic plates. These numbers were used to calculate the number of generations of the parent cells between each time points and the CFUs ratio of mutant versus parent cells at each time point. This ratio varies exponentially with the number of generations. The coefficient of this exponential is a good estimation of the rate of dimer formation [19]. Following this method, we estimated the rate of dimer formation for each mutant in three independent experiments. In Figure 3C, the relationship between the rate of dimer formation and the logarithm of chromosome size has a very high R2 (>0.9) with no significant departure from linearity (P value = 0.1827), which indicates a strong linear relationship between the two variables. The slope is significantly different from zero (P value<0.0001) and the confidence interval for the slope is 95%.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. SuwantoAKaplanS 1989 Physical and genetic mapping of the Rhodobacter sphaeroides 2.4.1 genome: presence of two unique circular chromosomes. J Bacteriol 171 5850 5859
2. CasjensS 1998 The diverse and dynamic structure of bacterial genomes. Annu Rev Genet 32 339 377
3. EganESFogelMAWaldorMK 2005 Divided genomes: negotiating the cell cycle in prokaryotes with multiple chromosomes. Mol Microbiol 56 1129 1138
4. HeidelbergJFEisenJANelsonWCClaytonRAGwinnML 2000 DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406 477 483
5. ReenFJAlmagro-MorenoSUsseryDBoydEF 2006 The genomic code: inferring Vibrionaceae niche specialization. Nat Rev Microbiol 4 697 704
6. DryseliusRKurokawaKIidaT 2007 Vibrionaceae, a versatile bacterial family with evolutionarily conserved variability. Res Microbiol 158 479 486
7. Le RouxFZouineMChakrounNBinesseJSaulnierD 2009 Genome sequence of Vibrio splendidus: an abundant planctonic marine species with a large genotypic diversity. Environ Microbiol 11 1959 1970
8. OkadaKIidaTKita-TsukamotoKHondaT 2005 Vibrios commonly possess two chromosomes. J Bacteriol 187 752 757
9. DemarreGChattorajDK 2010 DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. PLoS Genet 6 e1000939 doi:10.1371/journal.pgen.1000939
10. DryseliusRIzutsuKHondaTIidaT 2008 Differential replication dynamics for large and small Vibrio chromosomes affect gene dosage, expression and location. BMC Genomics 9 559
11. DuigouSKnudsenKGSkovgaardOEganESLobner-OlesenA 2006 Independent control of replication initiation of the two Vibrio cholerae chromosomes by DnaA and RctB. J Bacteriol 188 6419 6424
12. EganESWaldorMK 2003 Distinct replication requirements for the two Vibrio cholerae chromosomes. Cell 114 521 530
13. FogelMAWaldorMK 2005 Distinct segregation dynamics of the two Vibrio cholerae chromosomes. Mol Microbiol 55 125 136
14. FogelMAWaldorMK 2006 A dynamic, mitotic-like mechanism for bacterial chromosome segregation. Genes Dev 20 3269 3282
15. PalDVenkova-CanovaTSrivastavaPChattorajDK 2005 Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J Bacteriol 187 7167 7175
16. RasmussenTJensenRBSkovgaardO 2007 The two chromosomes of Vibrio cholerae are initiated at different time points in the cell cycle. EMBO J 26 3124 3131
17. SrivastavaPChattorajDK 2007 Selective chromosome amplification in Vibrio cholerae. Mol Microbiol 66 1016 1028
18. SrivastavaPFeketeRAChattorajDK 2006 Segregation of the replication terminus of the two Vibrio cholerae chromosomes. J Bacteriol 188 1060 1070
19. ValMEKennedySPEl KarouiMBonneLChevalierF 2008 FtsK-dependent dimer resolution on multiple chromosomes in the pathogen Vibrio cholerae. PLoS Genet 4 e1000201 doi:10.1371/journal.pgen.1000201
20. YamaichiYFogelMAWaldorMK 2007 par genes and the pathology of chromosome loss in Vibrio cholerae. Proc Natl Acad Sci U S A 104 630 635
21. SchmidMFernandez-BadilloAFeichtingerWSteinleinCRomanJI 1988 On the highest chromosome number in mammals. Cytogenet Cell Genet 49 305 308
22. DuigouSYamaichiYWaldorMK 2008 ATP negatively regulates the initiator protein of Vibrio cholerae chromosome II replication. Proc Natl Acad Sci U S A 105 10577 10582
23. YamaichiYDuigouSShakhnovichEAWaldorMK 2009 Targeting the replication initiator of the second Vibrio chromosome: towards generation of vibrionaceae-specific antimicrobial agents. PLoS Pathog 5 e1000663 doi:10.1371/journal.ppat.1000663
24. EganESLobner-OlesenAWaldorMK 2004 Synchronous replication initiation of the two Vibrio cholerae chromosomes. Curr Biol 14 R501 502
25. Lobner-OlesenASkovgaardOMarinusMG 2005 Dam methylation: coordinating cellular processes. Curr Opin Microbiol 8 154 160
26. JulioSMHeithoffDMProvenzanoDKloseKESinsheimerRL 2001 DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect Immun 69 7610 7615
27. KahngLSShapiroL 2001 The CcrM DNA methyltransferase of Agrobacterium tumefaciens is essential, and its activity is cell cycle regulated. J Bacteriol 183 3065 3075
28. RobertsonGTReisenauerAWrightRJensenRBJensenA 2000 The Brucella abortus CcrM DNA methyltransferase is essential for viability, and its overexpression attenuates intracellular replication in murine macrophages. J Bacteriol 182 3482 3489
29. WrightRStephensCShapiroL 1997 The CcrM DNA methyltransferase is widespread in the alpha subdivision of proteobacteria, and its essential functions are conserved in Rhizobium meliloti and Caulobacter crescentus. J Bacteriol 179 5869 5877
30. KochBMaXLobner-OlesenA 2010 Replication of Vibrio cholerae chromosome I in Escherichia coli: dependence on dam methylation. J Bacteriol 192 3903 3914
31. DraperGCGoberJW 2002 Bacterial chromosome segregation. Annu Rev Microbiol 56 567 597
32. LeonardTAMoller-JensenJLoweJ 2005 Towards understanding the molecular basis of bacterial DNA segregation. Philos Trans R Soc Lond B Biol Sci 360 523 535
33. LesterlinCBarreFXCornetF 2004 Genetic recombination and the cell cycle: what we have learned from chromosome dimers. Mol Microbiol 54 1151 1160
34. CampoNDiasMJDaveran-MingotMLRitzenthalerPLe BourgeoisP 2004 Chromosomal constraints in Gram-positive bacteria revealed by artificial inversions. Mol Microbiol 51 511 522
35. EsnaultEValensMEspeliOBoccardF 2007 Chromosome structuring limits genome plasticity in Escherichia coli. PLoS Genet 3 e226 doi:10.1371/journal.pgen.0030226
36. GuijoMIPatteJdel Mar CamposMLouarnJMRebolloJE 2001 Localized remodeling of the Escherichia coli chromosome: the patchwork of segments refractory and tolerant to inversion near the replication terminus. Genetics 157 1413 1423
37. HillCWGrayJA 1988 Effects of chromosomal inversion on cell fitness in Escherichia coli K-12. Genetics 119 771 778
38. LesterlinCMercierRBoccardFBarreFXCornetF 2005 Roles for replichores and macrodomains in segregation of the Escherichia coli chromosome. EMBO Rep 6 557 562
39. LiuGRLiuWQJohnstonRNSandersonKELiSX 2006 Genome plasticity and ori-ter rebalancing in Salmonella typhi. Mol Biol Evol 23 365 371
40. LouarnJMBoucheJPLegendreFLouarnJPatteJ 1985 Characterization and properties of very large inversions of the E. coli chromosome along the origin-to-terminus axis. Mol Gen Genet 201 467 476
41. MieselLSegallARothJR 1994 Construction of chromosomal rearrangements in Salmonella by transduction: inversions of non-permissive segments are not lethal. Genetics 137 919 932
42. RebolloJEFrancoisVLouarnJM 1988 Detection and possible role of two large nondivisible zones on the Escherichia coli chromosome. Proc Natl Acad Sci U S A 85 9391 9395
43. RochaEPDanchinA 2003 Gene essentiality determines chromosome organisation in bacteria. Nucleic Acids Res 31 6570 6577
44. SegallAMahanMJRothJR 1988 Rearrangement of the bacterial chromosome: forbidden inversions. Science 241 1314 1318
45. CuiTMoro-okaNOhsumiKKodamaKOhshimaT 2007 Escherichia coli with a linear genome. EMBO Rep 8 181 187
46. ItayaMTanakaT 1997 Experimental surgery to create subgenomes of Bacillus subtilis 168. Proc Natl Acad Sci U S A 94 5378 5382
47. KolisnychenkoVPlunkettG3rdHerringCDFeherTPosfaiJ 2002 Engineering a reduced Escherichia coli genome. Genome Res 12 640 647
48. VolffJNViellPAltenbuchnerJ 1997 Artificial circularization of the chromosome with concomitant deletion of its terminal inverted repeats enhances genetic instability and genome rearrangement in Streptomyces lividans. Mol Gen Genet 253 753 760
49. HendricksonHLawrenceJG 2006 Selection for chromosome architecture in bacteria. J Mol Evol 62 615 629
50. LouarnJMKuempelPCornetF 2005 The terminus region of the E. coli chromosome, or, all's well that ends well. HigginsNP The Bacterial Chromosome Washington, D.C. ASM press 251 273
51. RochaEP 2008 The organization of the bacterial genome. Annu Rev Genet 42 211 233
52. VesthTWassenaarTMHallinPFSnipenLLagesenK 2010 On the origins of a Vibrio species. Microb Ecol 59 1 13
53. WeisbergRAGottesmannMEHendrixRWLittleJW 1999 Family values in the age of genomics: comparative analyses of temperate bacteriophage HK022. Annu Rev Genet 33 565 602
54. SauerB 1996 Multiplex Cre/lox recombination permits selective site-specific DNA targeting to both a natural and an engineered site in the yeast genome. Nucleic Acids Res 24 4608 4613
55. TuranSKuehleJSchambachABaumCBodeJ 2010 Multiplexing RMCE: versatile extensions of the Flp-recombinase-mediated cassette-exchange technology. J Mol Biol 402 52 69
56. GottesmanMEWeisbergRA 1971 Prophage Insertion and Excision. HersheyAD Lambda: Cold Spring Harbor Laboratory 113 138
57. YamaichiYFogelMAMcLeodSMHuiMPWaldorMK 2007 Distinct centromere-like parS sites on the two chromosomes of Vibrio spp. J Bacteriol 189 5314 5324
58. AustinSNordstromK 1990 Partition-mediated incompatibility of bacterial plasmids. Cell 60 351 354
59. MarinusMGCasadesusJ 2009 Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol Rev 33 488 503
60. PeralsKCornetFMerletYDelonILouarnJM 2000 Functional polarization of the Escherichia coli chromosome terminus: the dif site acts in chromosome dimer resolution only when located between long stretches of opposite polarity. Mol Microbiol 36 33 43
61. SteinerWWKuempelPL 1998 Sister chromatid exchange frequencies in Escherichia coli analyzed by recombination at the dif resolvase site. J Bacteriol 180 6269 6275
62. CoxMMGoodmanMFKreuzerKNSherrattDJSandlerSJ 2000 The importance of repairing stalled replication forks. Nature 404 37 41
63. CromieGALeachDR 2000 Control of crossing over. Mol Cell 6 815 826
64. MichelBRecchiaGDPenel-ColinMEhrlichSDSherrattDJ 2000 Resolution of holliday junctions by RuvABC prevents dimer formation in rep mutants and UV-irradiated cells. Mol Microbiol 37 180 191
65. CouturierERochaEP 2006 Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Mol Microbiol 59 1506 1518
66. CambrayGMutalikVKArkinAP 2011 Toward rational design of bacterial genomes. Curr Opin Microbiol
67. GibsonDGBendersGAAndrews-PfannkochCDenisovaEABaden-TillsonH 2008 Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319 1215 1220
68. LartigueCVasheeSAlgireMAChuangRYBendersGA 2009 Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325 1693 1696
69. CarrPAChurchGM 2009 Genome engineering. Nat Biotechnol 27 1151 1162
70. GuoXFloresMMavinguiPFuentesSIHernandezG 2003 Natural genomic design in Sinorhizobium meliloti: novel genomic architectures. Genome Res 13 1810 1817
71. Le RouxFBinesseJSaulnierDMazelD 2007 Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl Environ Microbiol 73 777 784
72. GuerinECambrayGSanchez-AlberolaNCampoySErillI 2009 The SOS response controls integron recombination. Science 324 1034
73. CherepanovPPWackernagelW 1995 Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158 9 14
74. DatsenkoKAWannerBL 2000 One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 6640 6645
75. IidaTSuthienkulOParkKSTangGQYamamotoRK 1997 Evidence for genetic linkage between the ure and trh genes in Vibrio parahaemolyticus. J Med Microbiol 46 639 645
76. CooperSHelmstetterCE 1968 Chromosome replication and the division cycle of Escherichia coli B/r. J Mol Biol 31 519 540
77. MichelsenOTeixeira de MattosMJJensenPRHansenFG 2003 Precise determinations of C and D periods by flow cytometry in Escherichia coli K-12 and B/r. Microbiology 149 1001 1010
78. SkarstadKSteenHBBoyeE 1985 Escherichia coli DNA distributions measured by flow cytometry and compared with theoretical computer simulations. J Bacteriol 163 661 668
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Poly(ADP-Ribose) Polymerase 1 (PARP-1) Regulates Ribosomal Biogenesis in Nucleoli
- Microenvironmental Regulation by Fibrillin-1
- Parallel Mapping and Simultaneous Sequencing Reveals Deletions in and Associated with Discrete Inherited Disorders in a Domestic Dog Breed
- Two-Component Elements Mediate Interactions between Cytokinin and Salicylic Acid in Plant Immunity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy