
Pch2 Acts through Xrs2 and Tel1/ATM to Modulate Interhomolog Bias and Checkpoint Function during Meiosis
Proper segregation of chromosomes during meiosis requires the formation and repair of double-strand breaks (DSBs) to form crossovers. Repair is biased toward using the homolog as a substrate rather than the sister chromatid. Pch2 is a conserved member of the AAA+-ATPase family of proteins and is implicated in a wide range of meiosis-specific processes including the recombination checkpoint, maturation of the chromosome axis, crossover control, and synapsis. We demonstrate a role for Pch2 in promoting and regulating interhomolog bias and the meiotic recombination checkpoint in response to unprocessed DSBs through the activation of axial proteins Hop1 and Mek1 in budding yeast. We show that Pch2 physically interacts with the putative BRCT repeats in the N-terminal region of Xrs2, a member of the MRX complex that acts at sites of unprocessed DSBs. Pch2, Xrs2, and the ATM ortholog Tel1 function in the same pathway leading to the phosphorylation of Hop1, independent of Rad17 and the ATR ortholog Mec1, which respond to the presence of single-stranded DNA. An N-terminal deletion of Xrs2 recapitulates the pch2Δ phenotypes for signaling unresected breaks. We propose that interaction with Xrs2 may enable Pch2 to remodel chromosome structure adjacent to the site of a DSB and thereby promote accessibility of Hop1 to the Tel1 kinase. In addition, Xrs2, like Pch2, is required for checkpoint-mediated delay conferred by the failure to synapse chromosomes.
Published in the journal:
. PLoS Genet 7(11): e32767. doi:10.1371/journal.pgen.1002351
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002351
Summary
Proper segregation of chromosomes during meiosis requires the formation and repair of double-strand breaks (DSBs) to form crossovers. Repair is biased toward using the homolog as a substrate rather than the sister chromatid. Pch2 is a conserved member of the AAA+-ATPase family of proteins and is implicated in a wide range of meiosis-specific processes including the recombination checkpoint, maturation of the chromosome axis, crossover control, and synapsis. We demonstrate a role for Pch2 in promoting and regulating interhomolog bias and the meiotic recombination checkpoint in response to unprocessed DSBs through the activation of axial proteins Hop1 and Mek1 in budding yeast. We show that Pch2 physically interacts with the putative BRCT repeats in the N-terminal region of Xrs2, a member of the MRX complex that acts at sites of unprocessed DSBs. Pch2, Xrs2, and the ATM ortholog Tel1 function in the same pathway leading to the phosphorylation of Hop1, independent of Rad17 and the ATR ortholog Mec1, which respond to the presence of single-stranded DNA. An N-terminal deletion of Xrs2 recapitulates the pch2Δ phenotypes for signaling unresected breaks. We propose that interaction with Xrs2 may enable Pch2 to remodel chromosome structure adjacent to the site of a DSB and thereby promote accessibility of Hop1 to the Tel1 kinase. In addition, Xrs2, like Pch2, is required for checkpoint-mediated delay conferred by the failure to synapse chromosomes.
Introduction
Meiosis is a specialized cell division program to produce haploid gametes. To achieve faithful chromosome segregation during meiosis I (MI), cells utilize meiotic recombination to establish physical connections through the formation of chiasmata or crossing-over at the DNA level between homologous chromosomes [1].
In budding yeast, meiotic recombination is initiated by programmed double-strand breaks (DSBs) catalyzed by a topoisomerase II-like enzyme Spo11 [2]. The 5′ ends of DSBs are resected to produce 3′ single-stranded DNA, at which Dmc1 and Rad51 load to mediate strand exchange with a homologous DNA sequence [3], [4]. Unlike in vegetative cells where sister chromatids are preferred templates for DSB repair, most meiotic programmed DSBs are repaired using homologous non-sister chromatids [5], [6], [7]. A subset of DSBs is repaired to form crossovers (CO) through a double Holliday junction (dHJ) pathway [8], [9], [10]. CO formation and distribution is highly regulated during meiosis; each homolog must receive at least one CO to sustain reductional segregation in meiosis I [11].
Interhomolog bias is established and maintained by regulatory proteins associated with chromosome axis structures, including Hop1 and Mek1. In response to DSBs, the meiotic chromosome axis protein Hop1 is phosphorylated by Tel1/Mec1 (ATM/ATR homologs) [12]. Phosphorylated Hop1 promotes dimerization and auto-activation of Mek1 kinase [13], [14], [15], [16]. A Hop1 mutant that is refractory to Tel1/Mec1 phosphorylation fails to activate Hop1-dependent Mek1 phosphorylation and results in the loss of interhomolog bias [12]. Mek1 kinase plays dual roles by promoting interhomolog bias and checkpoint signaling in the presence of recombination intermediates [13].
The presence of unrepaired DSBs is monitored by DNA damage checkpoint proteins Mec1, Rad17, Rad24, Tel1, and the MRX (Mre11-Rad50-Xrs2) complex [17]. Mutants defective in the repair of meiosis-induced DSBs activate one or more pathways involving these proteins [17]. Different lesions appear to activate different checkpoint pathways. For example, unresected DSBs appear to activate a checkpoint requiring Tel1 (ATM homolog) while unrepaired resected breaks activate a Mec1 (ATR) pathway [18], [19].
Pch2 is a member of the AAA+-ATPase family of proteins and is implicated in a number of meiosis-specific processes in budding yeast Saccharomyces cerevisiae, including meiotic recombination, chromosome axis formation, checkpoint signaling, crossover control and interhomolog bias [20], [21], [22], [23], [24]. Pch2 participates in one branch of a bifurcated pathway that defines the recombination checkpoint: One branch is regulated by Rad17 and Mec1, likely in response to ssDNA [19]. A second branch is regulated by Pch2, however, the activating lesion has not been defined [20]. In mouse the Pch2 homolog TRIP13 plays roles in axis morphogenesis and early steps of recombination [25], [26], [27]. In Caenorhabditis elegans and Drosophila melanogaster, PCH-2 plays a role in a checkpoint that monitors synapsis and/or axis formation [28], [29], [30]. Whether these seemingly disparate roles of Pch2 share mechanisms in common is an open question.
Pch2 was originally identified by mutation as a suppressor of the arrest/delay phenotype conferred by the deletion of ZIP1 [31], which encodes the transverse element of the synaptonemal complex (SC) [32], [33]. Suppression of the zip1Δ delay phenotype by pch2Δ is enigmatic since the zip1Δ delay is also suppressed by deletion of RAD17 [20]. Multiple roles for Zip1 during meiosis are indicated by the pleiotropic phenotypes associated with the deletion mutation [1], [34], therefore it is possible that Pch2 might signal more than one lesion during a challenged meiosis.
Our data support these key findings: 1. Pch2 and Rad17 contribute to suppression of intersister recombination through independent pathways with partially overlapping functions. 2. Pch2 and Tel1 function in the same epistasis pathway to regulate meiotic recombination checkpoint signaling, independent of Rad17 and Mec1. 3. Pch2 functions to signal the presence of unresected breaks leading to the phosphorylation of Hop1. 4. Pch2 physically interacts with the N-terminal region of Xrs2 containing putative BRCT repeats. Deletion of this non-essential region of Xrs2 leads to a defect in Pch2-dependent checkpoint signaling. 5. Xrs2 and Pch2 play a role in the synapsis checkpoint while Tel1 does not. These findings link multiple roles of Pch2 in budding yeast to the ATM homolog Tel1 and/or the MRX component Xrs2. We propose that phosphorylation of the meiotic chromosome axis protein Hop1 depends on two partially redundant pathways: one pathway involving Tel1, Pch2 and Xrs2 that responds to the presence of unprocessed DSBs and another pathway involving Mec1 and Rad17 that responds to the presence of resected DSB intermediates of homologous recombination.
Results
Pch2 and Rad17 prevent intersister repair during meiotic recombination
Deletion of both PCH2 and RAD17 causes a synergistic reduction in spore viability and accelerated meiotic progression compared to either single mutant or wild type. Spore inviability is suppressed in a spo13 mutant background suggesting that programmed DSBs are repaired, most likely using the sister chromatid as a template [20]. These combined phenotypes led us to suggest that Pch2 and Rad17 function in redundant pathways to suppress the use of sister chromatids to repair meiotic programmed DSBs. To test this, we monitored the presence of intersister (IS) and interhomolog (IH) joint molecules that form as intermediates of meiotic DSB repair at the HIS4LEU2 hot spot in pch2Δ, rad17Δ and pch2Δ rad17Δ at various time points during meiotic progression in a synchronized cell culture using two-dimensional gel electrophoresis (Figure 1A, 1B). To detect maximal levels of these intermediates we used an ndt80Δ mutant background to block the resolution of dHJs to crossover products [8], [35]. While the ndt80Δ pch2Δ mutant gave ∼10% higher levels of IH-dHJ compared to the ndt80Δ strain, the levels in ndt80Δ rad17Δ and ndt80Δ pch2Δ rad17Δ were reduced by ∼60% and 67%, respectively. By contrast, while the ndt80Δ pch2Δ mutant gave ∼9% lower levels of IS-dHJ compared to the ndt80Δ strain, this species was increased in ndt80Δ rad17Δ and ndt80Δ pch2Δ rad17Δ mutants by ∼13% and 52%, respectively (based on averages of measurements from two independent time course experiments). Together these results suggest that Pch2 and Rad17 have independent and partially overlapping functions in promoting interhomolog bias.

In an independent test, we measured DSB levels in the dmc1Δ mutant background where DSBs form and are resected but their repair is blocked [3]. If the process of upholding interhomolog bias is compromised then breaks can be repaired using sister chromatids [7]. We found that steady-state DSB levels were decreased over two-fold in dmc1Δ pch2Δ rad17Δ compared to dmc1Δ pch2Δ and dmc1Δ rad17Δ (5 hours after transfer to SPM; Figure 1C and 1D). The observed decrease in DSBs in dmc1Δ pch2Δ rad17Δ (compare t = 3 hours and t = 5 hours), but not in sae2Δ pch2Δ rad17Δ where DSBs are not processed [20], suggests that repair of DSBs occurs using a sister chromatid. These results suggest that Pch2 and Rad17 are required to uphold the barrier to sister chromatid recombination.
Pch2 and Rad17 promote Hop1 phosphorylation and Mek1 activation
From the findings above, we reasoned that Pch2 and Rad17 might independently promote phosphorylation of Hop1 in response to DSBs. In wild-type cells, Hop1 was transiently phosphorylated starting at about t = 3.5, as revealed by slow-migrating bands in a western blot using an α-Hop1 antibody (Figure 2A). Slow-moving Hop1 isoforms were abundant in both pch2Δ and rad17Δ single mutants but dramatically reduced in the pch2Δ rad17Δ double mutant. These results suggest that Pch2 and Rad17 function in different pathways leading to Hop1 phosphorylation.
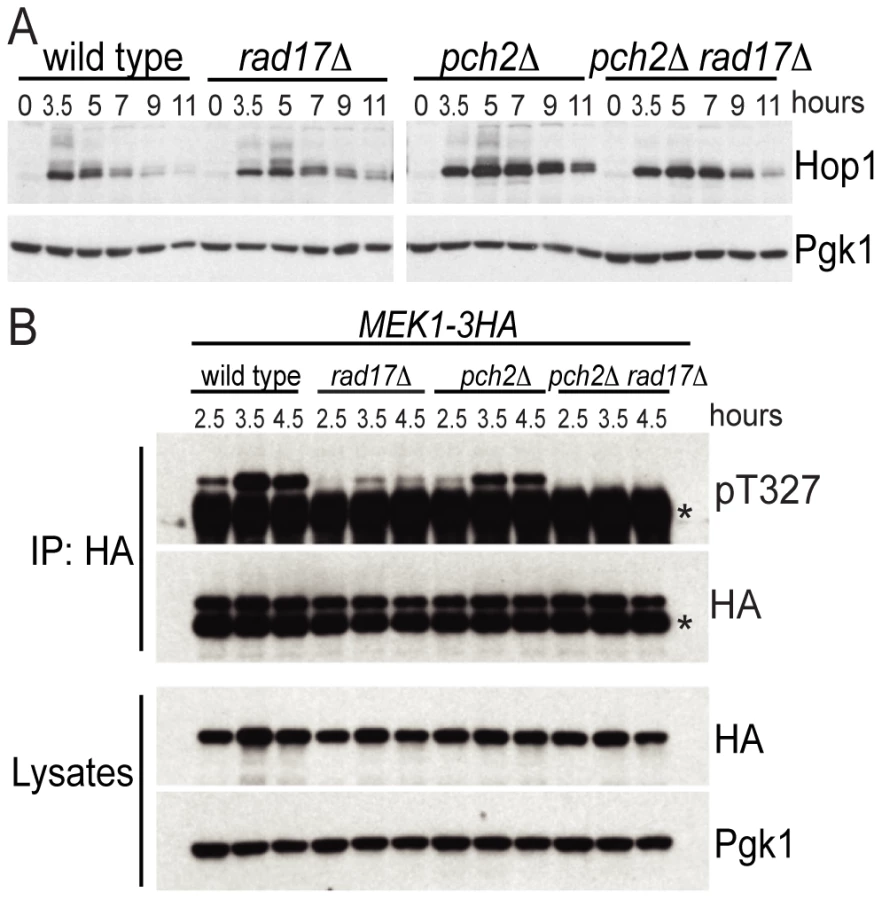
We next examined the phosphorylation status of Mek1 using an α-Akt-substrate antibody to the T327 residue in the activation loop [36]. While phosphorylation of the T327 residue was present in the pch2Δ and rad17Δ single mutants, it was completely abolished in the pch2Δ rad17Δ double mutant, similar to the results seen above for Hop1 (Figure 2B). The reduction in Mek1–3HA phosphorylation in rad17Δ was more dramatic than the reduction of Hop1 phosphorylation in the same background. One interpretation of this result is that Rad17 not only regulates signaling upstream of Hop1 but also impacts the Hop1-dependent autophosphorylation of Mek1. Consistent with this notion, rad17Δ shows aberrant SC formation [37] perhaps indicating aberrantly formed axial elements. Together, these results demonstrate that two independent pathways defined by Pch2 and Rad17, respectively, regulate the activation status of the meiotic chromosome axis proteins Hop1 and Mek1. The failure to phosphorylate Hop1 and Mek1 in the absence of both Pch2 and Rad17 may account for the loss of interhomolog bias in the pch2Δ rad17Δ double mutant background.
Pch2 acts together with Tel1 to promote spore viability and normal MI division timing
A hallmark of mutants defective in interhomolog bias is the formation of largely inviable spore products due to reduced levels of interhomolog crossovers [1]. Consistent with this pattern, the pch2Δ rad17Δ double mutant gives <0.1% viable spores, while each single mutant gives higher levels (37.1% for rad17Δ and 92.2% for pch2Δ; Table 1) [20]. Like Rad17 and Pch2, the ATR/ATM homologs Mec1 and Tel1 have also been shown to play partially redundant roles in meiotic interhomolog recombination by phosphorylating Hop1 [12]. Since RAD17 and MEC1 are in the same epistasis group that mediates checkpoint signaling in the presence of ssDNA [19], [37], one possibility is that Pch2 functions with Tel1 in a separate pathway, perhaps in response to unresected DSBs [18]. To test this, we examined spore viability in mutants containing pair-wise combinations of pch2Δ, rad17Δ, tel1Δ and mec1Δ mutations. In the cases where we predicted the two genes would act in the same pathway (e.g. pch2Δ tel1Δ and rad17Δ mec1Δ), there was no decrease in spore viability compared to the single mutants (Table 1). By contrast, in the cases where we predicted the two genes would function in different pathways we observed a synergistic decrease in spore viability in the double mutants (2.9% for pch2Δ mec1Δ and <0.1% for rad17Δ tel1Δ).

In a similar line of reasoning, checkpoint activation leads to a delay in MI division and can be triggered by loss of either Pch2 or Rad17, but not both. We showed previously that MI division kinetics in the pch2Δ rad17Δ double mutant is faster than in wild type, yet is delayed in the two single mutant strains [20]. From a first approximation, the epistasis pattern described above for spore inviability holds true: i) the MI delay conferred by pch2Δ was suppressed by mec1Δ to give divisions even faster than WT; and ii) the delay phenotype conferred by rad17Δ was suppressed by tel1Δ (Figure 3C and 3D). Notably, the MI delay in the pch2Δ tel1Δ double mutant was more severe than either single mutant, suggesting that each protein may function in additional pathways that do not involve the other.

To further confirm the epistasis relationship observed above, we examined Hop1 phosphorylation in the pch2Δ tel1Δ, rad17Δ mec1Δ, pch2Δ mec1Δ and rad17Δ tel1Δ double mutant combinations. As expected, we observed abundant Hop1 phosphorylation in pch2Δ tel1Δ and rad17Δ mec1Δ, while only a low level of Hop1 phosphorylation was seen in pch2Δ mec1Δ and rad17Δ tel1Δ which showed very low spore viability and fast meiotic progression (Figure 3E, 3F and Figure 4A). Together, these results suggest Pch2 acts together with Tel1 to promote an essential meiotic process, perhaps by ensuring interhomolog bias through Hop1 phosphorylation.

Pch2 is involved in signaling unprocessed DSBs
Tel1 is required to signal the presence of unprocessed DSBs during meiosis [17], [18]. Specifically, deletion of TEL1 eliminates the signaling of unresected DSBs to Hop1 [12]. To test if signaling of unprocessed DSBs also requires Pch2, we examined Hop1 phosphorylation in both pch2Δ and tel1Δ mutants in a sae2Δ mutant background where breaks are unprocessed to give blunt ends. Hop1 was phosphorylated in a sae2Δ mutant but not in sae2Δ pch2Δ or sae2Δ tel1Δ (Figure 4B), as expected if Tel1 and Pch2 are specifically required for unprocessed DSBs signaling. By contrast, Rad17 was not required for Hop1 phosphorylation in the sae2Δ background (Figure 4C), which is also expected since Rad17 is involved in signaling resected DSBs. As a control, we measured Hop1 phosphorylation in the dmc1Δ mutant background where DSBs are resected to give ssDNA. Hop1 phosphorylation was not affected in dmc1Δ pch2Δ and dmc1Δ tel1Δ (Figure 4D).
We noticed that Hop1 protein levels were elevated in the dmc1Δ pch2Δ double mutant (Figure 4D) compared to the dmc1Δ single mutant. On the other hand, sae2Δ pch2Δ showed no increase in Hop1 levels compared to the sae2Δ single mutant (Figure 4B). We reasoned that this effect of pch2Δ does not relate to the role of Pch2 in promoting Hop1 phosphorylation per se since pch2Δ only affected Hop1 phosphorylation in the sae2Δ background (where Hop1 levels were not altered) but not in the dmc1Δ background (where Hop1 levels were increased). The tel1Δ strain did not show such an effect either, again suggesting this aspect of Pch2 function is independent of its role in Tel1 signaling to Hop1. We speculate that the increase in Hop1 levels (or reduced Hop1 protein turnover) shown here by western blotting likely reflects altered Hop1 abundance/distribution shown previously by immunostaining [21] and is related to Pch2′s role in axis organization and CO control. Interestingly, this effect is manifested at a “post resection” stage of DSB repair since increased Hop1 levels were observed in dmc1Δ but not in sae2Δ. CO designation is also thought to occur around this stage of meiotic prophase [38], [39].
Pch2 physically interacts with the region of Xrs2 containing putative BRCT repeats
The ATM homolog Tel1 physically interacts with Xrs2 and promotes the phosphorylation of Sae2 and Hop1 [12], [40], [41]. We thus tested if Pch2 also interacts with components of the MRX complex using pair-wise bait-prey combinations of Pch2 with Mre11, Rad50 and Xrs2 for yeast two-hybrid analysis. In this trial, Pch2 interacted with Xrs2, but not Mre11 or Rad50 (Figure 5A). Mre11 and Rad50 two-hybrid constructs were functional since we detected interaction between Rad50-Mre11 (Figure 5B) and Mre11-Xrs2 (Figure 5C). We narrowed the Pch2-binding region of Xrs2 to a 187 amino acid region in the Xrs2(126–313)-Gal4AD construct (Figure 5D – 5F). This region contains two putative BRCT repeats, similar to the human ortholog Nbs1 [42]. Point mutations created to abolish FHA domain function present in Xrs2(1–313)-Gal4AD did not abolish interaction with LexA-Pch2 [43] (Figure 5F).

xrs2ΔN recapitulates pch2Δ effects on Hop1 phosphorylation and spore viability
The first 313 amino acids of Xrs2 are dispensable for the formation of normal levels of DSBs and crossover recombination products yet DSB turnover and MI division are delayed [44]. We created the allele xrs2ΔN-13myc that deleted the first 313 amino acid coding region of XRS2 and found it delayed MI division (Figure 5G and 5H), presumably due to the slow turnover of DSBs as in the pch2Δ mutant [20], [21], [45]. We wondered if xrs2ΔN-13myc, like pch2Δ, would suppress the MI delay conferred by rad17Δ (and vice versa). We found this to be the case with MI division timing in xrs2ΔN-13myc rad17Δ occurring earlier than either single mutant (Figure 5H). By contrast, MI division was delayed in xrs2ΔN-13myc pch2Δ (Figure 5G).
Spore viability of xrs2ΔN-13myc rad17Δ (1.4%; Table 1) was dramatically decreased compared to xrs2ΔN-13myc and XRS2-13myc rad17Δ (89.7% and 26.5%, respectively), while xrs2ΔN-13myc pch2Δ gave only a modest reduction of spore viability compared to XRS2-13myc pch2Δ (74.5% and 94.5%, respectively).
To test if xrs2ΔN-13myc affects checkpoint signaling in a similar manner to pch2Δ, we examined the effect of this mutation on Hop1 phosphorylation in xrs2ΔN-13myc rad17Δ and xrs2ΔN-13myc pch2Δ double mutants as well as in sae2Δ and dmc1Δ backgrounds. We found Hop1 phosphorylation was greatly reduced in xrs2ΔN-13myc rad17Δ but not in xrs2ΔN-13myc pch2Δ (Figure 6A). Furthermore, xrs2ΔN-13myc only abrogated Hop1 phosphorylation in sae2Δ but not in dmc1Δ backgrounds (Figure 6B and 6C). The absence of Hop1 phosphorylation in sae2Δ xrs2ΔN-13myc was not due to reduced DSB levels (Figure 6D). Notably, as with dmc1Δ pch2Δ, dmc1Δ xrs2ΔN-13myc accumulated more Hop1 protein (Figure 6C). Taken together, these results suggest that the interaction of Pch2 with the N-terminal region of Xrs2, and perhaps the putative BRCT repeats specifically, is required for Pch2′s role(s) in the recombination checkpoint and axis organization during meiosis.

xrs2ΔN but not tel1Δ recapitulates pch2Δ effects on the zip1Δ-induced MI delay
Budding yeast pch2Δ was originally isolated in the BR background as a mutation that suppresses the meiotic arrest that occurs in the absence of Zip1 [31] and Pch2 has been thought to be involved in a “synapsis checkpoint” [46]. In SK1, zip1Δ caused a meiosis I delay that is partially suppressed by pch2Δ [20]. We found that xrs2ΔN-13myc, but not tel1Δ, suppressed the zip1Δ-induced meiotic delay, suggesting that interaction with Xrs2 may also be required for Pch2′s role in the synapsis checkpoint (Figure 6E). In fact MI delays conferred by tel1Δ and zip1Δ are independent since the double mutant exhibits a more severe delay. It is thus possible that Xrs2-Pch2 interaction is required for most, if not all, functions of Pch2; while Pch2 and Tel1 may perform independent functions besides their concerted role in the recombination checkpoint.
Pch2 and Tel1 regulate spore viability independently when DSBs are reduced
Deletion of PCH2 has been shown to sensitize strains carrying hypomorphic alleles of spo11 to give lower levels of spore viability [22], . Through our studies we found that the deletion of TEL1 also gave a modest reduction of spore viability in a spo11-HA/spo11Y135F-HA background (50.1% versus 68.3%; Table 1), but not to the extent of pch2Δ (35.8%). When both Pch2 and Tel1 are absent, spore viability was dramatically reduced in this background (3.8%). Similar effects were also observed in the spo11-HA homozygous mutant background (Table 1). These data suggest that Pch2 and Tel1 independently influence an essential meiotic process that is sensitive to DSB levels. Identification of this process will require analysis of the pch2Δ tel1Δ spo11-HA/spo11Y135F-HA strain for defects in other meiotic chromosome events including DSB repair, crossover control, chromosome axis morphogenesis and/or synapsis.
Discussion
This work presents evidence defining new functions for Pch2 and Xrs2 in promoting proper chromosome segregation during meiosis: First, Pch2 and Xrs2 function in the same epistasis pathway as Tel1 (ATM) to activate the recombination checkpoint in response to unprocessed DSBs. Second, Pch2 and Xrs2 function together in the synapsis checkpoint, independent of Tel1. Third, Pch2 interacts with the N-terminus of Xrs2 containing tandem BRCT repeats. The N-terminus of Xrs2 is required to activate both the recombination and synapsis checkpoints. Finally, a role for Pch2 in preventing intersister recombination events is revealed when one branch of the recombination checkpoint is abolished by deletion of RAD17. The separate roles for Pch2 and Rad17 in mediating the recombination checkpoint via sequential phosphorylation of Hop1 and Mek1 may account for their combined role in preventing intersister repair of DSBs. These findings may help to address the seemingly disparate roles Pch2 plays among synaptic organisms, including meiotic recombination, chromosome axis formation, checkpoint signaling and crossover control [21], [47].
Recombination checkpoint
We propose that phosphorylation of the meiotic chromosome axis protein Hop1 is regulated by two partially redundant pathways: one pathway requires Tel1, Pch2 and Xrs2 and responds to the presence of unprocessed DSBs; a second pathway requires Mec1 and Rad17 and responds to the presence of resected DSB intermediates of homologous recombination (Figure 6F). This model is directly analogous to the different roles of Tel1/Xrs2 and Mec1/Rad17 in the DNA damage response during vegetative growth [18] with Pch2 providing a regulatory feature specific to meiotic chromosomes that coordinate the events of meiotic recombination with axis organization.
The physical association of Pch2 with Xrs2 suggests a mechanism to promote interhomolog bias near sites of DSBs by bringing the Tel1/ATM kinase near its substrate Hop1, a component of the chromosome axis. Pch2 might utilize the binding and/or hydrolysis of ATP to promote conformational changes in axis structure that enable the phosphorylation of Hop1 by Tel1. Alternatively or in addition, Pch2 interaction with Xrs2 might function to stabilize the association of the MRX complex at the chromosome axis analogous to the interaction of Mdc1 protein (Mediator of DNA damage Checkpoint) with the BRCT repeats of the mammalian Xrs2 ortholog, Nbs1 [48]. In this case, Mdc1 stabilizes the association of Nbs1 at sites of DNA damage, thus creating a microenvironment to promote phosphorylation of H2AX by ATM [49].
It is not clear if Pch2 interacts with Xrs2 (Nbs1) in other organisms. The mouse ortholog of Pch2, TRIP13, is implicated in early recombination steps that follow DSB resection but precede Rad51 focus formation [25]. It is possible that TRIP13-NBS1 interaction could establish a precondition that facilitates a later step of recombination. Indeed in yeast, deletion of PCH2 results in the slow turnover of resected DSBs [20], [21], [45]. In Drosophila, NBS is required for DSB repair [50], but its role in meiotic recombination has not been explored to date. While Xrs2/Nbs1 proteins are conserved from vertebrates to fungi, there is no apparent ortholog in C. elegans. It remains possible that Pch2 plays a role in a recombination checkpoint in C. elegans that has not yet been uncovered experimentally.
Synapsis checkpoint
Pch2 orthologs in worm and fly are implicated in a checkpoint activated by the failure to synapse chromosome and/or by disruptions in axis formation. The synapsis checkpoint functions in these instances even in absence of DSB formation [28], [30]. Although synapsis is dependent on DSB formation in budding yeast, several examples implicate Pch2 in a synapsis checkpoint that responds to defects in synapsis and/or axis structure in situations where DSBs are efficiently repaired [13], [51]. Strong evidence in support of a synapsis checkpoint comes from our previous observation that MEK1-GST, an artificially activated form of MEK1, acts as a genetic enhancer of zip1Δ by causing MI arrest [13]. Since DSBs are efficiently repaired in this situation [13], this result suggests that synapsis and/or axis defects trigger the arrest, not the persistence of unrepaired DNA breaks. Deletion of PCH2, but not TEL1, can bypass this arrest, suggesting an independent role for Pch2 in a synapsis checkpoint (unpublished data). Similarly, deletion of PCH2 suppresses the meiosis I arrest phenotype that is activated by the presence of aberrantly synapsed chromosomes caused by the non-null allele zip1-4LA, which also repairs DNA breaks efficiently [51]. We found here that xrs2ΔN-13myc, similar to pch2Δ, partially suppressed zip1Δ delay, suggesting that Xrs2, perhaps through association with Pch2, is required to execute the synapsis checkpoint. By contrast, Tel1 does not seem to be involved in this branch of Pch2′s function. Borner and colleagues argued previously that Pch2 might mediate Mec1/ATR activity with respect to sensing “structure-dependent interchromosome interactions” [21]. It is possible that a Pch2/Xrs2/Mec1 pathway functions in this program.
Further understanding of the differential requirements for Xrs2 and Tel1 for Pch2 function in the recombination checkpoint versus the synapsis checkpoint (and possibly crossover control) may help to identify common mechanisms shared among synaptic organisms, where pairing and SC formation are not always coupled to recombination [52].
Materials and Methods
Strains
All strains are derivatives of SK1 except the strain used for yeast two hybrid spot assay is L40 (MATa trp1 leu2 his3 LYS2::lexA-HIS3 URA3::lexA-lacZ) [53]. Deletion mutants were generated by PCR-based gene disruption [54], [55]. All the mec1Δ strains also carried sml1Δ to suppress inviability. MEK1-3HA and XRS2-13myc were made by using pFA6a-3HA-kanMX6 and pFA6a-13myc-kanMX6 modules, respectively [56]. xrs2ΔN-13myc was created by two-step allele replacement. Briefly, a PCR-amplified URA3 was used to replace the region encoding amino acid 1–313 of Xrs2-13myc. Then a PCR-generated fragment containing 385 bp upstream the first coding ATG fused to 375 bp downstream the ATG encoding amino acid 314 of Xrs2 was used to replace the URA3, resulting in xrs2ΔN-13myc expressing 13myc-tagged Xrs2(314–854) under the native promoter of XRS2. spo11-HA and spo11Y135F-HA are a gift from Scott Keeney and crossed into our strain background.
SBY strain numbers are listed in Table 1. Additional strains used in this study are: strains isogenic to SBY1903 (MATa/MATα ho::hisG/″ leu2::hisG/″ ura3(ΔSma-Pst)/″ his4-X::LEU2-(NBam)-URA3/HIS4::LEU2-(NBam)) except the indicated mutations: SBY3055 (ntd80Δ); SBY3280 (ntd80Δ pch2Δ); SBY3277 (ntd80Δ rad17Δ); SBY3274 (ntd80Δ pch2Δ rad17Δ); SBY2591 (dmc1Δ); SBY2597 (dmc1Δ pch2Δ); SBY2594 (dmc1Δ rad17Δ); SBY2606 (dmc1Δ pch2Δ rad17Δ); SBY3800 (dmc1Δ tel1Δ); SBY2611 (sae2Δ); SBY2625 (sae2Δ pch2Δ); SBY3843 (sae2Δ tel1Δ); SBY2616 (sae2Δ rad17Δ); SBY4684 (sae2Δ xrs2ΔN-13myc); SBY3589 (MEK1-3HA); SBY3595 (MEK1-3HA pch2Δ); SBY3592 (MEK1-3HA rad17Δ); SBY3598 (MEK1-3HA pch2Δ rad17Δ); strains isogenic to SBY4056 (MATa/MATα ho::hisG/″ lys2/″leu2::hisG/″ ura3Δ::hisG/″ trp1::hisG/″ GAL3/″) except the indicated mutations: SBY3560 (dmc1Δ), SBY4517 (dmc1Δ xrs2ΔN-13myc), SBY3644 (sae2Δ), SBY4514 (sae2Δ xrs2ΔN-13myc), SBY4445 (zip1Δ), SBY4451 (zip1Δ pch2Δ), SBY4448 (zip1Δ tel1Δ), SBY4404 (zip1Δ xrs2ΔN-13myc); tel1Δ, sml1Δ, and ndt80Δ are marked with hphMX; meclΔ is marked with natMX; all other mutations are marked with kanMX.
Sporulation conditions
Time courses were conducted by the SPS method [20]. Briefly, cells were patched on YPG plates (3% glycerol, 2% bactopeptone, 1% yeast extract, 2% bactoagar, 0.01% adenine sulphate, 0.004% tryptophan) for ∼14 hr and then stripped on YPD plates (2% glucose, 2% bactopeptone, 1% yeast extract, 2% bactoagar, 0.01% adenine sulphate, 0.004% tryptophan) and grown for 2 days. Single colonies were used to inoculate 5 ml YPD (plus 0.002% uracil if ura3 strains were used) liquid cultures and grown for at least 24 hr before diluted into SPS (1% potassium acetate, 0.5% yeast extract, 1% bactopeptone, 0.17% yeast nitrogen base, 0.5% ammonium sulphate, 1.02% potassium biphthalate) (plus 0.002% uracil if ura3 strains were used) at O.D.600 = 0.16. SPS cultures were grown for ∼15.5 hr, washed with H2O, and then resuspended into SPM (1% potassium acetate, 0.02% raffinose, 0.009% amino acid powder) at O.D.600 = 2–3. Spore viability data were obtained by sporulation on solid SPM media. All procedures were performed at 30°C.
DNA physical assay and meiotic progression analysis
DNA extraction, gel electrophoresis and southern blot were performed as previously described [9]. Meiosis I division timing was determined by calculating the percentage of post-MI cells at indicated time points. Briefly, meiotic cultures were fixed in 50% ethanol and stained with DAPI. Cells with more than 2 DAPI-stained nucleus bodies were counted as post-MI cells. 200 cells were counted for each time points.
Protein extraction, Western blotting, and immunoprecipitation
Denaturing whole-cell extracts were prepared as previously described [57] with modifications. Briefly, 1 mL meiotic cultures at indicated time points were spun down and resuspended in 1 mL ice-cold water with 1 mM PMSF, 10 mM sodium fluoride and 10 mM sodium diphosphate. 150 µL ice-cold 2 N NaOH / 8% 2-ME was then added and mixtures were incubated on ice for 10 min. After added 160 µL ice-cold 50% TCA and incubated on ice for 10 min, mixtures were spun for 8 min at 14000 rpm. Pellets were washed by 500 µL ice-cold acetone and spun for 5 min at 14000 rpm. Washed pellets were dried by spinning in the vacufuge for 8 min and then resuspended in 1X SDS sample buffer with PMSF, sodium fluoride and sodium diphosphate. A bath sonicator was used to facilitate resuspension in acetone and sample buffer. Proteins from denaturing whole-cell extracts were detected by Western blotting using α-Hop1 (S. Roeder), α-HA (Santa Cruz, sc-7392), α-phospho-Akt substrate (Cell signaling, #9614), and α-Pgk1 (Invitrogen, A-6457). Immunoprecipitation was performed as previously described [13] except α-HA antibody (Santa Cruz, sc-7392) was used.
Plasmids and yeast two hybrid analysis
Mre11, Rad50, and Xrs2 yeast two-hybrid plasmids are a gift from S. Keeney [58]. Xrs2 truncation plasmids were constructed by cloning PCR-generating fragments into the same plasmid for full-length Xrs2 (pACT2-2). Xrs2(1–313)-S47A H50A plasmid was created by QuikChange (Stratagene). LexA-Pch2 plasmid was made by cloning PCR amplified intronless PCH2 coding region into pCA1 plasmid. Y2H spot assay was performed by spotting 5 µL O.D.600 = 1 cultures onto SC-Leu-Trp plates and SC-Leu-Trp-His + 1mM 3AT plates and grown for 3–5 days.
Zdroje
1. ZicklerDKlecknerN 1999 Meiotic chromosomes: integrating structure and function. Annu Rev Genet 33 603 754
2. KeeneySGirouxCNKlecknerN 1997 Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88 375 384
3. BishopDKParkDXuLKlecknerN 1992 DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69 439 456
4. ShinoharaAOgawaHOgawaT 1992 Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69 457 470
5. KadykLCHartwellLH 1992 Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132 387 402
6. BzymekMThayerNHOhSDKlecknerNHunterN 2011 Double Holliday junctions are intermediates of DNA break repair. Nature 464 937 941
7. SchwachaAKlecknerN 1997 Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog-only pathway. Cell 90 1123 1135
8. AllersTLichtenM 2001 Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106 47 57
9. HunterNKlecknerN 2001 The single-end invasion: an asymmetric intermediate at the double-strand break to double-holliday junction transition of meiotic recombination. Cell 106 59 70
10. SchwachaAKlecknerN 1995 Identification of double Holliday junctions as intermediates in meiotic recombination. Cell 83 783 791
11. YoudsJLBoultonSJ 2011 The choice in meiosis - defining the factors that influence crossover or non-crossover formation. J Cell Sci 124 501 513
12. CarballoJAJohnsonALSedgwickSGChaRS 2008 Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132 758 770
13. WuHYHoHCBurgessSM 2010 Mek1 kinase governs outcomes of meiotic recombination and the checkpoint response. Curr Biol 20 1707 1716
14. NiuHWanLBaumgartnerBSchaeferDLoidlJ 2005 Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol Biol Cell 16 5804 5818
15. WanLde los SantosTZhangCShokatKHollingsworthNM 2004 Mek1 kinase activity functions downstream of RED1 in the regulation of meiotic double strand break repair in budding yeast. Mol Biol Cell 15 11 23
16. KimKPWeinerBMZhangLJordanADekkerJ 2010 Sister cohesion and structural axis components mediate homolog bias of meiotic recombination. Cell 143 924 937
17. HochwagenAAmonA 2006 Checking your breaks: surveillance mechanisms of meiotic recombination. Curr Biol 16 R217 228
18. UsuiTOgawaHPetriniJH 2001 A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7 1255 1266
19. LydallDNikolskyYBishopDKWeinertT 1996 A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature 383 840 843
20. WuHYBurgessSM 2006 Two distinct surveillance mechanisms monitor meiotic chromosome metabolism in budding yeast. Curr Biol 16 2473 2479
21. BornerGVBarotAKlecknerN 2008 Yeast Pch2 promotes domainal axis organization, timely recombination progression, and arrest of defective recombinosomes during meiosis. Proc Natl Acad Sci U S A 105 3327 3332
22. JoshiNBarotAJamisonCBornerGV 2009 Pch2 links chromosome axis remodeling at future crossover sites and crossover distribution during yeast meiosis. PLoS Genet 5 e1000557 doi:10.1371/journal.pgen.1000557
23. ZandersSAlaniE 2009 The pch2Delta mutation in baker's yeast alters meiotic crossover levels and confers a defect in crossover interference. PLoS Genet 5 e1000571 doi:10.1371/journal.pgen.1000571
24. ZandersSSonntag BrownMChenCAlaniE 2011 Pch2 Modulates Chromatid Partner Choice During Meiotic Double-Strand Break Repair in Saccharomyces cerevisiae. Genetics
25. RoigIDowdleJATothAde RooijDGJasinM 2010 Mouse TRIP13/PCH2 Is Required for Recombination and Normal Higher-Order Chromosome Structure during Meiosis. PLoS Genet 6 e1001062 doi.org/10.1371/journal.pgen.1001062
26. WojtaszLDanielKRoigIBolcun-FilasEXuH 2009 Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet 5 e1000702 doi:10.1371/journal.pgen.1000702
27. LiXCSchimentiJC 2007 Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet 3 e130 doi:10.1371/journal.pgen.0030130
28. BhallaNDernburgAF 2005 A conserved checkpoint monitors meiotic chromosome synapsis in Caenorhabditis elegans. Science 310 1683 1686
29. JoyceEFMcKimKS 2009 Drosophila PCH2 is required for a pachytene checkpoint that monitors double-strand-break-independent events leading to meiotic crossover formation. Genetics 181 39 51
30. JoyceEFMcKimKS 2010 Chromosome Axis Defects Induce a Checkpoint-Mediated Delay and Interchromosomal Effect on Crossing Over during Drosophila Meiosis. PLoS Genet 6 e1001059 doi:10.1371/journal.pgen.1001059
31. San-SegundoPARoederGS 1999 Pch2 links chromatin silencing to meiotic checkpoint control. Cell 97 313 324
32. DongHRoederGS 2000 Organization of the yeast Zip1 protein within the central region of the synaptonemal complex. J Cell Biol 148 417 426
33. SymMEngebrechtJARoederGS 1993 ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72 365 378
34. LynnASoucekRBornerGV 2007 ZMM proteins during meiosis: crossover artists at work. Chromosome Res 15 591 605
35. LaoJPOhSDShinoharaMShinoharaAHunterN 2008 Rad52 promotes postinvasion steps of meiotic double-strand-break repair. Mol Cell 29 517 524
36. NiuHLiXJobEParkCMoazedD 2007 Mek1 kinase is regulated to suppress double-strand break repair between sister chromatids during budding yeast meiosis. Mol Cell Biol 27 5456 5467
37. GrushcowJMHolzenTMParkKJWeinertTLichtenM 1999 Saccharomyces cerevisiae checkpoint genes MEC1, RAD17 and RAD24 are required for normal meiotic recombination partner choice. Genetics 153 607 620
38. BornerGVKlecknerNHunterN 2004 Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 117 29 45
39. BishopDKZicklerD 2004 Early decision; meiotic crossover interference prior to stable strand exchange and synapsis. Cell 117 9 15
40. NakadaDMatsumotoKSugimotoK 2003 ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev 17 1957 1962
41. Cartagena-LirolaHGueriniIViscardiVLucchiniGLongheseMP 2006 Budding Yeast Sae2 is an In Vivo Target of the Mec1 and Tel1 Checkpoint Kinases During Meiosis. Cell Cycle 5 1549 1559
42. BeckerEMeyerVMadaouiHGueroisR 2006 Detection of a tandem BRCT in Nbs1 and Xrs2 with functional implications in the DNA damage response. Bioinformatics 22 1289 1292
43. MatsuzakiKShinoharaAShinoharaM 2008 Forkhead-associated domain of yeast Xrs2, a homolog of human Nbs1, promotes nonhomologous end joining through interaction with a ligase IV partner protein, Lif1. Genetics 179 213 225
44. ShimaHSuzukiMShinoharaM 2005 Isolation and characterization of novel xrs2 mutations in Saccharomyces cerevisiae. Genetics 170 71 85
45. HochwagenAThamWHBrarGAAmonA 2005 The FK506 binding protein Fpr3 counteracts protein phosphatase 1 to maintain meiotic recombination checkpoint activity. Cell 122 861 873
46. MacqueenAJHochwagenA 2011 Checkpoint mechanisms: the puppet masters of meiotic prophase. Trends Cell Biol
47. JoyceEFMcKimKS 2011 Meiotic checkpoints and the interchromosomal effect on crossing over in Drosophila females. Fly (Austin) 5
48. WilliamsRSDodsonGELimboOYamadaYWilliamsJS 2009 Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 139 87 99
49. LukasCMelanderFStuckiMFalckJBekker-JensenS 2004 Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J 23 2674 2683
50. MukherjeeSLaFaveMCSekelskyJ 2009 DNA damage responses in Drosophila nbs mutants with reduced or altered NBS function. DNA Repair (Amst) 8 803 812
51. MitraNRoederGS 2007 A novel nonnull ZIP1 allele triggers meiotic arrest with synapsed chromosomes in Saccharomyces cerevisiae. Genetics 176 773 787
52. BhallaNDernburgAF 2008 Prelude to a division. Annu Rev Cell Dev Biol 24 397 424
53. VojtekABHollenbergSMCooperJA 1993 Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 74 205 214
54. GoldsteinALMcCuskerJH 1999 Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15 1541 1553
55. WachABrachatAPohlmannRPhilippsenP 1994 New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10 1793 1808
56. LongtineMSMcKenzieA3rdDemariniDJShahNGWachA 1998 Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14 953 961
57. YaffeMPSchatzG 1984 Two nuclear mutations that block mitochondrial protein import in yeast. Proc Natl Acad Sci U S A 81 4819 4823
58. AroraCKeeKMalekiSKeeneyS 2004 Antiviral protein Ski8 is a direct partner of Spo11 in meiotic DNA break formation, independent of its cytoplasmic role in RNA metabolism. Mol Cell 13 549 559
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Evidence-Based Annotation of Gene Function in MR-1 Using Genome-Wide Fitness Profiling across 121 Conditions
- De Novo Origins of Human Genes
- Capture of MicroRNA–Bound mRNAs Identifies the Tumor Suppressor miR-34a as a Regulator of Growth Factor Signaling
- TRY-5 Is a Sperm-Activating Protease in Seminal Fluid
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy