
The Importance of Imprinting in the Human Placenta
As a field of study, genomic imprinting has grown rapidly in the last 20 years, with a growing figure of around 100 imprinted genes known in the mouse and approximately 50 in the human. The imprinted expression of genes may be transient and highly tissue-specific, and there are potentially hundreds of other, as yet undiscovered, imprinted transcripts. The placenta is notable amongst mammalian organs for its high and prolific expression of imprinted genes. This review discusses the development of the human placenta and focuses on the function of imprinting in this organ. Imprinting is potentially a mechanism to balance parental resource allocation and it plays an important role in growth. The placenta, as the interface between mother and fetus, is central to prenatal growth control. The expression of genes subject to parental allelic expression bias has, over the years, been shown to be essential for the normal development and physiology of the placenta. In this review we also discuss the significance of genes that lack conservation of imprinting between mice and humans, genes whose imprinted expression is often placental-specific. Finally, we illustrate the importance of imprinting in the postnatal human in terms of several human imprinting disorders, with consideration of the brain as a key organ for imprinted gene expression after birth.
Published in the journal:
. PLoS Genet 6(7): e32767. doi:10.1371/journal.pgen.1001015
Category:
Review
doi:
https://doi.org/10.1371/journal.pgen.1001015
Summary
As a field of study, genomic imprinting has grown rapidly in the last 20 years, with a growing figure of around 100 imprinted genes known in the mouse and approximately 50 in the human. The imprinted expression of genes may be transient and highly tissue-specific, and there are potentially hundreds of other, as yet undiscovered, imprinted transcripts. The placenta is notable amongst mammalian organs for its high and prolific expression of imprinted genes. This review discusses the development of the human placenta and focuses on the function of imprinting in this organ. Imprinting is potentially a mechanism to balance parental resource allocation and it plays an important role in growth. The placenta, as the interface between mother and fetus, is central to prenatal growth control. The expression of genes subject to parental allelic expression bias has, over the years, been shown to be essential for the normal development and physiology of the placenta. In this review we also discuss the significance of genes that lack conservation of imprinting between mice and humans, genes whose imprinted expression is often placental-specific. Finally, we illustrate the importance of imprinting in the postnatal human in terms of several human imprinting disorders, with consideration of the brain as a key organ for imprinted gene expression after birth.
Introduction
Pronuclear transfer experiments in mice in the early 1980s showed that maternal and paternal genetic contributions were non-equivalent and that both were indispensable for normal development [1], [2]. The introduction of reciprocal translocations into mice, creating regions of uniparental disomy, showed that discrete areas of the mouse genome were subject to differential parental regulation [3]. In parallel with this fascinating mouse work, it was observed that several non-Mendelian human syndromes showed similar inheritance to phenotypes seen in the disomic mice [4]. The mapping of deletions causative in Prader Willi (PWS) and Angelman (AS) syndromes, for example, permitted localisation of parentally non-equivalent genomic regions in humans [4]. In 1991, the first endogenous imprinted genes were identified [5]–[7]. This parent-of-origin, monoallelic gene expression, with its associated differential DNA methylation (first shown in 1993, [8]) became defined as genomic imprinting.
Genomic imprinting, found predominantly in eutherian mammals, is an epigenetic phenomenon whose evolution may be linked to a dichotomy between paternal and maternal resource allocation. This is potentially powerful enough to promote evolution of unequal gene expression between selected parental alleles. Parental-specific monoallelic expression thus balances fetal growth to the equal benefit of both parental genomes, in spite of the resulting potentially damaging haploinsufficiency [9].
The canonical example of allelic expression of imprinted genes balancing growth is evident with the paternally expressed Igf2 and maternally expressed Igf2r genes [5], [7], [10]. Igf2 is a potent enhancer of fetal growth and inappropriate expression disturbs normal growth in mice [10]. A reduction in Igf2 expression leads to growth restriction, whereas biallelic expression and the subsequent increase in the number of Igf2 transcripts leads to overgrowth [11], [12]. Maternally expressed Igf2r has the opposite effect on growth, as the Igf2r protein acts as a negative regulator of Igf2 by binding to the Igf2 protein, reducing its bioavailability and targeting it for lysosomal degradation [13]–[16]. Monoallelic expression of imprinted genes is controlled by allelic DNA methylation, added differentially to the imprinting control regions (ICRs) of parental germlines [17], [18]. The paternal allelic expression of murine Igf2 is also found in humans, and in both species monoallelic expression is mediated in cis by maternal DNA methylation at the H19 ICR, the differentially methylated domain (H19 DMD) [19]–[22].
IGF2 is also an important growth enhancer in humans, and its expression and subsequent phenotypic effects may be similarly impacted by dysregulation of imprinting. A loss of methylation at the H19 DMD in humans is found in a subset of Silver Russell syndrome (SRS) cases [23]. The main phenotype of SRS is severe intrauterine growth restriction (IUGR) that could be caused by a reduction in IGF2 transcription as a result of a loss of methylation at the H19 DMD [23]. Hypermethylation at the H19 DMD is found in 30% cases of Beckwith Wiedemann syndrome (BWS) [24], and the overgrowth macroglossia and organomegaly associated with this disorder may be caused by an increase in IGF2 transcription as a result of its biallelic expression. IGF2R imprinting in the human, in contrast, is polymorphic, rare, and most likely restricted to the placenta [25]. Recent evidence of a potential human orthologue of the murine ncRNA Air, responsible in the mouse for paternal Igf2r silencing, indicates that some key features of reciprocal murine Igf2/Igf2r imprinting may be present in humans [26].
Human Placental Development
The placenta, particularly the invasive trophoblast lineages, is an important focus for potential parental conflict. It is directly responsible for bringing maternal and fetal blood supplies into contact, facilitating nutrient exchange and determining resource allocation (Figure 1) [27].

Human embryos implant interstitially in a highly invasive manner. Leading edge trophoblast cells fuse to form a syncytium, resulting in a two layered structure of multinucleated syncytiotrophoblast and cellular cytotrophoblast. Protusions of syncytiotrophoblast interdigitate into the decidualised endometrium, forming contacts with the maternal blood supply (Figure 1). Extravillous cytotrophoblast, which may be analogous to the endoreduplicated murine giant cells, form columns from the tips of anchoring villae, attached to the basal plate, and extend through the syncytium. Invasive cells break away from these columns and migrate to colonise maternal spiral arteries. Interstitial trophoblast cells invade to expand the placenta from its edge outwards [28]. Invasion is partly controlled by the decidua, which expresses proteins, including a wide variety of IGF binding proteins, balancing invasion and fetal provision [29], [30]. Perturbation of this is evident in ectopic pregnancy, when invasion is far more extensive in the absence of the decidua [31].
Genomic Imprinting in the Human Placenta
The physiological importance of genomic imprinting in humans can be demonstrated by the diseases resulting from mutations or epimutations in imprinted genes. Human imprinting disorders are somewhat rare but comprise a large group of diverse pathologies, primarily involving growth or neurological development. Consistent with the growth phenotypes observed, many of the imprinted genes known to-date are expressed in the human placenta (Table 1) [32], [33].
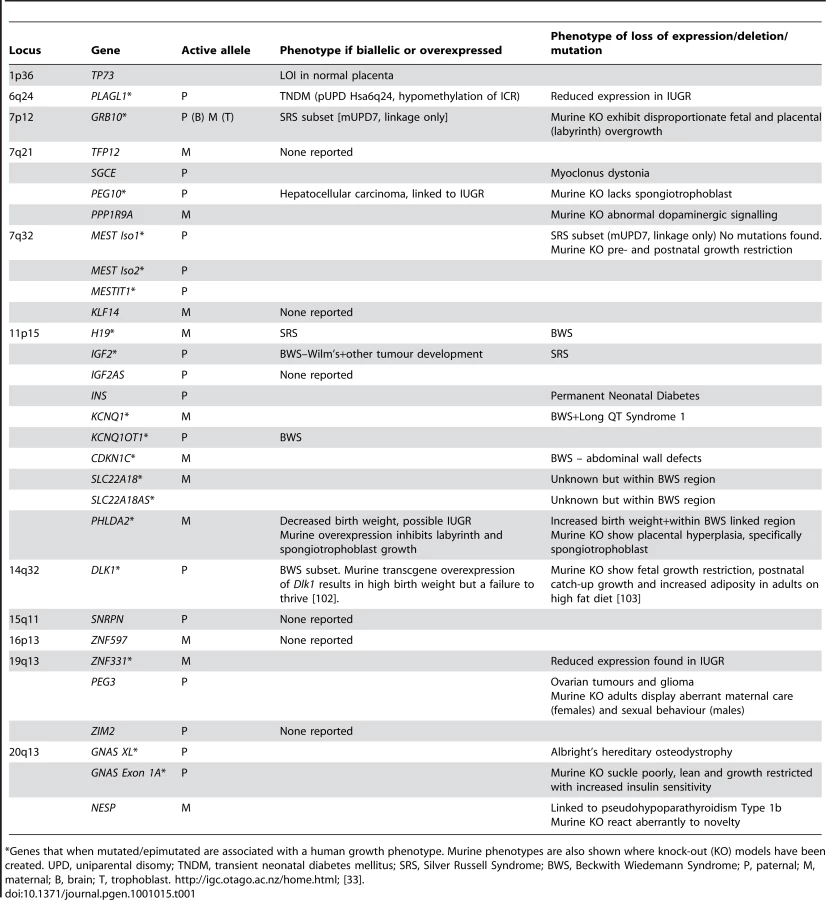
Disease pathologies resulting from inappropriate imprinted gene expression may each be due in part, or completely, to an aberrantly functioning placenta. The placenta is fundamental to fetal growth, and Table 1 highlights the imprinted genes expressed in the placenta that have been implicated in fetal growth disorders. As previously stated, IUGR is a defining characteristic of the imprinting disorder SRS. Up to half of all SRS cases may be caused by a reduction in IGF2 expression, as outlined above, but in the remainder the cause is unknown [23]. Whilst IUGR is often idiopathic, it is commonly accompanied by reduced blood flow through the placenta and limited invasion of the decidua and maternal blood vessels [34]. This phenotype is consistent with either the loss of expression of an imprinted gene involved in maximising recruitment of maternal resources (i.e., a paternally expressed gene), or an increase in expression of an imprinted gene acting to limit maternal input (i.e., a maternally expressed gene). A second disease associated with reduced placental perfusion is preeclampsia, whose matrilineal inheritance pattern has highlighted the possibility that imprinted genes might involved in its pathogenesis [35], [36]. In a recent study of 96 cases of BWS, seven resulted from maternally inherited CDKN1C mutations and of these, three pregnancies were complicated by preeclampsia, compared to three of the 89 BWS cases not related to CDKN1C mutations [37]. Interestingly, transgenic mice whose litters carry mutations of the maternal Cdkn1c copy display preeclampsia-like features, including hypertension, proteinuria, and abnormal trophoblast proliferation [38], [39]. These data suggest an important role for CDKN1C in a subset of preeclampsia cases, however, other imprinted susceptibility loci for this complication of pregnancy remain elusive [40].
The imprinted gene PHLDA2 on human Chromosome 11 (Hsa11) is expressed predominately in the placenta, and its expression in the placenta at term correlates negatively with fetal birth weight [41]. Given that PHLDA2 is maternally expressed, this trend is consistent with the parental resource conflict theory, that maternally expressed genes act to limit maternal resource provision. Further evidence that PHLDA2 expression levels in the human placenta might be important in regulating fetal growth comes from two studies comparing placentas from normal and IUGR pregnancies. Both studies found higher levels of PHLDA2 expression in placentas from IUGR pregnancies than placentas from non-IUGR pregnancies [33], [42].
Paternally expressed MEST is thought to play a role in angiogenesis in human trophoblast tissue and decidua, is highly expressed and robustly imprinted in the placenta [43]. MEST is located on Hsa7, and maternal uniparental disomy (mUPD) 7, is implicated in 7–10% of SRS cases. Additionally, one SRS case has been reported with a segmental mUPD for 7q31-qter, specifically implicating the MEST imprinting cluster in this instance, rather than any of the other imprinted genes on Hsa7 [44]. Currently, no direct evidence exists to link human MEST with disease, but mice deficient in Mest are pre - and postnatally growth restricted [45].
GRB10 is a growth factor binding protein, maternally expressed specifically in cytotrophoblast and biallelic elsewhere, located on Hsa7 [46]. The mUPD7 implicated in 7–10% SRS cases would lead to biallelic expression of GRB10 and may be linked to the growth restriction characteristic of SRS for this subset of patients. Currently, however, no evidence exists to directly link GRB10 expression levels with growth in humans [47], [48]. In mouse embryos, Grb10 is widely expressed from the maternally derived chromosome, and ablation of this copy causes embryonic overgrowth, such that neonates are 30% larger than wild-type littermates at birth [49]. This is accompanied by disproportionate overgrowth of the placental labyrinth [50].
Imprinting in the Mouse Placenta
Further clues regarding a role for imprinted genes in the placenta have been derived from studies in transgenic and knockout mice. Ablating the expression of individual imprinted genes leads to a range of pathologies, depending on the gene. Murine paternally expressed Igf2 has been shown to promote placental growth (see below), and loss of Mest or Peg3 causes placental growth restriction. Conversely, deletion of maternally expressed Igf2r, Cdkn1c, or Phlda2 results in placental hyperplasia [32].
The importance of genomic imprinting specifically in the murine placenta can be illustrated by the expression pattern of paternally expressed Igf2. Human and mouse IGF2/Igf2 can be expressed from several different promoters, but in the mouse, the transcripts from one promoter—Igf2P0—are placental-specific [51]. Deletion of the P0 promoter reduces placental size close to that of complete Igf2 KO, i.e., around 40% smaller than normal [51]. Humans also have an IGF2 P0 promoter, but it is not placental-specific, indicating only a partial conservation of imprinting of IGF2P0/Igf2P0 between mice and humans [52].
The main role of the placenta is the nutrition of the fetus. Murine Igf2P0 transcripts are expressed specifically in the labyrinthine trophoblast of the placenta [51], the cellular interface between the maternal blood supply and the fetal capillaries, and the surface across which nutrient exchange with the fetus takes place. Through increasing the surface area, Igf2P0 is thought to enhance passive permeability in the labyrinth, promoting nutrient exchange [51], [53]. In the Igf2P0-null model, fetal Igf2 expression is shown to regulate nutrient supply from the growth-restricted placenta in a paracrine manner [51]. The presence of circulating fetal Igf2 coincident with an imbalance between placental supply and fetal demand results in upregulation of nutrient transfer systems [54]. Placental transcription of glucose transporter Slc2a3 and paternally expressed amino acid transporter Slc38a4 are upregulated, followed by an increase in glucose and amino acid transport from the placenta to the fetus [54]. These data show that imprinted growth regulators may influence nutrient supply through action in the placenta, or by regulating demand from the fetus.
As previously discussed, the expression level of PHLDA2/Phlda2 correlates inversely with fetal growth in both humans and mice [33], [41], [55]. This role as a growth suppressor has recently been directly linked with the exchange of nutrients between mother and fetus in mice [56]. In a transgenic model, a two-fold increase in Phlda2 expression resulted in reduced placental weight, specifically in the junctional zone, and a decrease in glycogen stores and glycogen cell migration, important for fetal glucose supplies late in gestation [56]. This is the reverse of what is seen in the Phlda2 knockout mouse, and unlike the null, impacted on embryonic as well as placental development so that overexpression of Phlda2 led to 13% reduction in fetal weight [55], [56]. These data suggest that the regulation of fetal and placental growth by PHLDA2/Phlda2 might be effected through its potential role in nutrient transfer [56].
The KCNQ1/Kcnq1 Imprinting Cluster
Expression within the KCNQ1/Kcnq1 imprinting cluster on Hsa11/Mmu7 is only partially conserved between humans and mice [25]. Whilst the central six transcripts, covering 400 kb, maintain monoallelic expression in both species, the eight flanking genes are known to be maternally expressed in the mouse and bovine placenta, extending the imprinted domain to 780 kb [57]–[59]. In contrast, these flanking transcripts are biallelic in the human [25] (Figure 2).

The function of several of the genes in the KCNQ1/Kcnq1 cluster has been extensively characterised, and correspond with roles in embryonic and placental growth. ASCL2/Ascl2 is essential for early placental development, whilst CDKN1C/Cdkn1c is a growth suppressor, whose absence causes neonatal lethality in the mouse [60], [61]. Mutations or epimutations affecting CDKN1C result in one type of BWS in the human, commonly involving severe abdominal wall defects [62], [63]. Another group of BWS cases are due to mutations or epimutations immediately upstream of H19. These BWS patients have a high risk of tumours, especially compared to the CDKN1C region (epi)mutation group; see Table 1 [63]. KCNQ1 is imprinted at the majority of expressed sites in the human, except in the heart, the site of the defect in long-QT syndrome that are caused by mutations in KCNQ1 [64], [65]. PHLDA2, whose role has already been discussed, is also encoded at this locus.
A differentially methylated region (DMR) in intron 10 of KCNQ1 acts dually as the imprinting control region (ICR) for the cluster, called KvDMR, and the promoter of an antisense ncRNA KCNQ1OT1, which contributes to the regulation of imprinting at the domain [66], [67] (Figure 2). In the mouse, this ncRNA is imprinted and expressed from the paternally inherited chromosome where its transcription is required for the repression of the paternally inherited protein coding genes in cis [68], [69]. Kcnq1ot1 RNA may be linked to recruitment of the Eed-Ezh2 polycomb protein complex to the paternal chromosome, resulting in the enrichment of H3K27Me3 and H3K9Me2 and a repressed chromatin conformation conducive to allelic silencing [58]. Dnmt1−/− mice are deficient in DNA methytransferase DNMT1, the enzyme responsible for maintenance of DNA methylation. In these mice, histone modifications are able to maintain imprinting of the placental specific genes in the Kcnq1 region, indicating that maintenance methylation is not required for prolonged monoallelic expression of these genes in the placenta. Imprinting of the central six genes is lost in Dnmt1−/− mice [57], [58], [70], indicating that they do require maintenance methylation for monoallelic expression. Despite the absence of a requirement for maintenance methylation for the imprinting of a subset of genes in this cluster, the establishment of the germline DMR (by different enzymes, the de novo DNA methyltransferase, Dnmt3a [71]) remains essential for imprinting across the whole locus [70]. There is evidence that the murine Kcnq1ot1 RNA may form a silencing compartment in the nucleus, to which the repressed alleles are localised [72]. This compartment is larger in the murine placenta than in the fetus, perhaps reflecting the increased size of the repressed region in this tissue [73]. Given that imprinting of the KCNQ1 region in the human embryo and placenta both mirror that of the mouse embryo, if this model is correct it may be that such a transcriptional silencing compartment would be smaller in the human placenta, encompassing only the central seven transcripts.
Differences in the Placenta Between Humans and Mice
The placenta is the organ with the most varied morphology between mammalian species [74]. This is indicative of the different reproductive strategies employed by different species, where young may be precocial or altricial, and litter size and gestational length vary greatly. The lack of conservation of imprinting between humans and mice in the placenta, such as that of IGF2PO and the KCNQ1 region, has been suggested to be due to the marked differences between murine and human placentation and pregnancy [75], [76]. Mice have a labyrinthine interdigitation into the maternal decidua, compared to the villous structure of the exchange surface in the human. Mouse placentas have one or very few central maternal arteries, but in the human, several maternal spiral arteries provide the placenta with nutrients and oxygen. In the mouse, glycogen cells in the placenta become abundant between E13 and E18.5, invade the decidua basalis, and cluster at the base of the central maternal artery. They lyse just before term, possibly to provide energy for the final phase of prenatal growth [77]. Both species manipulate the maternal blood supply to maximise nutrient transfer. In the mouse, it is suggested that the primary cause of maternal artery transformation is the secretion of cytokines—i.e., by glycogen cells, which secrete Igf2 protein and express nuclear Cdkn1c and have been shown to be important for transformation of the central maternal artery [78], [79]. Artery transformation in the mouse is shallow and limited to the proximal decidua [75]. Conversely, human maternal arteries are extensively colonised by endovascular trophoblast cells. These cells relax the elastic artery walls and expand the lumen, allowing increased blood flow to the growing human fetus.
Differences in Imprinted Gene Expression Between Human and Mouse Placentas
In the mouse, 5–15 fetuses may be carried in utero at the same time, depending on the mouse strain, and one pregnancy can occur from two separate matings [80], [81]. This intra-brood competition forms the basis of one of the key features of the parental conflict theory because such a scenario would be predicted to increase parental conflict at the materno-placenta interface [82]. Different levels of conflict in the placenta between mice and humans may account for the divergence in imprinted gene expression profiles, with imprinted expression of certain genes not being required in the human. The transcriptional regulator Ascl2 is imprinted in the mouse placenta, and absolutely required for placentation, whereas in the human this gene is biallelically expressed, indicative of a less stringent requirement for dosage management in humans for this gene [60], [83], [84], or the utilisation of a different mechanism of dosage control in the human. Sheep, like humans, bear singletons and the sheep orthologue of placental specific Ascl2 (SASH2), is biallelically expressed whilst CDKN1C is maternally expressed [85]. To date, most genes that are imprinted in mouse but not in human, including those previously discussed, are imprinted specifically in the placenta of the mouse. Table 2 lists placental-specific imprinted genes identified in the mouse at several loci. With the exception of TFPI2 these genes are not imprinted in the human [25]. This observation lends support to the idea that the placenta could be at the centre of the differences in imprinting between mice and humans. Of the genes listed in Table 2, most are maternally expressed, consistent with an involvement of these genes in limiting placental and/or fetal growth [25]. Perhaps the mouse placenta manages parental conflict through a more limited invasion of the maternal decidua and blood vessels than that of the human placenta, with imprinted genes playing a role in modulating the process.

Total reproductive capability of mammalian females over a lifetime could also have an impact on parental conflict, and so possibly imprinted gene expression, since deleterious effects of pregnancy on the mother may be additive between pregnancies. It would therefore be illuminating to compare imprinting in monoseasonally oestrous species, such as the giant panda, with imprinting in mammals capable of many fertile oestrous cycles in their lifetimes, such as mice and humans.
Changes in Global Gene Expression in the Placenta during Gestation
Genome-wide expression analyses of early and late murine and human placentas show that early placentation events are more similar between mammalian species than later placental growth [86]. During early gestation and placental developmental stages—i.e., E8.5 to E10.5 in the mouse—the placenta utilises evolutionarily ancient genes, such as those involved in metabolism, the cell cycle, and RNA processing. During mid to late gestation (E10.5 to E15) a transition occurs where expression profiles become enriched for genes that evolved since the divergence of rodents and primates from their common ancestor. In rodents, from E15 to P0 genes specific to the rodent placenta are expressed, and in the human placenta primate-specific genes are all enriched compared to the mouse [86].
This striking selection for high expression of evolutionarily new, species-specific genes, during mid-gestation with specificity increasing as gestation continues, is indicative of the progressive divergence of placental physiology during development. Concomitantly, the conservation of genomic imprinting between humans and mice may be dynamic through pregnancy. Imprinting can be developmentally regulated by epigenetic regulators that are tissue-specific. Germline methylation can therefore be “read” differently in different cell types and at different stages in development, resulting in, for example, the highly tissue-specific imprinting at the GNAS locus on Hsa20/Mmu2 [87]. Differential reading of the germline methylation mark could depend on the presence of tissue-specific transcription factors or epigenetic effectors such as polycomb group proteins. For example, allelic histone modifications in the Kcnq1 region are required to maintain imprinting of placental-specific imprinted genes in the mouse placenta and are able to do so without maintenance of differential methylation at the KvDMR, which is not the case for the genes imprinted in the embryo that still require an intact KvDMR [57], [58]. As placental physiology diverged throughout gestation, differences in developmentally regulated imprinting may also have evolved. It is possible that placental-specific imprinting seen in the mouse (Table 2) may be present in the human placenta, but at a much earlier gestation than has so far been analysed, before differentiation resulted in biallelic expression of these genes. Similarly, in later gestation in humans, genes not imprinted in the mouse may be imprinted in the human placenta.
Imprinting in the Postnatal Human
After birth, resource allocation is distinct from that during pregnancy, and the interaction between offspring and mother is vastly changed. The placenta, and its function to transfer nutrients from the maternal bloodstream and pass them on to the fetus, is no longer present, and the neonate has developed strategies to function ex utero, leading to full independence after weaning. Key organs for imprinted expression postnatally include the brain and endocrine tissues, such as brown adipose tissue, which regulates non-shivering thermogenesis, a pre-weaning postnatal adaptation to independent life [88]. Genes whose imprinted expression was previously vital in the placenta, may cease to be important in some tissues, exemplified by the biallelic expression of IGF2 in human adult liver [89].
It is likely that parental conflict in mammals therefore continues after birth, albeit in an altered fashion (Figure 3) [90]. During the period between weaning and independence of children from their parents, the father has an increased role given his position as “breadwinner” that may be an investment of higher magnitude than that of the mother in older children [91], [92]. Postnatally, some aspects of several imprinting syndromes seem incompatible with the conflict theory in its simplest form. For example, PWS results from a loss of paternally expressed transcripts, yet PWS children are characteristically large. This can be reconciled with the concept of resource allocation by focussing on behaviour. Genes imprinted in the PWS/AS region, which are highly expressed in the brain, may act postnatally to modify behaviour to maximise resources (Figure 3). Emotional and behavioural cues could be utilised by the neonate to manipulate parents in order to provide adequate nutrition. In AS, caused by loss of expression of maternally expressed UBE3A, children prolong suckling and exhibit convivial behaviour that maximises maternal input [93]. In PWS, resulting from the loss of paternally expressed HBII-85 snoRNAs, children suckle badly and wean quickly but are hyperphagic after birth, arguably maximising utilisation of paternal resources and minimising usage of maternal ones [94], [95]. So, conflict exists after birth, but its arena might be considered to have moved from the placenta to the brain [92]. Whether this facet of imprinting displays consistency between humans and mice remains to be seen. Mouse models with targeted deletions of the MBII-85 snoRNA cluster display characteristic PWS features of hypotonia and a failure to thrive, followed by hyperphagia [96], [97]. The mice do not become obese, indicating some species-specific differences in metabolism, however the behavioural parallels between between PWS and the MBII-85-deleted transgenic mouse indicate that some aspects of postnatal conflict may be managed similarly between the two species [96], [97]. Imprinting in the brain is conserved between mice and humans at the GRB10/Grb10 locus, where transcripts are paternally expressed in the central nervous system through similar tissue specific chromatin modifications [46], [98]–[100]. Grb10 is a growth inhibitor and is maternally expressed in most tissues in mouse [49]. In utero, Grb10 negatively regulates fetal and placental growth, whilst it is involved in glucose homeostasis in adult muscle and adipose tissue [101]. The function of Grb10 in brain and the purpose of its maternal suppression is unknown. The distinct mechanism of GRB10/Grb10 regulation observed in human and mouse brain [46], [100], and opposing allelic repression compared to other tissues, is suggestive of it having a distinct role in this tissue, perhaps in influencing postnatal behaviour in the father's favour [46]. Imprinting in the brain is a developing field, one that will provide new and exciting insights into human behaviour and the evolution of imprinting.

Summary
The biological function of reducing the diploid state to functional haploidy has to be questioned in terms of its evolutionary significance. A case needs to be made for the benefit of silencing of one parental allele balanced against the negative impact of a mutation at the remaining allele that would leave the cell with no gene product.
In humans, inappropriate expression of imprinted genes leads in many cases to severe syndromes. This shows that the monoallelic expression of this small subset of genes is indispensible for normal human development. Aberrant prenatal growth occurs frequently in imprinting syndromes. This shows that an important feature of imprinting is the regulation of growth and nutrient transfer between mother and fetus, for which the placenta is key. This regulation should be balanced to serve the interests of both parents equally.
There are several genes that are imprinted in mice but not in humans. This is suggestive of a difference in importance or function of these transcripts between these two species, possibly due to species-specific differences in their respective placental physiology. The lack of conservation in imprinted expression for some genes may also be linked to a reduction in conflict during human pregnancy compared to the mouse, as humans bear singletons rather than large litters, and so have little or no possibility of multiple paternity.
Whilst differences in the reproductive biology of mice and humans are evident, large distinctions in imprinting in organs unrelated to pregnancy have not yet been identified. Following birth, offspring are free from maternal growth constraints, no longer rely on the placenta and must now recruit input from both parents in order to maximise fitness. A resolution of parental conflict postnatally will therefore rely on specific behavioural and emotional cues, engaging organs such as the brain and endocrine axis.
Genomic imprinting in humans is clearly important. Analysis of imprinting disorders and information from closely related mammalian models allows us to define the importance of its conservation and the relevance of any absence of conservation. Through further focussed research into human imprinting, we will elucidate the specialised functions of this remarkable transcriptional mechanism in our species.
Zdroje
1. McGrathJ
SolterD
1984 Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37 179 183
2. SuraniMA
BartonSC
NorrisML
1984 Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308 548 550
3. CattanachBM
KirkM
1985 Differential activity of maternally and paternally derived chromosome regions in mice. Nature 315 496 498
4. NichollsRD
KnollJH
ButlerMG
KaramS
LalandeM
1989 Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 342 281 285
5. BarlowDP
StogerR
HerrmannBG
SaitoK
SchweiferN
1991 The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 349 84 87
6. BartolomeiMS
ZemelS
TilghmanSM
1991 Parental imprinting of the mouse H19 gene. Nature 351 153 155
7. DeChiaraTM
RobertsonEJ
EfstratiadisA
1991 Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64 849 859
8. Ferguson-SmithAC
SasakiH
CattanachBM
SuraniMA
1993 Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 362 751 755
9. HaigD
2000 The kinship theory of genomic imprinting. Annu Rev Ecol Syst 31 9 32
10. Ferguson-SmithAC
CattanachBM
BartonSC
BeecheyCV
SuraniMA
1991 Embryological and molecular investigations of parental imprinting on mouse chromosome 7. Nature 351 667 670
11. DeChiaraTM
EfstratiadisA
RobertsonEJ
1990 A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345 78 80
12. LeightonPA
IngramRS
EggenschwilerJ
EfstratiadisA
TilghmanSM
1995 Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 375 34 39
13. LauMM
StewartCE
LiuZ
BhattH
RotweinP
1994 Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev 8 2953 2963
14. WangZQ
FungMR
BarlowDP
WagnerEF
1994 Regulation of embryonic growth and lysosomal targeting by the imprinted Igf2/Mpr gene. Nature 372 464 467
15. LudwigT
EggenschwilerJ
FisherP
D'ErcoleAJ
DavenportML
1996 Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev Biol 177 517 535
16. Munier-LehmannH
MauxionF
HoflackB
1996 Function of the two mannose 6-phosphate receptors in lysosomal enzyme transport. Biochem Soc Trans 24 133 136
17. LiE
BeardC
JaenischR
1993 Role for DNA methylation in genomic imprinting. Nature 366 362 365
18. TremblayKD
SaamJR
IngramRS
TilghmanSM
BartolomeiMS
1995 A paternal-specific methylation imprint marks the alleles of the mouse H19 gene. Nat Genet 9 407 413
19. ThorvaldsenJL
DuranKL
BartolomeiMS
1998 Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev 12 3693 3702
20. FrevelMA
SowerbySJ
PetersenGB
ReeveAE
1999 Methylation sequencing analysis refines the region of H19 epimutation in Wilms tumor. J Biol Chem 274 29331 29340
21. CuiH
NiemitzEL
RavenelJD
OnyangoP
BrandenburgSA
2001 Loss of imprinting of insulin-like growth factor-II in Wilms' tumor commonly involves altered methylation but not mutations of CTCF or its binding site. Cancer Res 61 4947 4950
22. TakaiD
GonzalesFA
TsaiYC
ThayerMJ
JonesPA
2001 Large scale mapping of methylcytosines in CTCF-binding sites in the human H19 promoter and aberrant hypomethylation in human bladder cancer. Hum Mol Genet 10 2619 2626
23. GicquelC
RossignolS
CabrolS
HouangM
SteunouV
2005 Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet 37 1003 1007
24. CooperWN
LuhariaA
EvansGA
RazaH
HaireAC
2005 Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur J Hum Genet 13 1025 1032
25. MonkD
ArnaudP
ApostolidouS
HillsFA
KelseyG
2006 Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci U S A 103 6623 6628
26. YotovaIY
VlatkovicIM
PaulerFM
WarczokKE
AmbrosPF
2008 Identification of the human homolog of the imprinted mouse Air non-coding RNA. Genomics 92 464 473
27. FowdenAL
Sferruzzi-PerriAN
CoanPM
ConstanciaM
BurtonGJ
2009 Placental efficiency and adaptation: Endocrine regulation. J Physiol 587 3459 3472
28. LunghiL
FerrettiME
MediciS
BiondiC
VesceF
2007 Control of human trophoblast function. Reprod Biol Endocrinol 5 6
29. BowenJM
ChamleyL
KeelanJA
MitchellMD
2002 Cytokines of the placenta and extra-placental membranes: roles and regulation during human pregnancy and parturition. Placenta 23 257 273
30. GudeNM
RobertsCT
KalionisB
KingRG
2004 Growth and function of the normal human placenta. Thromb Res 114 397 407
31. von RangoU
KruscheCA
KertschanskaS
AlferJ
KaufmannP
2003 Apoptosis of extravillous trophoblast cells limits the trophoblast invasion in uterine but not in tubal pregnancy during first trimester. Placenta 24 929 940
32. CoanPM
BurtonGJ
Ferguson-SmithAC
2005 Imprinted genes in the placenta–a review. Placenta 26 Suppl A S10 S20
33. DiplasAI
LambertiniL
LeeMJ
SperlingR
LeeYL
2009 Differential expression of imprinted genes in normal and IUGR human placentas. Epigenetics 4
34. PardiG
MarconiAM
CetinI
2002 Placental-fetal interrelationship in IUGR fetuses–a review. Placenta 23 Suppl A S136 S141
35. GravesJA
1998 Genomic imprinting, development and disease–is pre-eclampsia caused by a maternally imprinted gene? Reprod Fertil Dev 10 23 29
36. OudejansCB
MuldersJ
LachmeijerAM
vanDM
KonstAA
2004 The parent-of-origin effect of 10q22 in pre-eclamptic females coincides with two regions clustered for genes with down-regulated expression in androgenetic placentas. Mol Hum Reprod 10 589 598
37. RomanelliV
BelinchonA
Campos-BarrosA
HeathKE
Garcia-MinaurS
2009 CDKN1C mutations in HELLP/preeclamptic mothers of Beckwith-Wiedemann Syndrome (BWS) patients. Placenta 30 551 554
38. TakahashiK
KobayashiT
KanayamaN
2000 p57(Kip2) regulates the proper development of labyrinthine and spongiotrophoblasts. Mol Hum Reprod 6 1019 1025
39. KanayamaN
TakahashiK
MatsuuraT
SugimuraM
KobayashiT
2002 Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Mol Hum Reprod 8 1129 1135
40. Iglesias-PlatasI
MonkD
JebbinkJ
BuimerM
BoerK
2007 STOX1 is not imprinted and is not likely to be involved in preeclampsia. Nat Genet 39 279 280
41. ApostolidouS
bu-AmeroS
O'DonoghueK
FrostJ
OlafsdottirO
2007 Elevated placental expression of the imprinted PHLDA2 gene is associated with low birth weight. J Mol Med 85 379 387
42. McMinnJ
WeiM
SchupfN
CusmaiJ
JohnsonEB
2006 Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta 27 540 549
43. MayerW
HembergerM
FrankHG
GrummerR
WinterhagerE
2000 Expression of the imprinted genes MEST/Mest in human and murine placenta suggests a role in angiogenesis. Dev Dyn 217 1 10
44. HannulaK
Lipsanen-NymanM
KontiokariT
KereJ
2001 A narrow segment of maternal uniparental disomy of chromosome 7q31-qter in Silver-Russell syndrome delimits a candidate gene region. Am J Hum Genet 68 247 253
45. LefebvreL
VivilleS
BartonSC
IshinoF
KeverneEB
1998 Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet 20 163 169
46. MonkD
ArnaudP
FrostJ
HillsFA
StanierP
2009 Reciprocal imprinting of human GRB10 in placental trophoblast and brain: evolutionary conservation of reversed allelic expression. Hum Mol Genet
47. MonkD
WakelingEL
ProudV
HitchinsM
bu-AmeroSN
2000 Duplication of 7p11.2–p13, including GRB10, in Silver-Russell syndrome. Am J Hum Genet 66 36 46
48. Abu-AmeroS
MonkD
FrostJ
PreeceM
StanierP
2008 The genetic aetiology of Silver-Russell syndrome. J Med Genet 45 193 199
49. CharalambousM
SmithFM
BennettWR
CrewTE
MackenzieF
2003 Disruption of the imprinted Grb10 gene leads to disproportionate overgrowth by an Igf2-independent mechanism. Proc Natl Acad Sci U S A 100 8292 8297
50. CharalambousM
CowleyM
GeogheganF
SmithFM
RadfordEJ
2010 Maternally-inherited Grb10 reduces placental size and efficiency. Dev Biol 337 1 8
51. ConstanciaM
HembergerM
HughesJ
DeanW
Ferguson-SmithA
2002 Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 417 945 948
52. MonkD
SanchesR
ArnaudP
ApostolidouS
HillsFA
2006 Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum Mol Genet 15 1259 1269
53. SibleyCP
CoanPM
Ferguson-SmithAC
DeanW
HughesJ
2004 Placental-specific insulin-like growth factor 2 (Igf2) regulates the diffusional exchange characteristics of the mouse placenta. Proc Natl Acad Sci U S A 101 8204 8208
54. ConstanciaM
AngioliniE
SandoviciI
SmithP
SmithR
2005 Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc Natl Acad Sci U S A 102 19219 19224
55. FrankD
FortinoW
ClarkL
MusaloR
WangW
2002 Placental overgrowth in mice lacking the imprinted gene Ipl. Proc Natl Acad Sci U S A 99 7490 7495
56. TunsterSJ
TyckoB
JohnRM
2010 The imprinted Phlda2 gene regulates extraembryonic energy stores. Mol Cell Biol 30 295 306
57. LewisA
MitsuyaK
UmlaufD
SmithP
DeanW
2004 Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 36 1291 1295
58. UmlaufD
GotoY
CaoR
CerqueiraF
WagschalA
2004 Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 36 1296 1300
59. ArnoldDR
LefebvreR
SmithLC
2006 Characterization of the placenta specific bovine mammalian achaete scute-like homologue 2 (Mash2) gene. Placenta 27 1124 1131
60. GuillemotF
CasparyT
TilghmanSM
CopelandNG
GilbertDJ
1995 Genomic imprinting of Mash2, a mouse gene required for trophoblast development. Nat Genet 9 235 242
61. YanY
FrisenJ
LeeMH
MassagueJ
BarbacidM
1997 Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 11 973 983
62. HatadaI
OhashiH
FukushimaY
KanekoY
InoueM
1996 An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet 14 171 173
63. EngelJR
SmallwoodA
HarperA
HigginsMJ
OshimuraM
2000 Epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. J Med Genet 37 921 926
64. WangQ
CurranME
SplawskiI
BurnTC
MillhollandJM
1996 Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12 17 23
65. LeeMP
HuRJ
JohnsonLA
FeinbergAP
1997 Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat Genet 15 181 185
66. MitsuyaK
MeguroM
LeeMP
KatohM
SchulzTC
1999 LIT1, an imprinted antisense RNA in the human KvLQT1 locus identified by screening for differentially expressed transcripts using monochromosomal hybrids. Hum Mol Genet 8 1209 1217
67. FitzpatrickGV
SolowayPD
HigginsMJ
2002 Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet 32 426 431
68. Mancini-DiNardoD
SteeleSJ
LevorseJM
IngramRS
TilghmanSM
2006 Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 20 1268 1282
69. ShinJY
FitzpatrickGV
HigginsMJ
2008 Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. Embo J 27 168 178
70. GreenK
LewisA
DawsonC
DeanW
ReinhartB
2007 A developmental window of opportunity for imprinted gene silencing mediated by DNA methylation and the Kcnq1ot1 noncoding RNA. Mamm Genome 18 32 42
71. KanedaM
OkanoM
HataK
SadoT
TsujimotoN
2004 Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429 900 903
72. TerranovaR
YokobayashiS
StadlerMB
OtteAP
vanLM
2008 Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell 15 668 679
73. RedrupL
BrancoMR
PerdeauxER
KruegerC
LewisA
2009 The long noncoding RNA Kcnq1ot1 organises a lineage-specific nuclear domain for epigenetic gene silencing. Development 136 525 530
74. CarterAM
2001 Evolution of the placenta and fetal membranes seen in the light of molecular phylogenetics. Placenta 22 800 807
75. CarterAM
2007 Animal models of human placentation–a review. Placenta 28 Suppl A S41 S47
76. EndersAC
2009 Reasons for diversity of placental structure. Placenta 30 Suppl A S15 S18
77. CoanPM
ConroyN
BurtonGJ
Ferguson-SmithAC
2006 Origin and characteristics of glycogen cells in the developing murine placenta. Dev Dyn 235 3280 3294
78. GeorgiadesP
WatkinsM
BurtonGJ
Ferguson-SmithAC
2001 Roles for genomic imprinting and the zygotic genome in placental development. Proc Natl Acad Sci U S A 98 4522 4527
79. PringleKG
RobertsCT
2007 New light on early post-implantation pregnancy in the mouse: roles for insulin-like growth factor-II (IGF-II)? Placenta 28 286 297
80. BakerRJ
MakovaKD
ChesserRK
1999 Microsatellites indicate a high frequency of multiple paternity in Apodemus (Rodentia). Mol Ecol 8 107 111
81. DeanMD
ArdlieKG
NachmanMW
2006 The frequency of multiple paternity suggests that sperm competition is common in house mice (Mus domesticus). Mol Ecol 15 4141 4151
82. HaigD
1999 Multiple paternity and genomic imprinting. Genetics 151 1229 1231
83. GuillemotF
NagyA
AuerbachA
RossantJ
JoynerAL
1994 Essential role of Mash-2 in extraembryonic development. Nature 371 333 336
84. MiyamotoT
HasuikeS
JinnoY
SoejimaH
YunK
2002 The human ASCL2 gene escaping genomic imprinting and its expression pattern. J Assist Reprod Genet 19 240 244
85. ThurstonA
TaylorJ
GardnerJ
SinclairKD
YoungLE
2008 Monoallelic expression of nine imprinted genes in the sheep embryo occurs after the blastocyst stage. Reproduction 135 29 40
86. KnoxK
BakerJC
2008 Genomic evolution of the placenta using co-option and duplication and divergence. Genome Res 18 695 705
87. LiuJ
YuS
LitmanD
ChenW
WeinsteinLS
2000 Identification of a methylation imprint mark within the mouse Gnas locus. Mol Cell Biol 20 5808 5817
88. PlaggeA
GordonE
DeanW
BoianiR
CintiS
2004 The imprinted signaling protein XL alpha s is required for postnatal adaptation to feeding. Nat Genet 36 818 826
89. DaviesSM
1994 Developmental regulation of genomic imprinting of the IGF2 gene in human liver. Cancer Res 54 2560 2562
90. MooreT
HaigD
1991 Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet 7 45 49
91. ColeTJ
PowerC
MooreGE
2008 Intergenerational obesity involves both the father and the mother. Am J Clin Nutr 87 1535 1536
92. UbedaF
2008 Evolution of genomic imprinting with biparental care: implications for Prader-Willi and Angelman syndromes. PLoS Biol 6 e208
93. BuntinxIM
HennekamRC
BrouwerOF
StroinkH
BeutenJ
1995 Clinical profile of Angelman syndrome at different ages. Am J Med Genet 56 176 183
94. HaigD
WhartonR
2003 Prader-Willi syndrome and the evolution of human childhood. Am J Hum Biol 15 320 329
95. SahooT
delGD
GermanJR
ShinawiM
PetersSU
2008 Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 40 719 721
96. SkryabinBV
GubarLV
SeegerB
PfeifferJ
HandelS
2007 Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet 3 e235
97. DingF
LiHH
ZhangS
SolomonNM
CamperSA
2008 SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 3 e1709
98. BlagitkoN
MergenthalerS
SchulzU
WollmannHA
CraigenW
2000 Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue - and isoform-specific fashion. Hum Mol Genet 9 1587 1595
99. HitchinsMP
MonkD
BellGM
AliZ
PreeceMA
2001 Maternal repression of the human GRB10 gene in the developing central nervous system; evaluation of the role for GRB10 in Silver-Russell syndrome. Eur J Hum Genet 9 82 90
100. ArnaudP
MonkD
HitchinsM
GordonE
DeanW
2003 Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Hum Mol Genet 12 1005 1019
101. SmithFM
HoltLJ
GarfieldAS
CharalambousM
KoumanovF
2007 Mice with a disruption of the imprinted Grb10 gene exhibit altered body composition, glucose homeostasis, and insulin signaling during postnatal life. Mol Cell Biol 27 5871 5886
102. da RochaST
CharalambousM
LinSP
GutteridgeI
ItoY
2009 Gene dosage effects of the imprinted delta-like homologue 1 (dlk1/pref1) in development: implications for the evolution of imprinting. PLoS Genet 5 e1000392
103. MoonYS
SmasCM
LeeK
VillenaJA
KimKH
2002 Mice lacking paternally expressed Pref-1/Dlk1 display growth retardation and accelerated adiposity. Mol Cell Biol 22 5585 5592
104. ReikW
LewisA
2005 Co-evolution of X-chromosome inactivation and imprinting in mammals. Nat Rev Genet 6 403 410
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- CHD7 Targets Active Gene Enhancer Elements to Modulate ES Cell-Specific Gene Expression
- Extensive DNA End Processing by Exo1 and Sgs1 Inhibits Break-Induced Replication
- Question and Answer: An Anniversary Interview with Jane Gitschier
- Multi-Variant Pathway Association Analysis Reveals the Importance of Genetic Determinants of Estrogen Metabolism in Breast and Endometrial Cancer Susceptibility
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy