
Intersticiální plicní onemocnění asociovaná s kouřením
Interstitial lung diseases associated with smoking
There are many different interstitial lung diseases associated with smoking. This short review describes officially recognized disorders (desquamative interstitial pneumonia, respiratory bronchiolitis and pulmonary Langerhans´cells histiocytosis) and entities with uncertain relationship to smoking, which have recently been published in the literature. Histopathological pictures and differential diagnosis of smoking-related diseases of the lungs are discussed.
Keywords:
emphysema – pneumonia – bronchiolitis – smoking – fibrosis – histiocytosis
Autoři:
Markéta Nová 1; Helena Hornychová 1; Radoslav Matěj 2,3
Působiště autorů:
Fingerlandův ústav patologie, Fakultní nemocnice a Lékařská fakulta Karlovy univerzity, Hradec Králové
1; Oddělení patologie a molekulární medicíny, Thomayerova nemocnice, Praha
2; Ústav patologie 1. LF UK a VFN, Praha
3
Vyšlo v časopise:
Čes.-slov. Patol., 52, 2016, No. 2, p. 100-105
Kategorie:
Přehledové články - Interscitiální plicní procesy
Souhrn
Intersticiálních plicních onemocnění spojených s kouřením je celá řada. V přehledném sdělení jsou popsány oficiálně uznané jednotky (deskvamativní pneumonie, bronchiolitida respiračních bronchiolů a plicní histiocytóza z Langerhansových buněk) i jednotky s diskutabilním zařazením, se kterými se lze v současné literatuře setkat. Největší pozornost je věnována zvláště morfologickému obrazu jednotlivých onemocnění a diferenciálně diagnostické rozvaze.
Klíčová slova:
emfyzém – pneumonie – bronchiolitida – kouření – fibróza – histiocytóza
Plicní patologie je velmi dynamický obor, v němž stále vznikají nové jednotky a subjednotky, často s komplikovanými a opravdu dlouhými jmény. Pro pulmopatology je tedy s výhodou používat zkratky, což je pro nespecialisty povětšinou relativně neatraktivní. Nejinak je tomu i v případě intersticiálních plicních onemocnění asociovaných s kouřením (SR-ILD). Vzhledem k tomu, že se zde zkratky a specializované pojmy užívají běžně a protože se jedná o vzácnější jednotky, s nimiž se i v běžné pulmopatologii nesetkáváme až tak často, uvádíme seznam užitých zkratek a pojmů místo tradičního konce na úplný začátek:
- kuřácký makrofág: alveolární makrofág s jemně eozinofilní až „špinavě hnědou“ cytoplazmou, lze vidět světle hnědý pigment v drobných granulích, který je pouze slabě pozitivní v barvení na železo (detail viz obr. 3)
- CPFE – kombinovaná plicní fibróza s emfyzémem
- DIP – deskvamativní intersticiální pneumonie
- GGO – ground glass opacity – jemné zastření plicního parenchymu na rentgenovém snímku přirovnávané k mléčnému sklu
- ILD – intersticiální plicní onemocnění
- IPF – idiopatická plicní fibróza
- NSIP – nespecifická intersticiální pneumonie
- PLCH – plicní histiocytóza z Langerhansových buněk
- RB – bronchiolitida respiračních bronchiolů
- SRIF – intersticiální fibróza spojená s kouřením
- SR-ILD - intersticiální plicní onemocnění asociované s kouřením
- UIP – obvyklá intersticiální pneumonie.
Negativní vliv kouření na lidský organismus, zvláště plíce, je všeobecně znám. Etiopatogeneza plicních nenádorových chorob není ještě zcela do detailů prozkoumaná, nicméně se obecně přijímá teorie významné účasti oxidativního stresu, kdy dochází k aktivaci a poškození makrofágů s následným spuštěním zánětlivé kaskády – což vede k uvolnění metaloproteináz, které jsou významným činitelem při destrukci plicního parenchymu. Kromě makrofágů je poškozován i epitel (zvýšená apoptóza) a aktivován je imunitní systém. Působení nikotinu a zplodin vzniklých při kouření na fibrogenezu je víceúrovňové – poškozují epitel, spouští produkci zánětlivých faktorů, ovlivňují makrofágy a fibroblasty, aktivují TGF beta. U pacientů s SR-ILD je signifikantně zvýšená hladina osteopontinu, látky, která ovlivňuje chemotaxi makrofágů, monocytů, dendritických buněk a Langerhansových histiocytů (1,2). Vliv kouření je jistě multifaktoriální a vzhledem k tomu, že projevy jsou u kuřáků různé (bez ohledu na věk, počet balíčkoroků, rodinnou anamnézu a pod), hraje velmi pravděpodobně významnou roli i genetická predispozice (2).
Onemocnění plic s prokázaným etiopatogenetickým vlivem kouření dělíme na nádorová a nenádorová.
V nenádorových onemocněních spojených s kouřením se vyčleňují dvě hlavní skupiny:
- chronická obstrukční bronchopulmonální choroba – CHOPN
- intersticiální plicní choroba spojená s kouřením – SR-ILD
SR-ILD se dále rozpadá do subtypů, které jsou svými autory považované za jasně definované a od sebe oddělené jednotky.
Nicméně oficiálně uznané jednotky dle hlavních světových pneumologických odborných společností - Americká hrudní společnost, Evropská respirační společnost, Japonská respirační společnost a Hrudní asociace Latinské Ameriky (ETS / ERSJRS / ALAT) z roku 2013 jsou zatím pouze dvě (3). Jedná se o bronchiolitidu respiračních bronchiolů asociovanou s intersticiálním plicním onemocněním (RB nebo RB-ILD) a deskvamativní intersticiální pneumonii (DIP). V roce 2014 byl publikován článek v European Respiratory Journal, oficiálním odborném časopisu Evropské respirační společnosti, kde je vztah mezi onemocněními plicního intersticia a kouřením ne zcela jednoznačně specifický a autoři konstatují, že pro zpřesnění tohoto vztahu bude potřeba ještě více objektivních pozorování (4).
V literatuře se však, kromě RB a DIP, setkáme s dalšími jednotkami ILD a tezemi, které daná onemocnění dávají více či méně do příčinné souvislosti s kouřením.
Vassallo a kolektiv rozdělili intersticiální plicní onemocnění ve spojitosti s kouřením do 4 skupin (tab.1) (5). V první skupině jsou onemocnění, jejichž kuřácká etiologie je jasně prokázána. V druhé skupině jsou akutní ILD, kde se zdá, že kouření hraje významnou roli, ovšem ne tak jednoznačnou v etiopatogenezi jako v první skupině. Třetí skupinu tvoří ILD, která se vyskytují významně častěji u kuřáků, nicméně etiologicky je kouření spíše podružné. Tyto jednotky však významně interferují v rámci histologické diagnostiky se skupinou první! Čtvrtá skupina obsahuje ILD, která se vyskytují u kuřáků méně často než u nekuřáků. Zde je diskutován „protektivní“ vliv kouření (nesmíme zapomenout, že protekce je tvrdě zaplacená nežádoucími účinky kouření!), zřejmě přes ovlivnění imunitní odpovědi – a to supresí T helperů. Na druhou stranu, pokud už kuřák např. sarkoidózu rozvine, je terapeutická odpověď mnohem horší než u nekuřáků (6).

SR-ILD jsou dle literárních údajů poměrně vzácná onemocnění, není přesně známa ani prevalence, ani incidence (7,8). Je však otázkou, jedná-li se opravdu o onemocnění vzácná, či jsou pouze významně poddiagnostikovaná.
ILD spojené s kouřením jsou klinicko-radiologicko-histopatologicky jasně definované jednotky, často je však jejich průběh a typický obraz modifikovaný léčbou, dalšími plicními i neplicními chorobami. Některé jednotky mohou v sebe přecházet či se kombinovat. Jejich jednoznačné zařazení je tedy možné pouze na základě velmi dobré, často i dlouhodobé a opakované multidisciplinární spolupráce. A přes veškerou snahu může nakonec histologické vyšetření skončit popisem s uvedením široké diferenciální rozvahy. Histopatologické zařazení nálezu do určité jednotky/jednotek dále limituje výtěžnost materiálu – reprezentativní množství tkáně. Zde platí stejné pravidlo jako pro diagnostiku všech nenádorových plicních onemocnění – reprezentativní vzorek by měl obsahovat celou respirační jednotku (čím víc jednotek tím lépe), tzn. periferii včetně pleury, bronchiolus a přilehlé alveoly. Optimální, a téměř vždy nutné, je vyšetření vzorků z radiologicky postižené a nepostižené plicní tkáně. Stěžejní význam histopatologické diagnostiky nespočívá ani tak v určení jednotlivých jednotek v rámci SR-ILD, ale v rozlišení jednotlivých fibrotizujících procesů, které mají zcela odlišnou prognózu a terapii – tzn. odlišení SR ILD od NSIP a UIP/IPF.
Intersticiální plicní choroby silně spojené s kouřením:
- RB, RB-ILD – bronchiolitida respiračního bronchiolu
- DIP – deskvamativní intersticiální pneumonie
- SRIF – intersticiální fibróza asociovaná s kouřením
- PLCH – plicní histiocytóza z Langerhansových buněk
- CPFE – kombinovavaná plicní fibróza a emfyzém
RB a DIP (a k tomu SRIF) často představují spojité spektrum téhož poškození plíce při kouření, v rámci vyšetření plicní tkáně jsou tedy obrazy těchto jednotek přítomny, avšak v různě vyjádřené míře (2). Dle dominantního projevu (klinického, radiologického, patologického) se řídí léčba, která je odlišná. U části pacientů můžeme najít navíc i PLCH.
RB, RB-ILD – BRONCHIOLITIDA RESPIRAČNÍHO BRONCHIOLU
Onemocnění je histologicky definováno jako kumulace kuřáckých makrofágů v bronchiolárním a peribronchiolárním prostoru – tzn. oblast respiračního a terminálního bronchiolu a přilehlých alveolů (obr. 1). Změny jsou ložiskové, nikoli difúzní.

Další histologické nálezy zahrnují možnou mírnou převážně chronickou zánětlivou infiltraci (včetně možných ojedinělých eozinofilů), nevýraznou peribronchiální fibrotizaci a bronchiální metaplázii v postižené oblasti. Nikdy nedochází k destrukci plicního parenchymu. Nález je přetrvávající u aktivních i pasivních kuřáků i u dlouholetých exkuřáků.
Klinické projevy odlišují RB (nález je iniciální, pacient nemá klinické obtíže, ani abnormální nález na zobrazovacích metodách) od RB-ILD (histologicky stejné jako u RB), pacient je v tomto případě symptomatický (dušnost, kašel, abnormální spirometrie) a vykazuje změny plicního parenchymu na zobrazovacích metodách.
Prognóza je velmi dobrá, bez známého progresivního chování (2).
Diferenciální rozvaha:
- Fibrotizace a zánět – obraz NSIP like, v malých vzorcích není snadné odlišit od fibrózní NSIP (fNSIP), zvláště ve vzorcích z periferních oblastí nebo ve vzorcích, kde není zřetelná kumulace kuřáckých makrofágů. U fNSIP by mělo být postižení plicního parenchymu difúzní (na rozdíl od kuřáckých změn, které jsou broncho - a bronchiolocentricky vázané), bývá i jiný a často závažnější klinický a radiologický nález.
Při kombinaci zánětu a fibrotizace by mělo být cíleně pátráno i po granulomech s cílem vyloučit nejen specifickou infekční etiologii či sarkoidózu, ale především chronickou hypersenzitivní pneumonii (HP). Granulomatózní formace u HP mohou být zcela diskrétní, s výhodou je užít imunohistochemický průkaz CD68, nicméně v malých vzorcích či v pokročilých fázích nemusí být granulomy přítomny – zde je nutná pečlivá klinicko-radiologicko histopatologická korelace s důrazem na charakteristickou distribuci změn, expoziční data a výsledky alergologického a imunologického vyšetření.
- Kumulace makrofágů – vždy provést barvení na železo. Pokud jsou patrna hrubá silně pozitivní granula, je nutné zvážit možnost alveolární hemoragie - často je hemosiderin i v přítomném vazivu či plicním intersticiu. Vhodné je provést barvení na elastická vlákna a vyloučit tak možnost vaskulitidy. Při difúzní kumulaci makrofágů v celé respirační jednotce je nutné zvážit možnost DIP.
DIP – DESKVAMATIVNÍ INTERSTICIÁLNÍ PNEUMONIE
Onemocnění je histologicky definováno jako kumulace kuřáckých makrofágů v alveolárních prostorech – difúzně nebo plošně (obr. 2,3). Další histologické nálezy zahrnují lehkou chronickou zánětlivou infiltraci (opět včetně nečetných eozinofilů), nevýraznou intersticiální fibrotizaci. Ani do obrazu DIP nepatří destrukce plicního parenchymu.


Klinické projevy jsou nekonstantní, zpravidla je přítomna zhoršující se dušnost, kašel, někdy systémové projevy – např. teploty. Je patrné zhoršení plicních funkcí. 90 % DIP se vyskytuje u kuřáků, nález však může být asociovaný s autoimunitními chorobami, léčbou, pneumokoniózami apod., někdy je obraz DIP idiopatický, vzácně může být asociován s genetickými defekty surfaktantových proteinů (9).
Radiologický nález odpovídá GGO, zvláště v subpleurálních lokacích a bazálně, někdy lze pozorovat i známky fibrotizace.
Prognóza je celkově velmi dobrá při ukončení kouření, bez léčby a bez přerušení nikotinismu dochází k progresi projevů včetně postupné fibrotizace, dobrá je reakce na imunosupresivní terapii.
Diferenciálně diagnostická rozvaha se odvíjí od dílčích histologických projevů:
- Fibrotizace – pokud je difúzní, tak je třeba zvažovat možnost fNSIP či HP (dif.dg. viz výše), pokud je ložisková s destrukcí parenchymu (voština), tak je třeba do diferenciální rozvahy zahrnout DIP like UIP.
- Makrofágy – vždy provést barvení na železo (a viz výše – v případě silné pozitivity zvážit alveolární hemoragii).
- Jaderné atypie makrofágů – vyloučit alveolární infiltraci disociovaně rostoucími tumory.
- Vícejaderné makrofágy – projev pneumokoniózy z tvrdých kovů (obrovskobuněčná intersticiální pneumonitida)
- Tzv. „DIP-like“ reakce - velmi často v okolí tumoru, u zánětů, po inhalaci toxických látek, poléková reakce. Zvláště při hodnoceních malých vzorků je nutná pečlivá klinicko patologická spolupráce, diagnóza DIP v malém vzorku je víceméně per exclusionem!
SRIF - INTERSTICIÁLNÍ FIBRÓZA ASOCIOVANÁ S KOUŘENÍM
SRIF byla prvně popsána Katzensteinovou a kol. v roce 2010 jako časté postižení plicního intersticia v plicních lobektomiích u chronických kuřáků (10). Přestože je SRIF velmi častá u kuřáků, studie Kawabaty dokonce popisuje přítomnost změn kompatibilních se SRIF u 6,5 % lehkých a u 21,1 % těžkých kuřáků, klinicky není příliš často diagnostikována. Pravděpodobně přítomnost významného emfyzému maskuje jen mírný stupeň restrikční ventilační poruchy a onemocnění je pak klinicky zahrnuto do syndromu chronické obstrukční plicní nemoci (CHOPN). Navíc funkční význam těchto změn není zcela jasný, protože progrese do klinicky významné plicní fibrózy nebyla v původní práci zaznamenána u žádného sledovaného pacienta, samozřejmě pouze v omezeném časovém intervalu od diagnózy (11).
Onemocnění je histologicky definováno jako uniformní hypocelulární septální fibróza s různým stupněm rozšíření alveolárních sept depozity kolagenu. Fibrotizace je přítomná zejména subpleurálně, kde je asociována s okolním emfyzémem, je však přítomná i hlouběji v parenchymu v centrolobulárních oblastech. Emfyzém je přítomen ve všech případech, často jde o nejnápadnější postižení plicního parenchymu, ale fibróza muže být i v plicním parenchymu s diskrétními emfyzematózními změnami. Zánětlivá infiltrace je minimální (obr. 4). Další histologické nálezy zahrnují změny charakteru RB, DIP či PLCH. V depozitech kolagenu mohou být svazky hladké svaloviny (12).

Klinické projevy jsou typicky dušnost a kašel. Nález je stacionární, na rozdíl od NSIP či UIP/IPF.
Radiologický nález popisuje difúzní nodularity a GGO.
Prognóza je nejistá, někteří autoři tvrdí, že stejná jako pro RB ILD, DIP a emfyzém. Je důležité sledovat, jaká je vlastně klinická relevance těchto fibrotizujících změn u kuřáků z hlediska dlouhodobé prognózy, jde-li o prekurzorovou lézi klinicky závažné plicní fibrózy, nebo se jedná o zcela samostatnou entitu.
Diferenciálně diagnostická rozvaha:
- Fibrózní NSIP – hlavní histologický rozlišovací znak je u SRIF typický vzhled kolagenu, přítomnost emfyzému a nepřítomnost zánětu, SRIF je ložiskový proces na rozdíl od NSIP.
- UIP – SRIF má morfologicky jiný typ vaziva, chybí voština (i když fibrotizace kolem emfyzematózních prostorů může voštinu napodobovat!) a nejsou přítomny fibroblastické fokusy. V akutní fázi SRIF se však lze s ojedinělými fibroblastickými fokusy setkat.Klinicky je SRIF stacionární stav, na rozdíl od UIP/IPF a NSIP. Bohužel část pacientů s UIP/IPF a NSIP jsou zároveň i (ex)kuřáci a pak mohou mít v histologickém vzorku, v závislosti na reprezentativnosti odběru, navíc i obraz SRIF (nebo pouze SRIF).
- CPFE – kombinovaná plicní fibróza a emfyzém (viz níže). Fibróza u SRIF má odlišný charakter, nejsou přítomny fibroblastické fokusy ani voština. V části plic jsou možné oba nálezy, tzn. že zvláště v malém vzorku je odlišení CPFE od SRIF téměř nemožné. Podstatná je korelace se zobrazovacími metodami a klinickým nálezem.
PLCH – PLICNÍ HISTIOCYTÓZA Z LANGERHANSOVÝCH BUNĚK
Plicní histiocytóza z Langerhansových buněk je klinicky zcela odlišné onemocnění než je systémová histiocytóza X, která se u malých dětí projevuje jako akutní diseminované onemocnění, tzv. Abtova-Lettererova-Siweova nemoc, u větších dětí může onemocnění probíhat pod obrazem multifokálních lézí jako, tzv. Handův-Schüllerův-Christianův syndrom. V rámci systémového onemocnění mohou být vzácně postiženy i plíce, prognóza je spíše špatná. PLCH je onemocnění plicní (bez postižení dalších orgánů, jen výjimečně je spojována s postižením kostí a CNS), typicky se vyskytuje u mladších dospělých, kuřáků a jeho prognóza je povětšinou velmi dobrá (13,14).
Onemocnění je histologicky definováno jako granulomy postihující a destruující distální bronchioly. Granulomy jsou vágně formované, kromě velkého množství histiocytů Langerhansova typu („ledvinovité jádro“, CD1a, CD207 (langerin) a S100 pozitivní), jsou v granulomech i lymfocyty, makrofágy, ložiskově i četnější eozinofily (obr. 5,6). Ve starších granulomech ubývá zánětlivé infiltrace i histiocytů, centrálně lze v lézi najít prázdný prostor – dilatované reziduální lumen postiženého bronchu. Staré léze mají vzhled typické hvězdicovité jizvy, ve kterých jsou nečetné makrofágy s pigmentovanou cytoplazmou, histiocyty jsou ojedinělé nebo dokonce chybí. V okolí jsou vyvinuté trakční emfyzematózní prostory až buly. Splynutá starší ložiska mohou napodobovat i voštinu. V některých lézích mohou být v rámci granulomu zavzaty i peribronchiální tepny se změnami typu vaskulitidy – avšak sekundární.
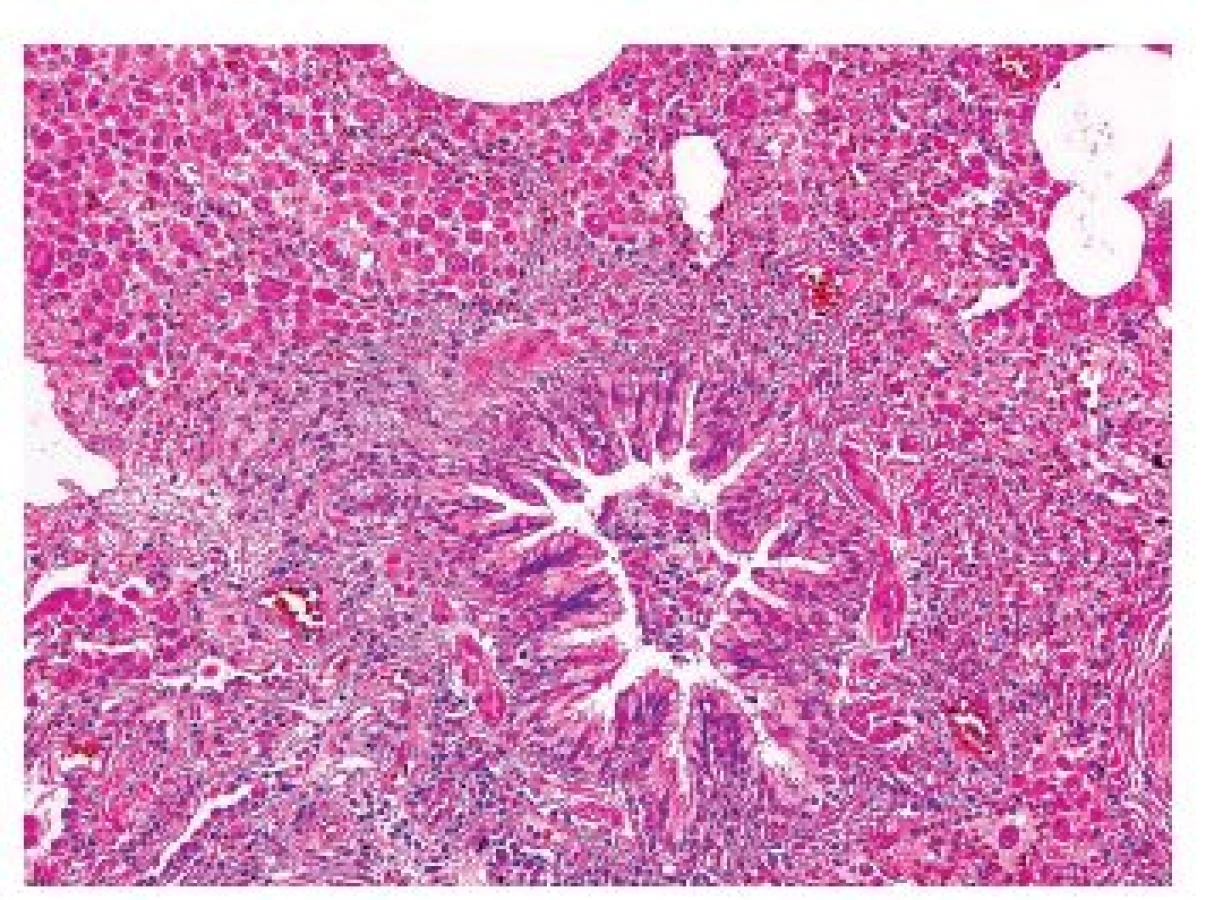

Další histologické nálezy zahrnují kombinace změn charakteru jiných onemocnění spojených s kouřením, velmi často nález charakteru RB, DIP, emfyzému a podobně.
Klinické projevy jsou odrazem chronického nikotinismu – lehká dušnost, kašel. Pacienti jsou typicky mladí kuřáci okolo 30. roku. Pacienti mohou být i zcela asymptomatičtí (i přes nález na zobrazovacích metodách), někdy může dojít i ke spontánní regresi nálezu. Typicky jsou klinické projevy (malé) v kontrastu s výrazným nálezem na zobrazovacích metodách. Bohužel 10 – 20 % pacientů má závažnou formu nemoci s rozsáhlým plicním postižením a recidivujícími pneumotoraxy a s tím souvisejícím respiračním selháním. Zbytek nemocných má perzistující formu, obvykle s cystami v plicní tkáni, avšak nemoc neprogreduje a nemocní nemají významné funkční omezení.
Radiologický nález je různý v závislosti na stádiu onemocnění, typicky je přítomna kombinace nodulů s centrálním lumen i bez něj, tenko i silnostěnných cyst (emfyzematózní buly), více postiženy jsou typicky horní laloky.
Prognóza je obecně velmi dobrá při přerušení kouření, není však známa kurabilní terapie. Část pacientů profituje z léčby kortikoidy, část pacientů vykáže spontánní regresi i bez léčby. Malá část pacientů má komplikovaný průběh s opakovanými pneumotoraxy a rozvojem cor pulmonale (14).
Diferenciálně diagnostická rozvaha závisí na vzhledu ložiska:
- Časné granulomy – v případě výraznějšího zánětu a četnějších eosinofilů je třeba vyloučit zvláště infekční původ granulomů (aspergilus), aspiraci a podobně. Jednoznačný je imunohistochemický průkaz Langerhansových histiocytů.
- Staré granulomy – vzhledu fibrózy. Zvláště v malých vzorcích není struktura granulomu patrná, často nejsou přítomny ani typické histiocyty. I bez přítomnosti histiocytů je vhodné PLCH do rozvahy zahrnout - zde je nutná korelace s klinickým nálezem (RTG obraz je poměrně specifický a histologická diagnostika není obvykle potřebná).
Obecně je vždy vhodné při anamnéze či histologických změnách spojených s kouřením (kuřácké makrofágy) a přítomnosti fibrózních fokusů (bez ohledu na celularitu) doplnit imunohistochemické vyšetření na průkaz Langerhansových histiocytů.
CPFE – KOMBINOVANÁ PLICNÍ FIBRÓZA A EMFYZÉM
CPFE je relativně nově popsaná jednotka. Jedná se o kombinaci fibrotizujícího intersticiálního plicního onemocnění nejčastěji typu UIP-IPF s emfyzémem. Cottin dokazuje, že se jedná o samostatnou jednotku, nikoli o dvě chorobné změny v rámci jednoho pacienta (16). Tento názor však část odborníků rozporuje s tím, že emfyzém u kuřáků (centrolobulární a paraseptální) je téměř vždy s fokální fibrotizací spojen a Cottinem popisované fibrózní partie v emfyzému považují za běžnou kuřáckou změnu. Vzhledem k těmto diskuzím se tedy v literatuře můžeme setkat s dalším pojmem – AEP – alveolar enlargement with fibrosis (17), což je emfyzém spojený s fibrotizací intersticia, neměl by však být přítomen obraz UIP.
CPFE je onemocnění histologicky definováno jako kombinace morfologických projevů fibrotizujícího intersticiálního procesu obrazu UIP nebo NSIP a emfyzému (obr. 7). Další histologické nálezy zahrnují histologické obrazy spojené s kouřením RB, DIP, PLCH, SRIF.

Klinické projevy jsou obdobné jako u IPF. Jedná se o starší pacienty, kteří mají v anamnéze nikotinismus, obvyklá je velmi rychle progredující dušnost, kašel, rozvoj těžké plicní hypertenze a respiračního selhání. Těžká plicní hypertenze je odpovědná za velmi špatnou léčebnou odpověď a určuje prognózu.
Radiologický nález je v rozvinutých fázích onemocnění typický - v dolních plicních lalocích je fibróza s obrazem voštiny (UIP pattern) nebo bez voštiny (NSIP pattern), predominantně v horních lalocích emfyzém.
Prognóza je velmi špatná, odpovídající IPF a to včetně antifibrotických novinek v léčbě (pirferidon, nintedanib). Roční mortalita je stále 60 % (16).
ZÁVĚR
Intersticiální plicní onemocnění spojená s kouřením tvoří skupinu klinicko-radiologicko-histopatologicky definovaných jednotek, jejichž průběh a typický obraz je však často modifikovaný léčbou a dalšími faktory. Navíc některé jednotky v sebe mohou přecházet či se různě kombinovat. Pro jednoznačné zařazení jednotlivých SR-ILD je bezpodmínečně nutná, tak jako v případě ostatních onemocnění plicního intersticia, dlouhodobá intenzivní multidisciplinární spolupráce pneumologa, radiodiagnostika a patologa. Avšak vzhledem k recentně publikovaným nejednoznačnostem o vztahu některých SR-ILD k délce a intenzitě kouření bude třeba ještě dalších podrobných pozorování k definitivnímu vyjasnění celého spektra působení kouření na plicní intersticium.
PODĚKOVÁNÍ
Poděkování patří našim kolegům (plicní kliniky, radiodiagnostická pracoviště, oddělení hrudní chirurgie) a hlavně kuřákům, bez nichž by byla patologie o tolik ochuzena. Práce vznikla za částečné podpory PRVOUK P37/11 a P27/LF1/1, OPPK CZ.2.16/3.1.00/24509 a BBMRI LM2010004.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
MUDr. Markéta Nová
Fingerlandův ústav patologie
Fakultní nemocnice Hradec Králové
Sokolská 581,
500 05 Hradec Králové
tel.: 495832238
e-mail: fup.marketa@gmail.com
Zdroje
1. Prasse A, Stahl M, Schulz G et al. Essential role of osteopontin in smoking-related interstitial lung diseases. Am J Pathol 2009; 174 : 1683–1691.
2. Hasleton P, Flieder DB. Spencer‘s pathology of the lung (6th edn). Cambridge University Press: Cambridge; 2013 : 385-390.
3. Travis WD, Costabel U, Hansell DM et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2013; 188 : 733–748.
4. Flaherty K, Fell Ch, Aubry M et al. Smoking-related idiopathic interstitial pneumonia. Eur Respir J 2014; 44 : 594–602.
5. Vassallo R, Ryu JH. Smoking-related interstitial lung diseases. Clin Chest Med 2012; 33 : 165–178.
6. Margaritopoulos G, Vasarmidi E, Jacob J et al. Smoking and interstitial lung diseases. Eur Respir Rev 2015; 24 : 428–435.
7. Jankowich MD, Rounds SI. Combined Pulmonary Fibrosis and Emphysema Syndrome: a review. Chest 2012; Jan 141(1): 222-231.
8. Hagmeyer L, Randerath W. Smoking-related interstitial lung disease. Dtsch Arztebl Int 2015; 112 : 43–50.
9. Bruder E, Hofmeister J, Aslanidis C et al. Ultrastructural and molecular analysis in fatal neonatal interstitial pneumonia caused by a novel ABCA3 mutation. Modern Pathology 2007; 20 : 1009–1018.
10. Katzenstein AL, Mukhopadhyay S, Zanardi C et al. Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum Pathol 2010; 41(3): 316-325.
11. Kawabata Y, Hoshi E, Murai K et al. Smoking-related changes in the background lung of specimens resected for lung cancer: a semiquantitative study with correlation to postoperative course. Histopathology 2008; 53 : 707-714.
12. Katzenstein A. Smoking-related interstitial fibrosis (SRIF), pathogenesis and treatment of usual interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Modern Pathology 2012; 25 : 68–78.
13. Tazi A. Adult pulmonary Langerhans‘ cell histiocytosis. Eur Respir J 2006; 27 : 1272-1285.
14. Suri HS, Yi ES, Nowakowski GS et al. Pulmonary langerhans cell histiocytosis.Orphanet Journal of Rare Diseases 2012; 7 : 16.
15. Cottin V, Nunes H, Brillet PY et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 2005; 26(4): 586-593.
16. Cottin V, Le Pavec J, Prévot G et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J 2010; 35 : 105–111.
17. Wright J, Tazelaar H, Churg A. Fibrosis with emphysema. Histopathology 2011; 58 : 517–524.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2016 Číslo 2
Nejčtenější v tomto čísle
- Diferenciální diagnostika granulomatózních procesů v plicích
- Intersticiální plicní onemocnění asociovaná s kouřením
- Idiopatická plicní fibróza - problematika multidisciplinární diagnostiky a léčby ve světle nových poznatků
- Histopatologické principy vyšetření intersticiálních plicních procesů
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy