
Amyloidózy kůže
Cutaneous Amyloidosis. A Review
The term amyloidosis refers to a heterogeneous group of disorders characterized by deposition of misfolded and undegradable proteinaceous material with typical staining properties. Skin is involved in both systemic and localized form of amyloidosis, primary and/or secondary. Amyloid types relevant for cutaneous pathology include amyloid derived from keratin, serum A amyloid, immunoglobulin light chains, β2microglobulin, and transthyretin. The review presents current knowledge about these diseases.
Keywords:
immunohistochemistry – amyloid – AA amyloid – AL amyloid – AK amyloid
Autoři:
E. Sticová 1,2
Působiště autorů:
Ústav patologie, 3. LF UK a FN Královské Vinohrady, přednosta prof. MUDr. Radoslav Matěj, Ph. D.
1; Kožní oddělení, Krajská nemocnice Liberec, primář MUDr. Dana Frydrychová
2
Vyšlo v časopise:
Čes-slov Derm, 98, 2023, No. 1, p. 5-12
Kategorie:
Souborné referáty (doškolování lékařů)
Souhrn
Amyloidózy představují heterogenní skupinu chorob charakterizovaných ukládáním patologicky poskládaného a obtížně degradovatelného proteinu s typickými tinkčními vlastnostmi. Kůže bývá postižena v rámci systémové amyloidózy, častěji se však setkáváme s formami lokalizované kožní amyloidózy, buď primární nebo sekundární. Mezi typy amyloidu relevantní pro kožní patologii patří zejména amyloid odvozený z keratinu, sérového amyloidového proteinu A, lehkých řetězců imunoglobulinů, β2mikroglobulinu a transthyretinu. Článek přináší stručný přehled současných znalostí o těchto onemocněních.
Klíčová slova:
imunohistochemie – amyloid – AA amyloid – AL amyloid – AK amyloid
ÚVOD
Amyloidóza představuje heterogenní skupinu poměrně vzácných chorob charakterizovaných ukládáním patologicky poskládaného a obtížně degradovatelného extracelulárního proteinu ve tkáních a orgánech. V současnosti je známo minimálně 38 amyloidogenních fibrilárních proteinů, z nichž některé jsou produkovány přímo v místě vzniku amyloidních depozit (lokalizovaná či orgánová amyloidóza), jiné cirkulují v krvi a vytvářejí depozita v různých tkáních a orgánech (systémová či generalizovaná amyloidóza). Navzdory variabilní chemické podstatě i klinické manifestaci mají různé typy amyloidu velmi blízkou ultrastrukturu, světelně mikroskopický vzhled i základní tinkční vlastnosti [2, 13].
Fyzikální a chemické vlastnosti amyloidu
Amyloid je z 95 % tvořen komponentou z nevětvených fibril o průměru 7,5–10 nm a dále minoritní nefibrilární P-komponentou, apolipoproteinem E a glykosaminoglykany. Pro fibrilární komponentu amyloidu je typické abnormální uspořádání se sekundární strukturou ve formě β-skládaného listu, tvořeného dvěma polypeptidovými řetězci stabilizovanými H-můstky. Tato patologická konformace je zodpovědná za odolnost amyloidu vůči různým rozpouštědlům i proteolýze a rovněž za jeho specifické tinkční vlastnosti [2, 18].
Detekce amyloidu
Makroskopicky se větší depozita amyloidu projevují špekovitým nebo voskovým zbarvením postiženého orgánu, který je křehčí a mívá tužší konzistenci. V minulosti prováděné makroskopické Virchowovy reakce s Lugolovým roztokem a H2SO4 daly na základě podobnosti s tinkčními vlastnostmi škrobu amyloidu jeho název (amylum – latinsky škrob), v současné době jsou však považovány za zcela obsoletní a diagnostika amyloidózy je založena především na mikroskopickém vyšetření vzorku tkáně.
Histopatologická diagnostika amyloidózy probíhá ve dvou krocích, a to detekcí vlastního amyloidu ve tkáni, a dále typizací amyloidu, která je rozhodující pro nastavení správného terapeutického režimu [11, 19].
Na světelně mikroskopické úrovni se amyloid jeví jako amorfní eozinofilní depozita, pozitivní v reakcích s konžskou červení (obr. 1, 2). V polarizovaném světle vykazuje amyloid v preparátech s řezy o tloušťce minimálně 5 μm barvenými konžskou červení typický barevný, obvykle zelený dvojlom (obr. 3). K detekci velmi malých depozit v preparátu barveném konžskou červení lze s výhodou použít rovněž fluorescenční mikroskop (obr. 4). Reakce s konžskou červení je považována za zlatý standard v průkazu přítomnosti amyloidních depozit, avšak vzhledem k riziku nestandardního výsledku barvení či existenci vzácného kongo-negativního typu amyloidu je doporučováno podpořit diagnózu amyloidózy ještě další barvící technikou. S výhodou lze použít saturnovou červeň, thioflavin, případně metachromatickou reakci s krystalovou violetí [2, 11, 19].
Amorfní eozinofilní depozita amyloidu v dermis a podkoží.
Barvení HE, původní zvětšení 20krát.
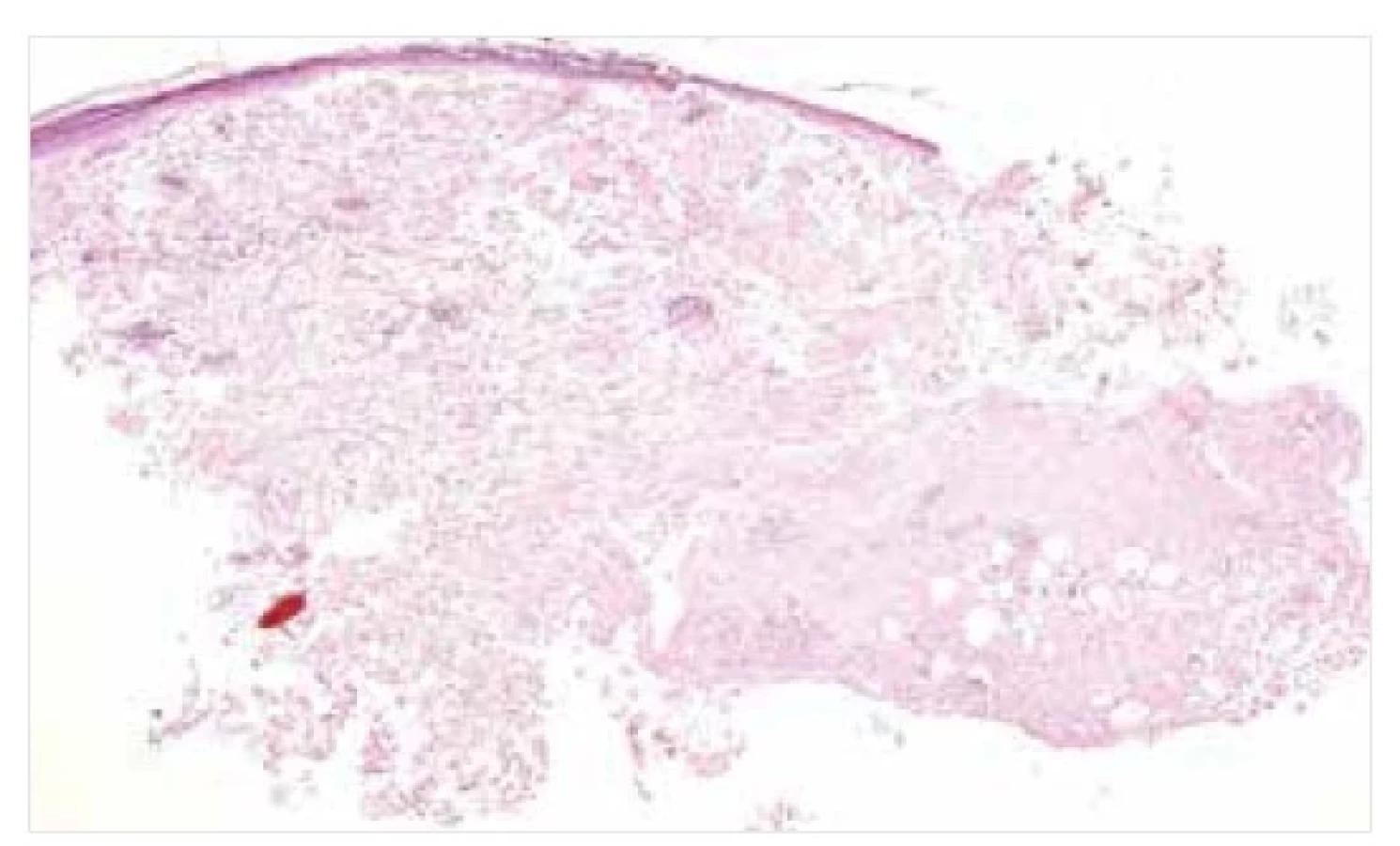
Depozita amyloidu v dermis a podkoží, pozitivní v reakcích
s konžskou červení. Původní zvětšení 20krát.

Původní zvětšení 600krát.

Drobná depozita ve stěně krevních cév s červenou
fluorescencí. Původní zvětšení 600krát.

K vlastní typizaci amyloidu se využívá imunohistochemické a imunofluorescenční vyšetření, případně imunoelektronová mikroskopie, tedy techniky založené na specifické reakci antigenu a protilátky. V běžné praxi se v současnosti používají zejména primární protilátky proti sérovému A amyloidu, lehkým řetězcům kappa a lambda, transthyretinu či vysokomolekulárnímu cytokeratinu (HMWCK, klon 34βE12). K získání optimálních výsledků je doporučováno provést imunofluorescenční vyšetření nativní tkáně, bez předchozí fixace formolem, která může významně alterovat antigenní determinanty detekovaných struktur [11, 19]. Slibnou technikou využívající současně výhod imunohistochemie a elektronové mikroskopie je imunoelektronová mikroskopie (obr. 5). V současnosti je však vzhledem ke značným nárokům na zpracování tkáně využití této metody v rutinní praxi limitované.
Původní zvětšení 12 000krát.

Imunohistologickými technikami lze při jejich správném provedení diagnostikovat nejběžnější typy amyloidu. K dalším metodám, které mohou pomoci s detekcí zejména nových či vzácně se vyskytujících forem, patří hmotnostní spektrometrie. Hlavním omezením této metody, zvláště v počátečních stadiích onemocnění s malými tkáňovými depozity amyloidu, je její relativně nízký rozlišovací limit [10, 18].
ZÁKLADNÍ TYPY AMYLOIDU
Klasifikace amyloidózy se opírá zejména o původ a typ amyloidogenního proteinu, který tvoří jádro amyloidních fibril. V kožní patologii patří mezi hlavní typy zejména amyloid odvozený z keratinu a z lehkých řetězců imunoglobulinů, vzácněji pak ze sérového amyloidového proteinu A, transthyretinu a β2mikroglobulinu [2, 4, 13].
Amyloid odvozený z keratinu (AK) je obvykle detekován u lokalizovaných forem kožní amyloidózy, zejména u lichen amyloidosus, makulózní amyloidózy, případně bifázických typů amyloidózy, mnohem vzácněji pak u tzv. nodulární kožní amyloidózy [2, 8, 12]. Způsob, jakým tento typ amyloidu vzniká, není zcela objasněn. AK amyloid často vykazuje pouze slabé reakce v barvení s konžskou červení a je spolehlivě detekován imunohistochemicky s primární protilátkou proti vysokomolekulárnímu cytokeratinu (HMWCK, klon 34βE12), případně proti komponentě komplementové kaskády C4d [25].
AL amyloidóza představuje typ amyloidózy z nadprodukce monoklonálních lehkých řetězců imunoglobulinů kappa či lambda a provází jako systémové postižení některá hematologická onemocnění, typicky plazmocytární myelom či monoklonální gamapatie [30]. AL amyloid bývá rovněž detekován u lokalizované formy, tzv. nodulární kožní amyloidózy a u tumoriformní amyloidózy (amyloidomu) [4, 26]. Lehké řetězce lambda jsou obecně považovány za více amyloidogenní než řetězce kappa. Vzácně je popisována amyloidóza vznikající z těžkých řetězců imunoglobulinů [2]. Amorfní depozita tvořená neamyloidogenními lehkými řetězci, negativními v reakcích s konžskou červení, byla vzácně popisována v kůži u tzv. nemoci z lehkých řetězců (light chain deposition disease) [10, 31].
AA amyloidóza je systémovým typem amyloidózy obvykle asociovaným s chronickými zánětlivými procesy, infekčními i neinfekčními, zpravidla autoimunitními a autoinflamatorními. Vzácně může být součástí projevů hereditární poruchy s nadprodukcí prozánětlivých cytokinů (viz níže). Prekurzorovým proteinem AA amyloidózy je N-terminální fragment sérového amyloidového proteinu A (SAA), játry produkovaný protein patřící do skupiny tzv. reaktantů akutní fáze zánětu [2, 20, 27].
U autozomálně recesivní familiární středomořské horečky je popisována aktivační mutace genu kódujícího protein pyrin, vedoucí ke konstituční nadprodukci interleukinu IL-1 a k přetrvávajícímu zánětu. Tento stav již dříve popsaným mechanismem nadprodukce SAA vede rovněž k rozvoji AA amyloidózy [15, 25]. Obdobný mechanismus geneticky podmíněné nadprodukce prozánětlivých cytokinů s ukládáním depozit A amyloidu byl popsán u Muckle-Wellsova syndromu, autozomálně dominantní poruchy s mutací genu pro kryopyrin, protein se shodnou N-terminální doménou jako u pyrinu [7].
U některých heredofamiliárních forem amyloidózy a u tzv. senilní amyloidózy je prekurzorovým proteinem transthyretin (ATTR), α-globulin tvořený v játrech, fungující za fyziologických podmínek jako transportní protein pro retinol a tyroxin. Pro ATTR amyloidózu bývá typické postižení srdce a neurologické příznaky se známkami senzoricko-motorické nebo vegetativní polyneuropatie [20, 23].
Amyloidní depozita formující se z β2 mikroglobulinu (Aβ2M) nacházíme u pacientů dlouhodobě dialyzovaných, u kterých se snižuje glomerulární filtrace Aβ2M, který není hemodialýzou efektivně odstraňován. Predilekčně bývá postižen osteoartikulární systém se známkami destruktivní artropatie velkých kloubů, spondylartropatie a syndromu karpálního tunelu. Postižení kůže a podkoží bývá popisováno spíše výjimečně [2, 20].
KLASIFIKACE AMYLOIDÓZ S KOŽNÍM POSTIŽENÍM
I přes prakticky identický světelně mikroskopický vzhled a tinkční vlastnosti představují amyloidózy značně heterogenní skupinu chorob. Kůže jako orgán může být postižena v rámci generalizované (systémové) amyloidózy, častěji se však setkáváme s lokalizovanou formou kožní amyloidózy, primární či sekundární – tabulka 1 [18, 20].

1. Systémové amyloidózy
AL amyloidóza
AL amyloidóza je nejčastějším typem systémové amyloidózy v Evropě a USA, objevující se u 5–10 % pacientů jako komplikace základního hematologického onemocnění, zpravidla plazmocelulární dyskrazie [27, 31].
AL amyloidóza mívá pestré klinické projevy, často s postižením více orgánů a orgánových systémů, zejména ledvin, srdce a zažívacího traktu. U části pacientů se může objevit syndrom karpálního tunelu a makroglosie. Kožními projevy AL amyloidózy jsou obvykle papuly až plaky voskového vzhledu v obličejové části hlavy, zejména periorbitálně, na skalpu, v oblasti krku a na genitáliích. Plaky se mohou objevit rovněž v oblasti rukou a na flexorových částech končetin. Poměrně časté je vzhledem k současnému postižení krevních cév prokrvácení projevů, buď spontánní či indukované malým traumatem (obr. 6) [30].

Mikroskopicky jsou detekována poměrně rozsáhlá depozita amyloidu v retikulární dermis, případně i v podkoží, kde amyloid vytváří prstence kolem jednotlivých adipocytů. Poměrně běžně se setkáváme s bulami, kdy dochází ke štěpení kolem či přímo ve větších depozitech acelulárního materiálu. Menší depozita amyloidu vázaná obvykle na stěnu krevních cév lze detekovat i ve vzorcích z makroskopicky nepostižené kůže a zejména podkoží (obr. 7) [27, 31].
Drobná depozita ve stěně krevních cév pozitivní v reakcích
s konžskou červení. Původní zvětšení 600krát.

Amyloidní elastóza je vzácnou formou systémové AL amyloidózy s depozity pokrývajícími elastická vlákna v kůži i serózních blanách. Klinicky se tento typ prezentuje ve formě mnohočetných papul na krku, břiše, v lumbosakrální oblasti a v místech flekčních rýh. Mikroskopicky nacházíme fragmentovaná elastická vlákna s drobnými protruzemi, krytá amyloidními mikrofibrilami [2].
AA amyloidóza
AA amyloidóza je systémová forma amyloidózy komplikující některé dlouhotrvající zánětlivé procesy. Vzácně se může jednat o primárně kožní zánět typu lepromatózní lepry, supurativní hidrosadenitidy/acne inversa, případně artropatické formy psoriázy [2, 27].
Drobná depozita pozitivní v imunohistochemických reakcích s protilátkou proti SAA vzácně prokážeme v papilární dermis, podkoží, ve stěně krevních cév či kolem potních žlázek ve vzorcích z makroskopicky nepostižené kůže [27].
Heredofamiliární formy amyloidózy
Klinická manifestace těchto typů systémových amyloidóz je poměrně variabilní a zahrnuje například urtikariální projevy u Muckle-Wellsova syndromu (amyloidóza, hluchota, chronická rekurentní kopřivka) [7] či trofické změny kůže s depozity amyloidu ve vzpřimovači chlupu či kolem vláken periferních nervů u heredofamiliární amyloidní polyneuropatie [9, 23].
2. Lokalizované formy kožní amyloidózy
a) Primární kožní amyloidóza
Lichen amyloidosus, makulózní amyloidóza a bifázické formy
Lichen amyloidosus (LA) a makulózní amyloidóza (MA) jsou považovány za různé klinické varianty téhož procesu, kde prokazujeme depozita AK amyloidu pouze v kůži, bez známek systémového postižení [12, 16]. V etiopatogenezi obou onemocnění může hrát úlohu chronický pruritus a opakované traumatizace kůže škrábáním [12, 25].
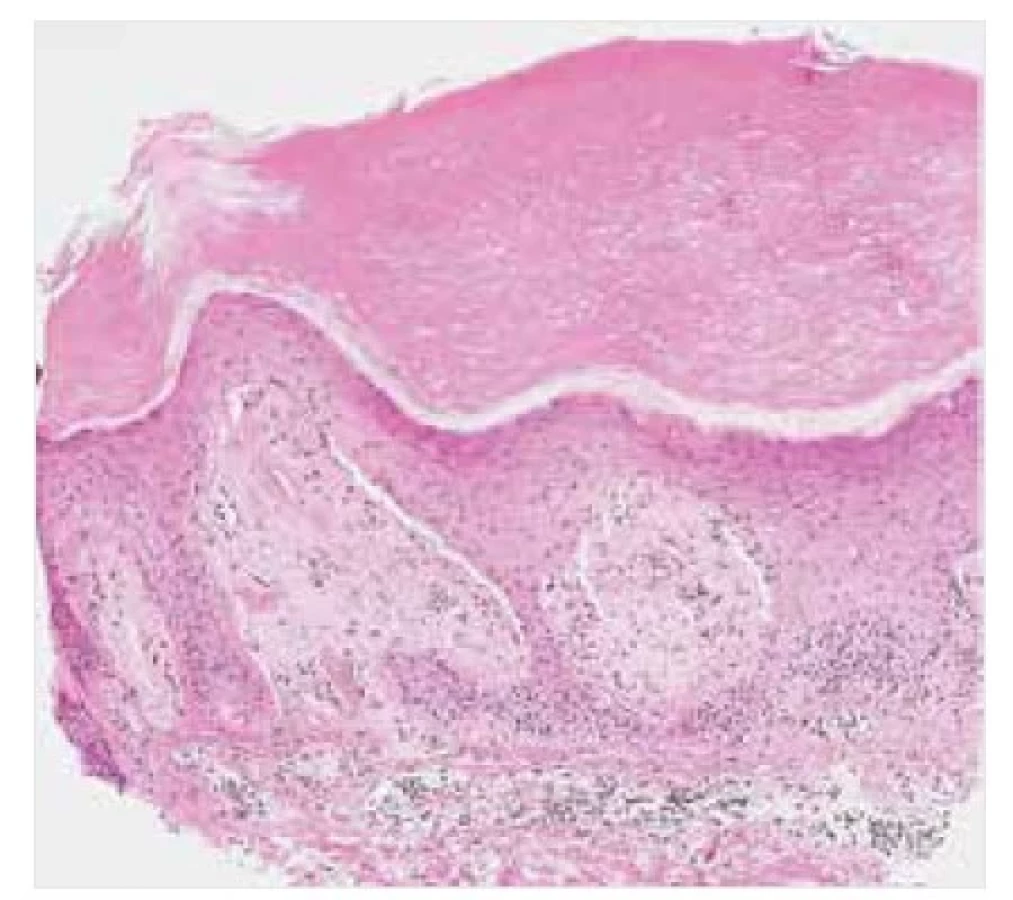
LA se klinicky manifestuje ve formě drobných, často hyperpigmentovaných svědivých voskových papul na extenzorových plochách dolních končetin, zejména v oblasti bérců, dále kolem kotníku, na hřbetech nohou a na stehnech (obr. 8). Dalšími možnými postiženými oblastmi mohou být extenzorové plochy horních končetin a trup [16, 21]. MA se projevuje nejčastěji ve formě špatně ohraničených síťovitých hyperpigmentovaných skvrn na trupu, s predilekčním postižením zad a mezilopatkové oblasti (obr. 9), dále v oblasti extenzorových ploch končetin a na obličeji, zejména periorbitálně. Projevy rovněž často svědí [16, 21].


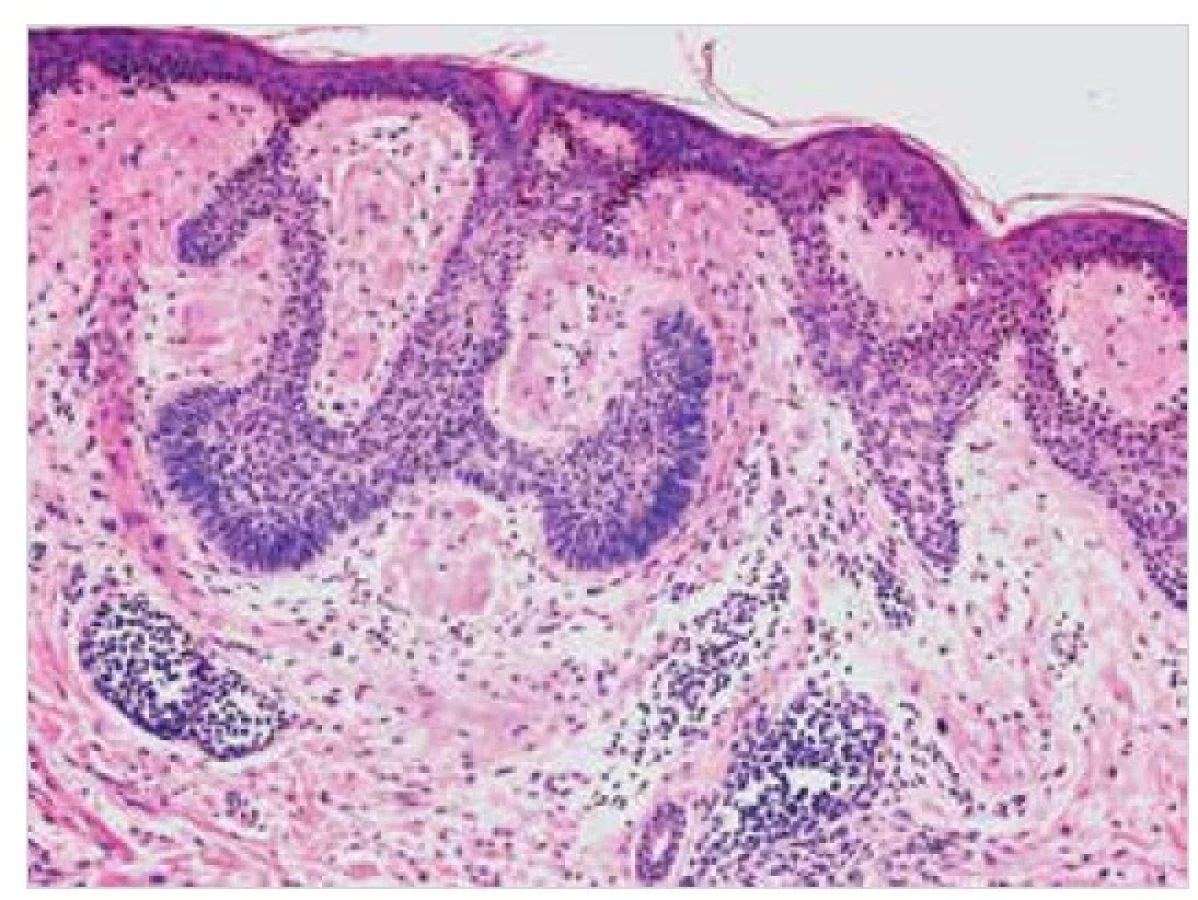
Obě tyto formy primární kožní amyloidózy jsou na mikroskopické úrovni charakterizovány přítomností malých globulárních depozit amorfního eozinofilního materiálu v papilární dermis, obvykle slabě pozitivního v reakcích s konžskou červení a s imunoexpresí vysokomolekulárního cytokeratinu (obr. 10, 11). V bazálních řadách epidermis mohou být přítomny známky vakuolární degenerace keratinocytů s apoptotickým zánikem individuálních epitelií a se zmnožením melanofágů v povrchových partiích koria, které je histologickým korelátem makroskopicky popisované hyperpigmentace projevů. U LA je epidermis zpravidla akantoticky rozšířená, často s hyperkeratózou [21, 23].
Barvení HE, původní zvětšení 100krát.
Drobná amorfní depozita amyloidu v papilární dermis (šipka) v barvení HE (A), pozitivní v imunohistochemických
reakcích s primární protilátkou proti HMWCK (B). Původní zvětšení 400krát (A) a 100krát (B).

Amyloidóza ušní konchy (aurikulární amyloidóza), dříve označovaná jako kolagenní papula ucha, je pravděpodobně variantou LA s amorfními depozity v rozšířených dermálních papilách [32].
V diferenciální diagnóze LA a MA mohou připadat v úvahu zejména lichen simplex chronicus či afekce ze skupiny lichen planus, dále pozánětlivé hyperpigmentace, notalgia paraesthetica, koloidní milium či parakoloid. Klíčem ke správné diagnóze bývá u LA a MA pozitivita amyloidních depozit v reakcích s konžskou či saturnovou červení s příslušným dvojlomem a pozitivita v imunohistochemických reakcích s HMWCK [13, 18].
Nodulární amyloidóza
Nodulární amyloidóza (NA) je poměrně vzácná forma kožní amyloidózy s přítomností jedné či více voskových papul či nodulů o průměru do 7 cm na dolních končetinách, dále na obličeji, krku, ve vlasaté části hlavy a v oblasti genitálu (obr. 12). V dermatoskopickém obraze mají depozita NA v centru žluto-oranžovou barvu, s přítomností vinutých dilatovaných cév [13]. Přibližně 7–15 % pacientů s původně lokalizovanou kožní formou vyvine systémové onemocnění [4, 12].

Chemicky se u většiny případů NA jedná o depozita lehkého řetězce lambda, vzácně byl prokázán β2mikroglobulin, zejména u dlouhodobě dialyzovaných pacientů, případně AK amyloid či inzulin [12, 18].
Mikroskopicky nacházíme u NA obvykle depozita amyloidu v dermis a podkoží, akcentovaná kolem větších krevních cév a kožních adnex (viz obr. 1, 2). Na periferii amorfních depozit bývá často monoklonální populace plazmatických buněk se známkami restrikce lehkých řetězců, někdy s dystrofickými kalcifikacemi a obrovskobuněčnou granulomatózní reakcí typu z cizích těles [12, 18].
Podkožní depozita pozitivní v imunohistochemických reakcích s inzulinem bývají vzácně popisována u pacientů s diabetem v místě opakované aplikace injekcí inzulinu [14].
V morfologické diferenciální diagnóze NA připadá v úvahu především lipoidní proteinóza (Urbach-Wiethe), vzácné autozomálně recesivní onemocnění s hyalinními depozity v kůži i na sliznicích, pozitivními v PAS (Periodic acid-Schiff) reakci a negativními v reakcích na průkaz amyloidu [18].
Tumoriformní amyloidóza
Tumoriformní amyloidóza (amyloidom) je vzácná forma lokalizované amyloidózy typu AL či AA, bez známek systémového postižení. Tento typ amyloidózy je nejčastěji popisován v bronchopulmonálním, urogenitálním a gastrointestinálním traktu, dále v nervovém systému, mléčné žláze a v měkkých tkáních [3, 17]. Amyloidom kůže a podkoží se vyskytuje zřídka a některými autory je považován za neobvyklou manifestaci kožního B lymfomu marginální zóny [26]. Typickou klinickou manifestací je solitární žlutohnědý plak na dolní končetině pacienta středního či vyššího věku. V mikroskopickém obraze obvykle nacházíme tumoriformní amorfní depozita stírající původní strukturu tkáně, provázená obrovskobuněčnou granulomatózní reakcí typu z cizích těles s dystrofickými kalcifikacemi a s řídkou lymfoplazmocelulární celulizací [17, 26].
Poikilodermatózní amyloidóza
Poikilodermatózní amyloidóza (PA) je vzácný typ kožní amyloidózy, manifestující se u části pacientů fotosenzitivitou, nízkým vzrůstem a přítomností palmoplantární keratodermie [2, 18]. Vzácně bývá PA asociována s některými autoimunitními či autoinflamatorními procesy, např. s lupus erythematodes či se sklerodermií [13].
Amyloidosis cutis dyschromica je považována za vzácný podtyp PA s generalizovanou retikulární hyperpigmentací kůže a s hypopigmentovanými okrsky [18].
Histopatologicky jsou u afekcí ze skupiny PA prokazována drobná depozita AK amyloidu ve stratum papillare a kolem drobných dermálních cév. Depozita provázená zmnožením melanofágů bývají pozorována v hyperpigmentovaných oblastech, v depigmentovaných lézích je patrný úbytek melanocytů [18].
Anosakrální amyloidóza
Vzácná forma primární kožní amyloidózy popisovaná zejména u Asiatů a projevující se ve formě světle hnědých lichenifikovaných perianálních plaků propagujících se do oblasti sakra. Chemicky se jedná o AK amyloid s mikroskopickými depozity v oblasti stratum papillare, provázenými mírnou akantózou a hyperkeratózou přilehlé epidermis, s inkontinencí pigmentu [5, 8].
Familiární primární kožní amyloidóza
Velmi vzácná genodermatóza s autozomálně dominantním typem přenosu, charakterizovaná přítomností keratotických papul se změnami pigmentace kůže končetin a vzácně trupu. Pro tento typ amyloidózy je typická transepidermální eliminace papilárních depozit amyloidu [8, 25].
b) Sekundární lokalizovaná kožní amyloidóza
Termín označuje depozita amyloidu ve stromatu různých kožních nádorů, obvykle bazaliomu (obr. 13), méně často dlaždicobuněčného karcinomu, melanocytárních lézí či některých vzácnějších adnexálních tumorů, dále na pozadí porokeratózy či v kůži vystavené UVA záření po aplikaci psoralenu. Prakticky ve všech případech se jedná o depozita AK amyloidu [8, 16].
Eozinofilní depozita ve stromatu bazaliomu. Barvení HE, původní
zvětšení 100krát.
TERAPIE
V případě systémových forem amyloidózy závisí léčebný přístup na základním onemocnění a typu amyloidu. U AL amyloidózy je zejména v počátečních stadiích onemocnění metodou volby chemoterapie či transplantace kmenových buněk, u pokročilých forem onemocnění se v poslední době staly součástí léčebných režimů monoklonální protilátky a inhibitory proteasomu [31]. V případě AA amyloidózy je klíčová léčba základního zánětlivého procesu. Dostupné jsou rovněž preparáty snižující sérové hladiny SAA blokací receptoru pro interleukin 6 [18, 29]. U transthyretinové amyloidózy je metodou volby transplantace jater, která eliminuje tvorbu mutovaného proteinu [23].
Terapie primární kožní amyloidózy je problematická a dosud nebyl stanoven žádný zlatý léčebný standard či doporučení optimálního terapeutického přístupu. Kromě lokalizace a rozsahu kožního nálezu musí dermatolog zvážit rovněž vedlejší nežádoucí efekt léčby a zátěž pro pacienta.
Obecně platí, že u LA a MA je základem úspěšné léčby eliminace základního onemocnění a s tím souvisejícího pruritu, vedoucího k opakované exkoriaci kůže. Slibné výsledky byly pozorovány po lokální aplikaci kalcipotriolu, po perorální aplikaci cyklofosfamidu, u fototerapie a u laserové terapie [1, 29]. K ošetření svědivých lichenifikovaných hyperkeratotických projevů lze využít metody dermabraze [22, 29]. Z dalších léčebných postupů je popisován částečný efekt lokálních i systémově podávaných retinoidů a lokálních kalcineurinových inhibitorů. Nejednoznačné výsledky byly zaznamenány po lokální aplikaci kortikosteroidů [29].
U nodulární kožní amyloidózy zůstává metodou volby chirurgická intervence s kompletním odstraněním ložiska, které je zároveň i prevencí progrese do systémového onemocnění. Komplikací chirurgického výkonu může být vzhledem k postižení krevních cév krvácení, které lze omezit kauterizací či použitím laseru s oxidem uhličitým [1].
Poděkování
Autorka děkuje za zapůjčení klinické fotodokumentace prof. MUDr. Jiřímu Štorkovi, CSc. (Dermatovenerologická Klinika 1. LF UK a Všeobecné fakultní nemocnice), prof. MUDr. Spyridonovi Gkalpakiotisovi, Ph.D., MBA (Dermatovenerologická klinika 3. LF UK a FN Královské Vinohrady), paní Monice Dubské a MUDr. Ondřeji Fabiánovi, Ph.D. (Pracoviště klinické a transplantační patologie, IKEM) a MUDr. Jakubovi Křížovi (ÚČOCH, Krajská nemocnice Liberec, a. s).
Prohlášení o střetu zájmů
Autorka v souvislosti s tématem práce v posledních 12 měsících nespolupracovala s žádnou farmaceutickou firmou.
Do redakce došlo dne 3. 1. 2023.
Adresa pro korespondenci:
doc. MUDr. Eva Sticová, Ph.D.
Ústav patologie 3. LF a FNKV
Šrobárova 1150/50
100 00 Praha 10
e-mail: eva.sticova@fnkv.cz
Zdroje
1. AHRAMIYANPOUR, N., AKBARI, Z., SARASYABI, M. S. et al. The therapeutic role of lasers in primary localized cutaneous amyloidosis: a systematic review. Lasers Med Sci., 2022, 37(2), p. 799–813.
2. BLACK, M. M., UPJOHN, E., ALBERT, S. Metabolic and Systemic diseases: Amyloidosis. Bolgonia. In: BOLOGNIA, J. L., JORIZZO, J. L., RAPINI, R. P. et al. Dermatology textbook. 2nd ed., Vol I. Mosby Elsevier, 2008, p. 623–631.
3. DESAI, S. S., RIZZO, M. G., RUSH, A. J., et al. Amyloidoma: a review and case report. Skeletal Radiol., 2021, 50(2), p. 437–444.
4. FERNANDEZ-FLORES, A. Cutaneous amyloidosis: a concept review. Am J Dermatopathol., 2012, 34(1), p. 1–14.
5. GERHARDT, C. A., CARDON, B., RODRIGUEZ-WAITKUS, P. et al. Anosacral amyloidosis in a Chinese-Caribbean male. JAAD Case Rep., 2022, 21, p. 46–48.
6. GUILLET, C., STEINMANN, S., MAUL, JT. et al. Primary Localized Cutaneous Amyloidosis: A Retrospective Study of an Uncommon Skin Disease in the Largest Tertiary Care Center in Switzerland. Dermatology, 2022, 238(3), p. 579-586.
7. HAAS, N., KÜSTER, W., ZUBERBIER, T. et al. Muckle - Wells syndrome: clinical and histological skin findings compatible with cold air urticaria in a large kindred. Br. J. Dermatol., 2004, 151(1), p. 99–104.
8. HAMIE, L., HADDAD, I., NASSER, N. et al. Primary Localized Cutaneous Amyloidosis of Keratinocyte Origin: An Update with Emphasis on Atypical Clinical Variants. Am. J. Clin. Dermatol., 2021, 22(5), p. 667–680.
9. HEMMINKI, K., LI, X., FÖRSTI, A. et al. Incidence of hereditary amyloidosis and autoinflammatory diseases in Sweden: endemic and imported diseases. BMC Med. Genet., 2013(14), p. 88.
10. HENDRICKS, C., FERNÁNDEZ FIGUERAS, M. T., LIERSCH, J. et al. Cutaneous Light Chain Deposition Disease: A Report of 2 Cases and Review of the Literature. Am. J. Dermatopathol., 2018, 40(5), p. 337–341.
11. HONSOVA, E., HERMANOVA, M., SVOBODOVA, I. et al. Doporučený postup pro diagnostiku systémových amyloidóz [online]. 2022. Dostupné na www: www.patologie.info/standardy/48.
12. KALTOFT, B., SCHMIDT, G., LAURITZEN, A. F. et al. Primary localised cutaneous amyloidosis – a systematic review. Dan. Med. J., 2013, 60(11), p. A4727.
13. KERL, H. Amyloidosis. In KERL, H. et al. Diagnostic Cutaneous Pathology, © e-Book 2021. Dermatopathology Books, p. 74–86.
14. KRANC, C., WAGNER, R., JOY, N. M. et al. Cutaneous insulin-derived amyloidosis presenting as hyperkeratotic nodules. Cutis, 2021, 107(1), p. E6–E9.
15. LANE, T., LOEFFLER, J. M., ROWCZENIO, D. M. et al. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum., 2013, 65(4), p. 1116–1121.
16. MEHROTRA, K., DEWAN, R., KUMAR, J. V. et al. Primary Cutaneous Amyloidosis: A Clinical, Histopathological and Immunofluorescence Study. J. Clin. Diagn. Res., 2017, 11(8), p. WC01–WC05.
17. PASTERNAK, S., WRIGHT, B. A., WALSH, N. Soft tissue amyloidoma of the extremities: report of a case and review of the literature. Am. J. Dermatopathol., 2007, 29(2), p. 152–155.
18. PATTERSON, J. W. Amyloidosis. In PATTERSON, J. W. et al. Weedon’s Skin Pathology, 5th Ed., © Elsevier 2019, p. 466–473. ISBN: 9780702075827.
19. PICKEN, M. M. Current practice in amyloid detection and typing among renal pathologists. Amyloid, 2011, 18(Suppl 1), p. 73–75.
20. PICKEN, M. M. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol., 2020, 143(4), p. 322–334.
21. SALIM, T., SHENOI, S. D., BALACHANDRAN, C. et al. Lichen amyloidosus: A study of clinical, histopathologic and Immunofluorescence findings in 30 cases. Indian J Dermatol. Venereol. Leprol., 2005, 71, p. 166–169.
22. SAVANT, S. S. Therapeutic regional dermabrasion in papular lichen amyloidosis of shins. Indian J. Dermatol. Venereol. Leprol., 1995, 61(4), p. 196–201.
23. SEKIJIMA, Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease - modifying treatments. J. Neurol. Neurosurg. Psychiatry, 2015, 86(9), p. 1036–1043.
24. SINHA, A., MANJUNATH, G. V., BASAVARAJ, V. Primary cutaneous amyloidosis: A clinicopathological, histochemical, and immunohistochemical study. Indian J. Pathol. Microbiol., 2021, 64(2), p. 323–328.
25. TANAKA, A., ARITA, K., LAI-CHEONG, J. E. et al. New insight into mechanisms of pruritus from molecular studies on familial primary localized cutaneous amyloidosis. Br. J. Dermatol., 2009, 161(6), p. 1217–1224.
26. WALSH, N. M., LANO, I. M., GREEN, P. et al. AL Amyloidoma of the Skin/Subcutis: Cutaneous Amyloidosis, Plasma Cell Dyscrasia or a Manifestation of Primary Cutaneous Marginal Zone Lymphoma? Am. J. Surg. Pathol., 2017, 41(8), p. 1069–1076.
27. WECHALEKAR, A. D., GILLMORE, J. D., HAWKINS, P. N. Systemic amyloidosis. Lancet, 2016, 387(10038), p. 2641–2654.
28. WISNIOWSKI, B., WECHALEKAR, A. Confirming the Diagnosis of Amyloidosis. Acta Haematol., 2020,143(4), p. 312–321.
29. WEIDNER, T., ILLING, T., ELSNER P. Primary Localized Cutaneous Amyloidosis: A Systematic Treatment Review. Am. J. Clin. Dermatol., 2017, 18(5), p. 629–642.
30. YAMAN, B., KUMBARACI, B. S., GÓMEZ GONZÁLEZ, C. A. et al. C4d as a Practical Marker for Cutaneous Amyloidosis. Am. J. Dermatopathol., 2022, 44(1), p. 28–32.
31. ZANELLI, M., PALICELLI, A., SANGUEDOLCE, F. et al. Cutaneous Involvement in Diseases with Plasma Cell Differentiation: Diagnostic Approach. Curr Oncol., 2022, 29(5), p. 3026–3043.
32. ZHOU, X., CHEN, Q., TIAN, X. Primary cutaneous amyloidosis of auricular concha. Indian J. Dermatol. Venereol. Leprol., 2020, 86(2), p. 230.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2023 Číslo 1
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Systémová léčba atopické dermatitidy konečně i u dětí
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
Nejčtenější v tomto čísle
- Amyloidózy kůže
- Dermatoskopie v „netypických“ lokalizacích: ploché pigmentové projevy na akrálních částech končetin
- Jak je to s prevalencí atopické dermatitidy v České republice?
- Hapteny pro epikutánní testování jsou k dispozici
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy