
Benigní maskující syndrom u pacientky s retinitis pigmentosa
Retinitis Pigmentosa Mimicking Uveitis. A Case Report
Purpose:
To describe a case report of a 23-year-old patient with retinitis pigmentosa (RP) misdiagnosed as uveitis.
Methods:
A comprehensive eye examination including automated visual field assessment, contrast sensitivity, colour vision discrimination, ultrasound examination (US), spectral domain optical coherence tomography (SD-OCT) and full-field electroretinography (ERG) was performed in a patient diagnosed elsewhere as having intermediate uveitis because of the observation of a cellular reaction in the anterior chamber, bilateral cystoid macular oedema and suspected left optic disc swelling.
Results:
The patient reported nyctalopia. The best corrected visual acuity in both eyes was 6/12. Concentric visual field constriction was detected bilaterally (less than 25 degrees in the right eye and 15 degrees in the left eye). Fundus examination revealed a few pigment clumps and cystoid macular edema in both eyes confirmed by SD-OCT. Contrast sensitivity was decreased to 1,20 in the right and 0,9 in the left. No colour vision disturbance was present. The B scan ultrasound showed left optic disc drusen. Rod ERG responses were bilaterally not detectable and cone ERGs were abnormally reduced. Based on the examination results, a diagnosis of nonsyndromic RP was made.
Conclusion:
Clinicians should be aware of various manifestations of RP, including mild inflammation, to avoid possible confusin with uveitis.
Key words:
benign masquerade syndrome, retinitis pigmentosa
Autoři:
E. Szabó; M. Brichová; P. Lišková; P. Svozílková; E. Říhová
Působiště autorů:
Oční klinika, 1. lékařská fakulta, Univerzita Karlova v Praze a Všeobecná fakultní nemocnice v Praze, přednosta doc. MUDr. Bohdana Kalvodová, CSc.
Vyšlo v časopise:
Čes. a slov. Oftal., 69, 2013, No. 1, p. 32-36
Kategorie:
Kazuistika
Souhrn
Cíl:
Cílem sdělení je prezentace 23leté pacientky s retinitis pigmentosa (RP), která byla nejprve mylně diagnostikována jako uveitida.
Metodika:
Bylo provedeno komplexní oční vyšetření včetně perimetrického vyšetření, stanovení kontrastní citlivosti a barvocitu, vyšetření fundu v mydriáze, ultrazvukového vyšetření (UZ), spektrální optické koherentní tomografie (SD-OCT) a elektroretinografie (ERG). V diferenciální diagnostice byla odesílajícím pracovištěm zvažována intermediální uveitida pro nález buněčné reakce v přední komoře oční oka levého OL, oboustranně vláknitě zkaleného sklivce, cystoidního makulárního edému a edému terče levého zrakového nervu.
Výsledky:
Pacientka byla šeroslepá. Při vyšetření jsme zjistili nejlepší korigovanou centrální zrakovou ostrost oboustranně 6/12. Perimetrické vyšetření odhalilo koncentrické zúžení zorného pole na pravém oku (OP) k 25°, na levém oku k 15°. Na fundu byly patrné v periterii ojedinělé shluky pigmentu a v centru cystoidní makulární edém potvrzený SD-OCT. Kontrastní citlivost byla oboustranně snížená (OP 1,20 OL 0,9), zatímco barvocit byl v normě. UZ vyšetření potvrdilo přítomnost drúzy terče zrakového nervu vlevo. ERG vyšetření ukázalo nevýbavné skotopické fenomény oboustranně, kdežto fotopické a flicker fenomény byly ještě na hranici výbavnosti. Na základě těchto výsledků byla diagnostikována retinitis pigmentosa.
Závěr:
Zánětlivá reakce může představovat jeden ze vzácných projevů manifestace RP a tím vést k záměně za uveitidu. Jedná se tedy o benigní maskující syndrom.
Klíčová slova:
benigní maskující syndrom, retinitis pigmentosa
Úvod
Maskující syndromy (MS) jsou nezánětlivá oční onemocnění, která se klinicky mohou manifestovat jako uveitida. MS dělíme na maligní, které ohrožují zrak i život pacienta, a benigní – např. cévní anomálie, Schwartzův syndrom, nitrooční tělísko, syndrom pigmentové disperze, dystrofie a degenerace sítnice, oční ischemický syndrom, juvenilní xantogranulom, choroidální osteom, poléková a postvakcinační uveitida [13, 24]. Nussenblatt et al. řadí k benigním maskujícím syndromům dále chronickou pooperační infekci [22].
Kazuistika a výsledky
V říjnu 2010 byla odeslána do Centra pro diagnostiku a léčbu uveitid 1. LF UK a VFN v Praze 23letá pacientka pro suspektní intermediální uveitidu nejspíše parazitárního typu. Důvodem pro podezření na tuto nemoc byl třítýdenní pobyt v Thajsku asi rok před manifestací onemocnění.
Z osobní anamnézy jsme se dozvěděli, že pacientka je celkově zdráva. V rodině se kromě katarakty a glaukomu u babičky jiné oční choroby nevyskytly. Zajímavější je již oční anamnéza pacientky. Udává, že od dětství viděla hůře než její vrstevníci (např. nikdy neviděla hvězdy), je šeroslepá a vnímá zúžené zorné pole (stav přirovnává ke klapkám na očích u koně). Pacientka je sledována na oční ambulanci v místě bydliště, odkud byla pro nález zadní subkapsulární katarakty odeslána k operačnímu řešení do soukromého centra. V září 2010 byla pacientce na tomto pracovišti provedena operace katarakty levého oka (OL) s implantací monofokální nitrooční čočky (IOČ). Dle dokumentace byl v pooperačním období nález komplikován cystoidním makulárním edémem (CME) a edémem papily zrakového terče. Za 2 týdny po operaci byl pacientce aplikován parabulbárně betamethason, provedena Nd-YAG laserová kapsulotomie OL a doporučeno celkové vyšetření u praktického lékaře a konzultace na naší uveální ambulanci.
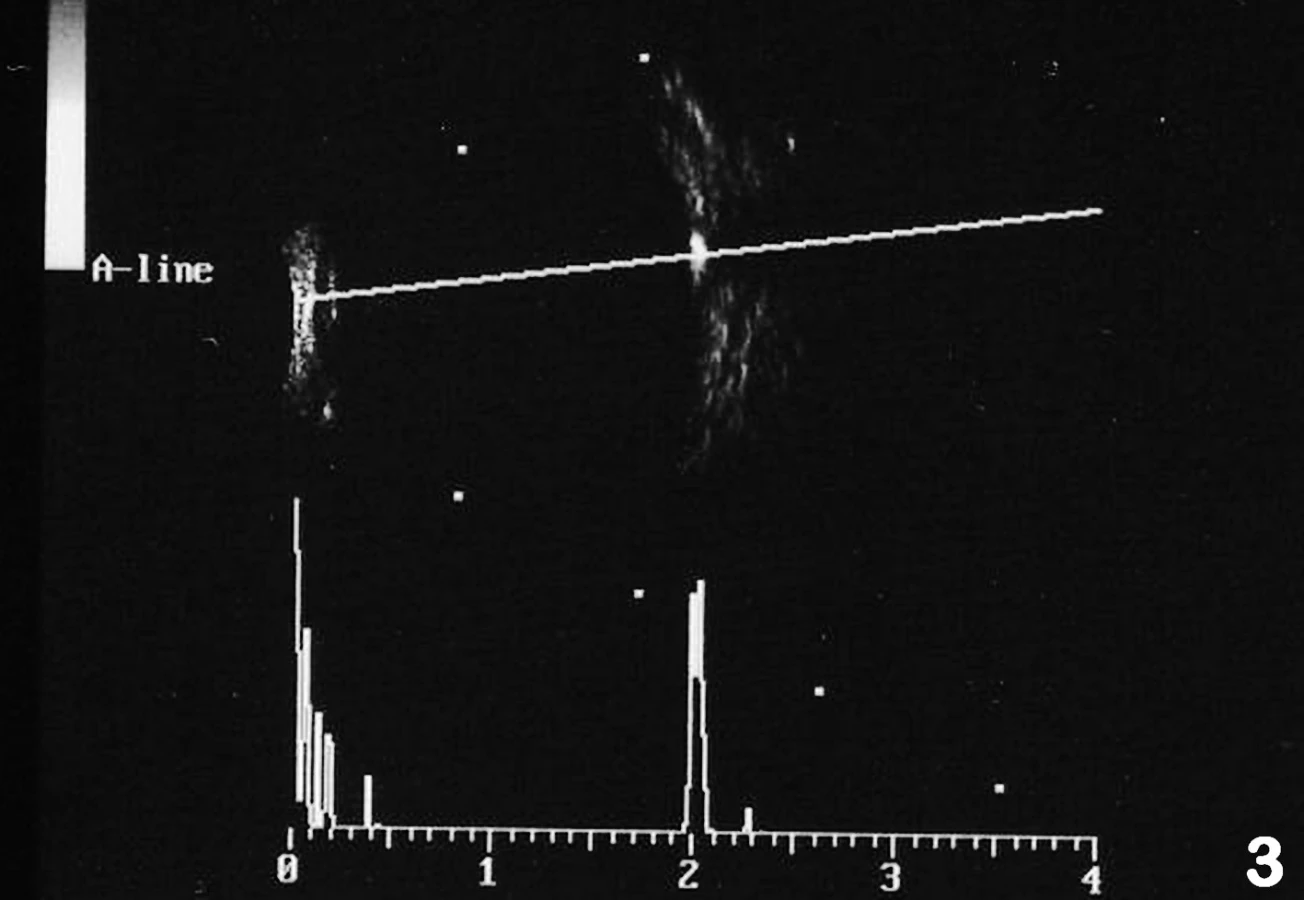
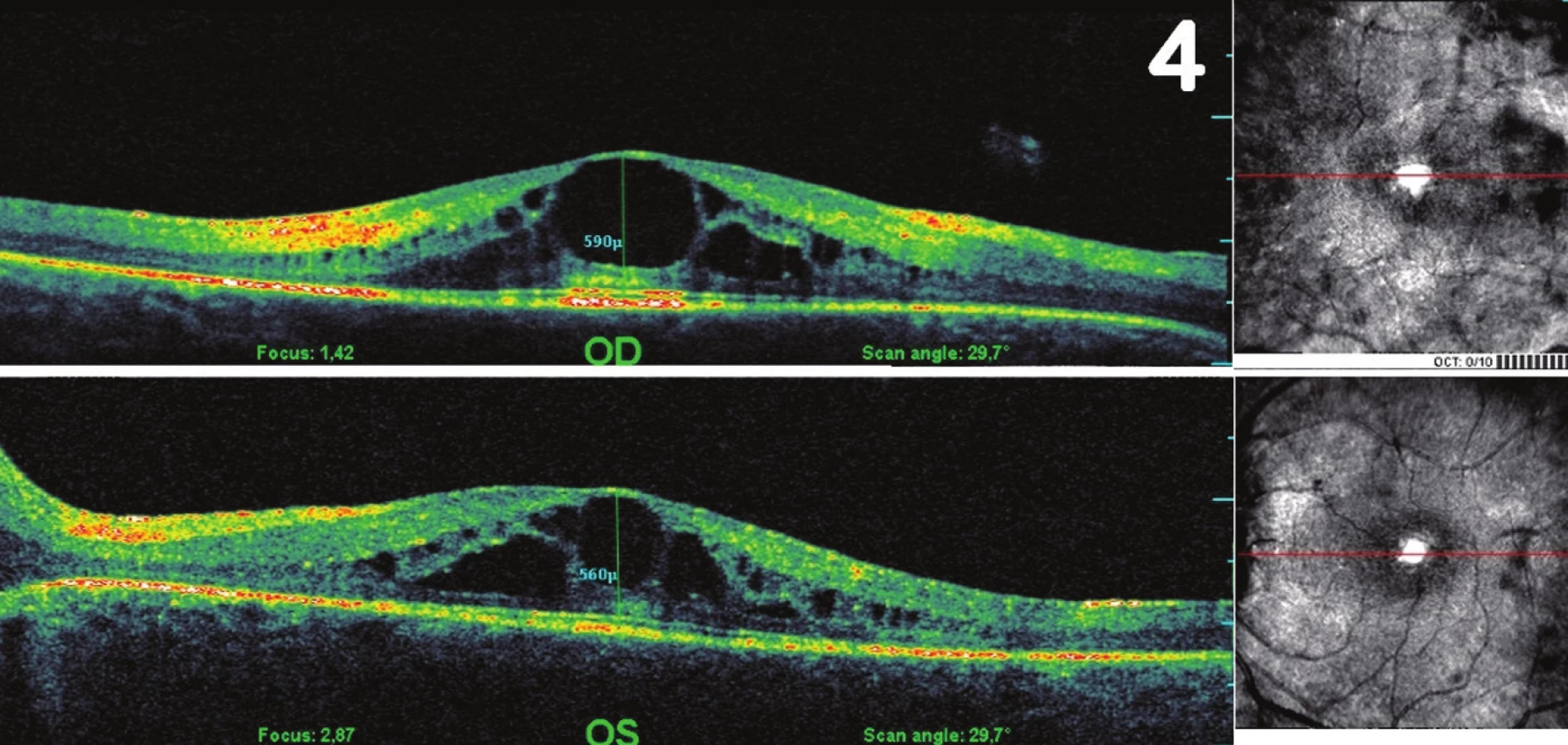
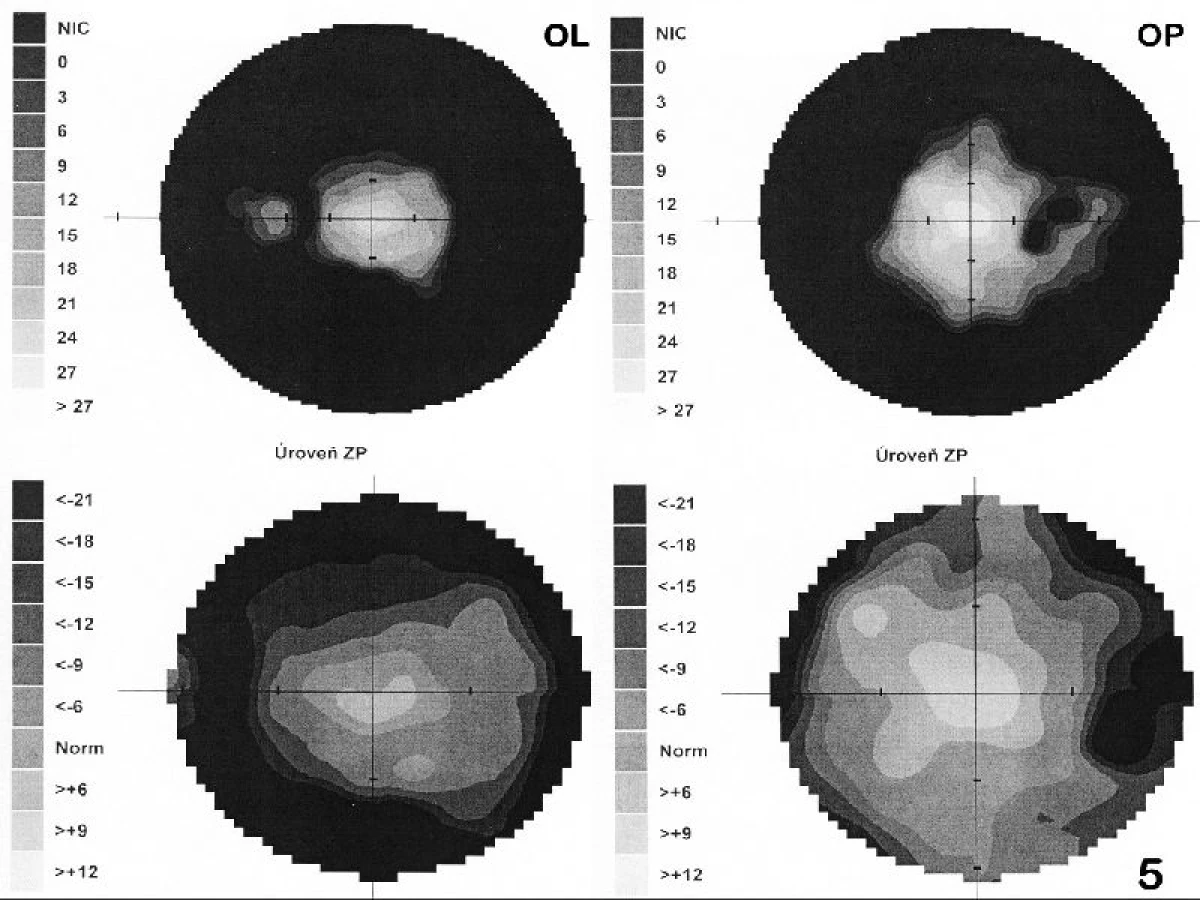
Při prvním vyšetření v naší ambulanci byla centrální zraková ostrost (CZO) oka pravého (OP) 6/12 s -0,5 cyl ax 180°, J.č.1, a OL 6/12 s +3,5 Dsf = - 2,5 Dcyl ax 180°, J.č.3 s addicí +3,0 Dsf, nitrooční tlak aplanačně 9 torrů oboustranně. Na klidném předním segmentu OP jsme zaznamenali zadní subkapsulární kataraktu (obr. 1a), na OL byly patrné buňky v přední komoře 1+, flare +/-, IOČ in situ a fenestrum v zadním pouzdře po kapsulotomii (obr. 1b). Na fundu OP byla růžová papila se žlutavým haló nejvíce v horním kvadrantu terče, makula s edémem, zúžené retinální artérie a v periferii ojediněle shluky pigmentu (obr. 2a). Na fundu OL byl nález obdobný, pouze byla navíc přítomna drúza na terči zrakového nervu (obr. 2b), kterou jsme ozřejmili ultrazvukovým vyšetřením (UZ – obr. 3). Edém v makule (obr. 4) jsme verifikovali optickou koherenční tomografií se spektrální doménou (SD-OCT, Spectral OCT/SLO, OTI Ophthalmic Technologies Inc., Kanada). Vyšetřením zorného pole (obr. 5) bylo prokázáno oboustranné koncentrické zúžení (OP ke 25°, na OL k 15°). Kontrastní citlivost na tabuli Pelli-Robson byla oboustranně snížená: OP 1,20, OL 0,9. Barvocit (15-Hue test) byl v normě.


Kontaktovali jsme ošetřující oční lékařku, která pacientku na operaci šedého zákalu odeslala. Dle její dokumentace bylo zúžení zorného pole přítomno již delší dobu. Pacientka pro suspektní nález na terčích podstoupila neurologické vyšetření k vyloučení demyelinizačního onemocnění centrální nervové soustavy, jehož výsledek včetně elektroencefalografie byl negativní.
Doporučili jsme pacientce elektroretinografické vyšetření (ERG – Ratiscan KL95/Roland konsult, Německo), které prokázalo nevýbavné skotopické fenomény a na hranici výbavnosti fotopické a flicker fenomény (obr. 6) a potvrdilo pracovní diagnózu retinitis pigmentosa (RP). Nasadili jsme lokální a celkovou antiedematózní terapii (indometacinum gtt. 4xd, dexamethasonum gtt. 4xd, escinum alfa tbl. 3x2) a vzhledem k iatrogenní anisometropii a astigmatismu jsme pacientku zacvičili v nošení torické kontaktní čočky.

Po 3 měsících došlo ke zhoršení CZO OL na 6/18 s +3,5 Dsf = -1,5 Dcyl ax 180°, J.č. 4 s addicí +3,0 D (obtížně), nitrooční tlak aplanačně 9 torrů. Na klidném předním segmentu byla patrná hutná membrána za IOČ (obr. 1c). Dle nálezu na kontrolním SD-OCT jsme konzervativní terapií CME OPL nijak neovlivnili. Proto byla pacientce provedena Nd-YAG membranotomie, po níž byl VOL zlepšen na 6/12 (+2) s korekcí, J.č. 2 s addicí +3,0 (obtížně). Ke zhoršení nálezu v makule OL po laserovém zákroku nedošlo.
S odstupem jednoho roku od operace byl vízus na obou očích nezměněn, došlo ovšem k dalšímu zúžení zorného pole (obr. 5b – OP k 20°, temporálně a dole k 15°, OL k 10° pouze nasálně do 15°). Doplnili jsme oční vyšetření sestry pacientky (včetně perimetrického vyšetření a ERG), které bylo v normě.
Diskuse
RP je geneticky heterogenní skupina progresivních onemocnění. Pokud dojde k záměně klinických projevů RP za uveitidu, řadíme ji k benigním maskujícím syndromům.
Diferenciálně diagnosticky u naší pacientky připadala vzhledem k uveitidě a CME v úvahu také pooperační komplikace po extrakci katarakty. Ta však nevysvětluje CME na druhém, neoperovaném oku. Zvažována by mohla být také pigmentová paravenózní retinochoroidální atrofie, která je však získané onemocnění po prodělaném infektu (např. po meningoencefalitidě, tuberkulóze, syfilis či rubeole) a pacientka jej negovala. Klinický obraz podobný nálezu u naší pacientky by mohl být dále způsoben vrozenou pigmentací sítnice s obrazem medvědích stop na fundu, která je zapříčiněna hypertrofií pigmentového epitelu sítnice, ovšem nález na perimetru ani vizus pacientky tomuto onemocnění neodpovídaly. Jednou z nejčastějších příčin vzniku pigmentací na očním pozadí je traumatická retinopatie, při které může dojít ke generalizované ztrátě RPE a k migraci melaninu do retinálních vrstev. Nález je však výhradně jednostranný a pacientka negovala jakékoliv trauma. Autoimunitní paraneoplastická retinopatie jako oční projev vzdáleného tumoru, nejčastěji malobuněčného karcinomu plic či karcinomu děložního čípku, se také projevuje šeroslepostí a zúženým zorným polem, ale má mnohem rychlejší progresi a vyskytuje se u starších pacientů. Toxická retinální degenerace po užívání chlorochinu, chlorpromazinu či fenothiazinů byla vyloučena vzhledem k negativní anamnéze. Podobné shluky pigmentu na sítnici jako jsou přítomny u RP mohou být vyvolány i kongenitální rubeolou (většinou spojena i s hluchotou), spalničkami či vrozenou syfilis [14, 12, 23]. Vše bylo u naší pacientky anamnesticky vyloučeno.
U RP je dominujícím znakem degenerace tyčinek, která bývá většinou následována degenerací čípků a může vést až k úplné slepotě. Typickým rysem je přítomnost tzv. kostních buněk, které však mohou zvláště v raných stadiích chybět. Onemocnění má značnou fenotypovou variabilitu (unilaterální RP, sektorová RP, RP sine pigmento, retinopathia punctata albescens, cone-rod dystrophy) [10]. RP se dělí na syndromovou (především Usherův syndrom) a nesyndromovou. Prevalence nejčastější nesyndromové RP se v běžné populaci odhaduje na 1 : 4000. Onemocnění se projevuje progredující šeroslepostí a zúžením zorného pole. V pozdějších fázích si pacienti stěžují i na horší CZO. Příčinou poklesu vízu může být podobně jako u naší pacientky rozvoj subkapsulární katarakty (u 35–50 %) či CME (u 15 až 23 %), dále přítomnost makulární atrofie (43 %), méně často epiretinální membrány [9].
Objektivně u pacientů vídáme klidný přední segment, může být přítomna opacifikace pod zadním pouzdrem čočky. Sklivcový prostor bývá čirý, papila voskově bledá, v časnějších fázích může být i růžová, neohraničená, se žlutavým haló v okolí. U 10 % pacientů jsou patrné drúzy terče zrakového nervu, jako jsme je zachytili i my u naší pacientky [11]. Makula zůstává dlouho intaktní, později dochází ke změnám popsaným výše. Cévy jsou úzké, napřímené a od střední periferie bývají patrné shluky pigmentu (kostní buňky). Vzácně mohou být přítomny neovaskularizace terče zrakového nervu či sítnice.
Diagnózu ověříme pomocí ERG vyšetření, které může prokázat patologický nález i při zcela negativním nálezu na fundu a perimetru. Nedílnou součástí diagnostiky je vyšetření příbuzných probanda, které jsme provedli u sestry pacientky. Dovolí-li to okolnosti, je vhodné i genetické vyšetření.
Přestože existuje řada klinických studií prováděných u pacientů s progresivními degenerativními onemocněními sítnice, účinná léčba RP v praxi dosud neexistuje. Jediné, co nyní můžeme pro pacienty udělat, je včas (tedy při dostatečně zachovalé CZO) doporučit profesionální rehabilitační služby pro osoby s postižením zraku. Vidoucí pacienti si rychleji osvojí každodenní úkoly, jež musí překonávat nevidomí. Když daný moment slepoty nastane, dokáží se o sebe sami postarat.
Ke zpomalení progrese je národním očním institutem USA doporučován vitamin A. Protože však hodnocení pacientů na této terapii neprokázalo z pohledu některých dalších nezávislých odborníků jednoznačný výsledek, není toto doporučení obecně přijímáno. Dílčí klinické studie byly dále provedeny na účinnost docosahexanové kyseliny, omega-3 mastných kyselin a luteinu, nedospěly ovšem ke zcela jasným závěrům [4, 5, 6, 7, 18, 21].
Kauzální léčba RP tedy v praxi dosud neexistuje. Velké naděje jsou vkládány do genové terapie [19], pro úspěšnou léčbu je však nutné zachování alespoň zbytku zrakových funkcí pacienta [25]. Na univerzitě v Tübingenu byla provedena pilotní studie s implantací sítnicových mikročipů, jejichž součástí jsou fotosenzitivní diody (až 1500), které dokáží stimulovat vnitřní části sítnice (gangliové buňky) a přenášet signál do zrakové kůry [8, 19]. Na univerzitě John Hopkins v Baltimoru podstoupilo 21 pacientů s RP studii s epiretinální protézou Arcus II (Arcus II, Second Sight Medical Products, Sylmar, California). Nikdo z pacientů neměl lepší centrální zrakovou ostrost než světlocit. Studie prokázala zlepšení jemné motoriky ruky za zrakové kontroly [3]. Ve stadiu laboratorního výzkumu je i využití kmenových buněk. Na rozdíl od genové terapie toto je naděje pro již slepé pacienty [9, 19]. Další novinkou jsou neurotropické faktory (CNF), jež zpomalují degeneraci tyčinek a tím pomáhají uchování vizu [9, 16, 19]. Aplikují se intraviteálně či subretinálně zatím pouze experimentálně na myších.
Nagpal a spol. [20] analyzovali procentuální zastoupení jednotlivých očních chorob, které byly primárně diagnostikovány jako zadní uveitida. Z celkového počtu 32 pacientů s benigním maskujícím syndromem byla RP nalezena u 3 z nich (9,3 %). Nejprve byla u těchto 3 pacientů diagnostikována zhojená chorioretinitida (pro nález pigmentových jizev na sítnici). Věk se pohyboval mezi 7 a 55 lety a centrální zraková ostrost byla 6/12 až počítání prstů před okem. Všichni byli šeroslepí, s pozitivním ERG nálezem. U jednoho pacienta byla dokonce přítomna pigmentová paravenózní atrofie, která může imitovat pigmentové zhojené jizvy po chorioretinitidě.
V našem souboru pacientů s uveitidou jsme diagnostikovali benigní maskující syndrom u 62 pacientů, z čehož cévní anomálie se vyskytovala u 63 %, Schwartzův syndrom u 13 % a poléková a postvakcinační uveitida u 6,5 % z nich. 5 % pacientů trpělo pigmentovou disperzí, dalších 5 % dystrofií a degenerací sítnice, zbylých 7,5 % je rovnoměrně rozděleno mezi pacienty s očním ischemickým syndromem, pacienty po kontuzi očního bulbu, pacienty s juvenilním xantogranulomem, osteomem či pooperační chronickou endoftalmitidou (nepublikovaná data, viz graf, tabulka).


Možná je i koincidence RP s uveitidou: s Fuchsovou heterochromní iridocyklitidou [15, 17], Behcetovou chorobou [1] či Coatsovou chorobou [2]. Jedná se však o raritní nálezy.
Willermain a spol. publikovali případ pacientky s birdshot chorioretinopatií HLA-A29 pozitivní, u které po 6 letech léčení imunosupresivy došlo ke vzácně popisované hyperpigmentaci krémových lézí imitujících tak nález kostních buněk při RP [26].
Závěr
RP mylně považovanou na uveitidu řadíme k benigním maskujícím syndromům. Může se u pacienta projevit horší CZO, zúžením zorného pole a šeroslepostí, tak jako u naší pacientky. Nález oboustranné subkapsulární katarakty, makulárního edému, napřímených cév a koncentrického zúžení zorného pole tuto diagnózu podporuje. Pro konečné potvrzení je vhodné doplnit ERG nebo molekulárně genetické vyšetření detekující příčinnou mutaci.
Do redakce doručeno dne 25. 9. 2012
Do tisku přijato dne 25. 2. 2013
MUDr. Eva Szabó
Oční klinika,
1. lékařská fakulta UK a VFN
U Nemocnice 2
128 08 Praha 2
e-mail: Eva.Szabo@vfn.cz
Zdroje
1. Baklouti, K., Mghaieth, F., Mhiri, N. et al.: Iridocyclitis in a patient with Behćet's disease and a familial form of retinitis pigmentosa. J Fr Ophtalmol. 2007 Oct; 30(8): e25.
2. Bansal, S., Saha, N., Woon, WH.: The management of "coats' response" in a patient with x-linked retinitis pigmentosa-a case report. ISRN Surg. 2011; 2011 : 970361. Epub 2011 Apr 20.
3. Barry, MP., Dagnelie, G.: Use of the Argus II Retinal Prosthesis to Improve Visual Guidance of Fine Hand Movements. Invest Ophthalmol Vis Sci. 2012 Jun 1.
4. Berson, EL., Rosner, B., Sandberg MA. et al.: Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup analyses. Arch Ophthalmol. 2004 Sep; 122(9): 1306-1314.
5. Berson, EL., Rosner, B., Sandberg, MA. et al.: A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993; 111(6): 761–772.
6. Berson, EL., Rosner, B., Sandberg, MA. et al.: Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Arch Ophthalmol. 2004; 122(9): 1297–1305.
7. Berson, EL., Rosner, B., Sandberg, MA. et al.: Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol. 2010; 128(4): 403–411.
8. Besch, D., Sachs, H., Szurman, P., et al.: Extraocular surgery for implantation of an active subretinal visual prosthesis with externalconnections: feasibility and outcome in seven patients. Br J Ophthalmol. 2008 Oct; 92 (10): 1361–8. Epub 2008 Jul 28.
9. Hamel, C.: Retinitis pigmentosa, Orphanet J Rare Dis. 2006 Oct 11; 1 : 40.
10. Hamel, CP.: Cone rod dystrophies. Orphanet J Rare Dis. 2007 Feb 1; 2 : 7.
11. Heidemann, DG., Beck, RW.: Retinitis pigmentosa. A mimic of neurologic disease. Surv Ophthalmol. 1987 Jul-Aug; 32(1): 45–51.
12. Kanski, J. J.: "Clinical Ophthalmology: A Systematic Approach", Butteworth-Heinemann 2007, s 150–155, ISBN 978070-2040931.
13. Kubicka-Trzaska, A., Romanowska-Dixon, B.: Non-malignant uveitis masquerade syndromes. Klin Oczna. 2008; 110 (4–6): 203–6.
14. Kuchynka, P. et al.: Oční lékařství, Grada Publishing, a.s., 2007, s. 279-281, SBN 978-80-247-1163-8.
15. L I van den Born, M J van Schooneveld, P T de Jong et al.: Fuchs' heterochromic uveitis associated with retinitis pigmentosa in a father and son. Br J Ophthalmol. 1994 June; 78(6): 504–505.
16. Liang, FQ., Aleman, TS., Dejneka, NS., et al.: Long-term protection of retinal structure but not function using RAAV. CNTF in animal models of retinitis pigmentosa. Mol Ther. 2001 Nov; 4(5): 461–72.
17. Lichtinger, A., Chowers, I., Amer, R.: Usher syndrome associated with Fuchs' heterochromic uveitis. Graefes Arch Clin Exp Ophthalmol. 2010 Oct; 248(10): 1481–5. Epub 2010 Jun 24.
18. Marmor, MF.: A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa [reply letter]. Arch Ophthalmol. 1993; 111(11): 1460–1461.
19. Musarella, A. M., MacDonald, I. M.: Current Concepts in the Treatment of Retinitis Pigmentosa. J Ophthalmol. 2011; 2011 : 753547. Epub 2010 Oct 11.
20. Nagpal, A., Biswas J.: Pseudouveitis - analysis of cases misdiagnosed as posterior uveitis. Ocul Immunol Inflamm. 2006 Feb; 14(1): 13–20.
21. Norton, EWD.: A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa [letter to the editor]. Arch Ophthalmol. 1993;111(11): 1460.
22. Nussenblatt, R.B., Whitcup, S.M., Palestine, A.G.: Masquerade syndromes. In: Uveitis: Fundamentals and clinical practice. Mosby-Year Book, Inc., St. Louis, 1996 : 385–395.
23. Otradovec, J.: Klinická neurooftalmologie, Grada Publishing, a.s., 2003, s. 159-164, ISBN: 80-247-0280-0.
24. Říhová, E. et al.: Uveitidy, Grada Publishing, a.s., 2009, s. 102-105, ISBN 978-80-247-2897-1.
25. Tan, MH., Smith, AJ., Pawlyk, B. et al.: Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: Effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum Mol Genet. 2009 Jun 15; 18 (12): 2099-114. Epud 2009 Mar 19.
26. Willermain, F., Greiner, K., Forrester, JV.: Atypical end-stage birdshot retinochoroidopathy. Ocul Immunol Inflamm. 2003 Dec; 11(4): 305–7.
Štítky
OftalmologieČlánek vyšel v časopise
Česká a slovenská oftalmologie

2013 Číslo 1
- Selektivní laserová trabekuloplastika nesnižuje nitroční tlak více než argonová laserová trabekuloplastika
- Progresi glaukomu je třeba hodnotit strukturálními i funkčními parametry
- Ztráta centrálního vidění po filtrujících operacích glaukomu
- Od PGF-2 alfa-isopropyl esteru k latanoprostu: přehled vývoje Xalatanu
- Compliance u pacientů s glaukomem
Nejčtenější v tomto čísle
- Klinické nálezy u členů české rodiny s retinitis pigmentosa podmíněnou mutací v ORF15 genu RPGR
- Oboustranná neuroretinitis jako projev nemoci kočičího škrábnutí u devítiletého chlapce
- Benigní maskující syndrom u pacientky s retinitis pigmentosa
- Evaluace parametrů jednoduchého binokulárního vidění na synoptoforu u zdravé dospělé populace
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy