
Klinická rozmanitosť Bestovej choroby
Clinical Variability of Best’s Disease
Retrospective view of the various phenotypes 20 persons affected by classic solitary form of vitelliform macular dystrophy, in 3 pedigrees with autosomal dominant transmission and in 4 single cases. Long-term monitoring allows to observe the variability of expression, from classic course to peculiarity of the clinical expression in the disc development and their corresponding functions of the central retina.
Key words:
solitary vitelliform macular dystrophy, variability of phenotypic expression, diagnostic
Autoři:
T. Streicher; J. Špirková; Marie Tichá
![]()
Působiště autorů:
Očné oddelenie NsP, Bojnice
primárka MUDr. Ida Simonidesová
Vyšlo v časopise:
Čes. a slov. Oftal., 68, 2012, No. 5, p. 189-194
Kategorie:
Původní práce
Souhrn
Retrospektívny pohľad na rozmanitý fenotyp 20 pacientov postihnutých klasickou obojstrannou solitárnou formou viteliformnej makulárnej dystrofie v 3 rodokmeňoch s autozomálne dominantným prenosom a 4 samostatných prípadov. Dlhodobé sledovanie umožňuje pozorovať variabilitu expresivity, od klasického priebehu jednotlivých štádií až po zvláštnosti vo vývoji viteliformného disku a im odpovedajúce funkcie centrálnej sietnice.
Kľúčové slová:
solitárna viteliformná dystrofia makuly, variabilita fenotypickej expresivity, diagnostika.
ÚVOD
Uplynulo 62 rokov od doby, kedy Zanen a Rausin referovali na belgickej oftalmologickej spoločnosti a následne opísali zaujímavú léziu na centrálnej sietnici. Dali jej priliehavý názov „Kyste vitelliforme congenital de la macula“ (31,32). Podobnosť vaječného žĺtka vyklopeného na dosku „Ięaspect dęun jaune dęoeuf sur un plat“, alebo „an egg with sunny side up“, je tak typická, že nemôže vzniknúť pochybnosť v diferenciálnej diagnostike. Je pravdepodobné, že ochorenie bolo známe už dávnejšie. Prvý opis podľa nákresov v publikácii pochádza od J. E. Adamsa z roku 1883 (cit. podľa 13). Väčšiu zásluhu na objavení tejto choroby mal ale Friedrich Best v roku 1905, keď opísal príbuzenstvo s výskytom tohto ochorenia u 8 z 59 vyšetrovaných členov a spoznal jej dedičnú povahu. Best síce v príbuzenstve nevidel viteliformný disk ako najcharakteristickejšiu léziu, iba jeho vývojové štádiá, ale spoznal dedičnú povahu choroby, a preto nesie táto jednotka oprávnene doteraz jeho meno. Bestov rodokmeň ďalej sledovali, referovali a ilustrovali Vossius 1921, Wiesel 1922, Jung 1937 a zistili 22 členov z 300 vyšetrovaných osôb (cit. podľa 7). Keď Jaeger a Bischoff (14) vyšetrili príbuzných III. a IV. generácie pôvodného rodokmeňa, našli včasné štádiá tejto dedičnej choroby. Stanovili, že Bestova choroba a viteliformná dystrofia makuly sú synonymá.
Pojem „cysta vitelliformis“ sa presadil do retinálnej patológie pre všetky lézie na sietnici, hlavne na makule, ktoré mali vzhľad pripomínajúci vaječný žĺtok. Pritom termín „cysta“ sa v týchto súvislostiach chápe v klinickom zmysle, pretože nemá úplnú výstelku vlastných buniek svojej steny. Preto sa v takýchto prípadoch uprednostňovali pojmy ako pseudocysta, disk, terč alebo lézia. Rovnako sa nesprávne uvádzal patologický pochod s ohľadom na etiológiu procesu a zamieňali sa pojmy degenerácia s dystrofiou. V súčasnosti sa chápe degenerácia ako deteriorizačný proces, do ktorého môžu vyústiť patologické stavy rôznej etiológie (cievnej, toxickej, traumatickej, zápalovej), ale aj dystrofickej v konečnom štádiu vývoja a priebehu choroby. Akékoľvek zamieňanie týchto pojmov sa nepovažuje za správne, a preto uprednostňujeme zatiaľ označenie „viteliformná lézia“, pokiaľ nepoznáme bližšie povahu choroby.
VLASTNÉ POZOROVANIE
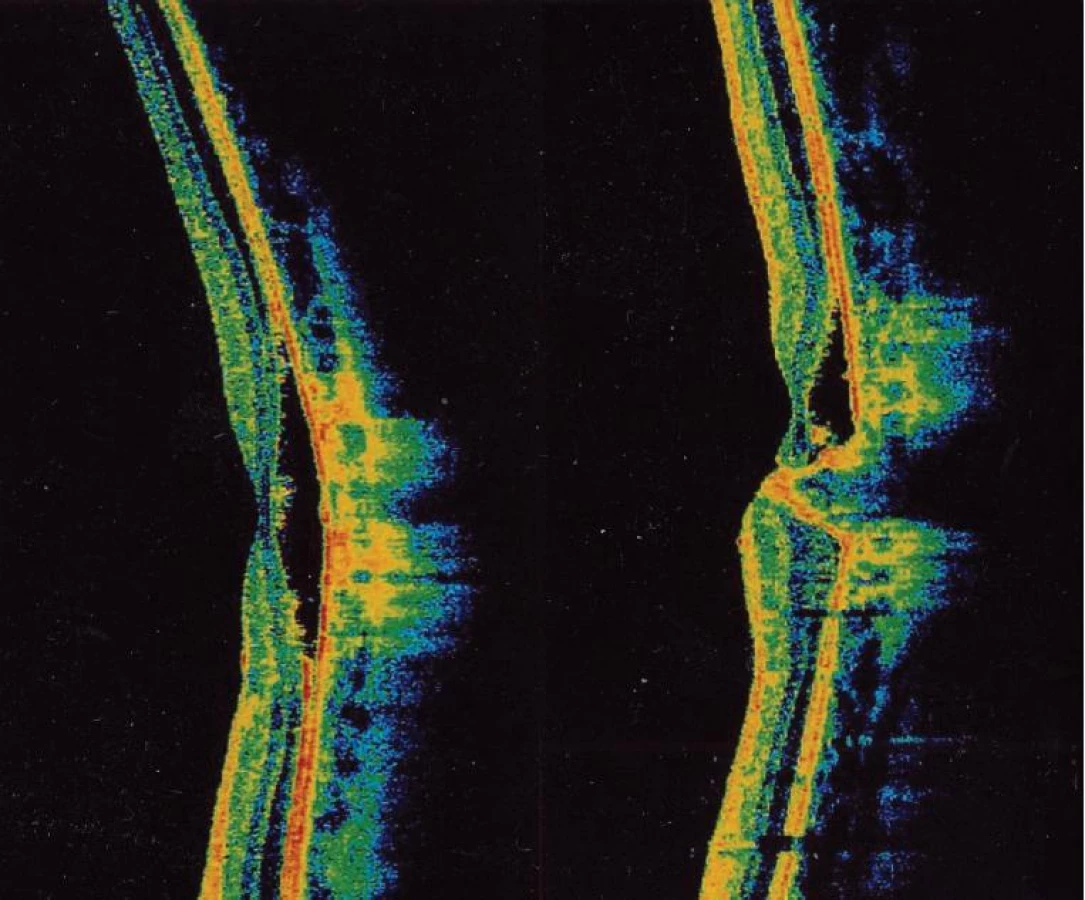
Naše poznatky o tejto dystrofii makuly sa zakladajú na pozorovaní 20 pacientov z troch rodokmeňov: č. 1 v V generáciách 47 rokov (12 pacientov), č. 2 v III generáciách 15 rokov (6 pacientov), č. 3 v II generáciách 14 rokov (2 pacienti). Ďalší 4 pacienti boli samostatné prípady bez dosiaľ dokázateľnej choroby v príbuzenstve. Všetci pacienti mali len obojstranné postihnutie makuly, väčšinou v rôznych štádiách vývoja viteliformného disku. U dostupných členov sme urobili základné oftalmologické vyšetrenia (centrálny vizus, biomikroskopia, zorné pole, farbocit, očné pozadie, fotografie fundu) a podľa potreby a súhlasu postihnutých aj viaceré kontrolné vyšetrenia. 17-krát sme doplnili vyšetrenia fluoresceínovou angiografiou fundu (FAG), 11-krát elektrofyziologickými testmi (ERG, EOG) a 6-krát optickou koherentnou tomografiou (OCT). V dvoch rodokmeňoch a v jednom samostatnom prípade sme zaznamenali u postihnutých obojstranné štádium viteliformného disku, v prvej dekáde v 3 prípadoch, v druhej dekáde u probandky na jednom oku (27). Pôvodný rodokmeň č. 1 z roku 1967 (27) sa doplnil neskôr o chorých v IV. a V. generácii a o extramatrimoniálneho syna III/14 (obr. 1). Jeho oftalmoskopický a FAG nález bol obojstranne pravidelný, ale mal patologický EOG. Jeho syn IV/7 v ôsmich rokoch pri našom FAG vyšetrení mal obojstranne obraz viteliformného disku v štádiu resorpcie žĺtkovej hmoty (obr. 2a, b). Priemerná veľkosť solitárneho viteliformného disku makuly bola v rozsahu 2–3 PD. Výnimkou boli len traja členovia II. generácie rodokmeňa č. 2, kde disk zaberal rozsah takmer celého zadného pólu (obr. 3a, b). V troch prípadoch sme zaznamenali krvácanie do disku z neovaskularizácie choriokapillaris, ktoré predstavovali najhoršie výsledky zrakových funkcií. U pacienta IV/7 v rodokmeni č. 1 v 27 rokoch na ľavom oku (obr. 4) a na OCT (obr. 5) vidieť na horizontálnom reze atrofiu vrstvy retinálneho pigmentového epitelu (RPE) subfoveolárne s prítomnými minimálnymi zvyškami hyperreflektívneho materiálu pred Bruchovou membránou. Medzi Bruchovou membránou a neuroretinou je opticky prázdny hyporeflektívny priestor. V tomto rozsahu je stenčenie vonkajšej jadrovej vrstvy. Na vertikálnom reze je zhora viditeľný opticky prázdny hyporeflektívny priestor typu ablácie neuroretiny, v ktorom sú viditeľné ostrovčeky hyperreflektívneho materiálu. Zdola je naopak viditeľná nápadná akumulácia hyperreflektívneho materiálu v podobe kužeľa, ktorá zasahuje až do vnútorných vrstiev neuroretiny k membrana limitans interna.





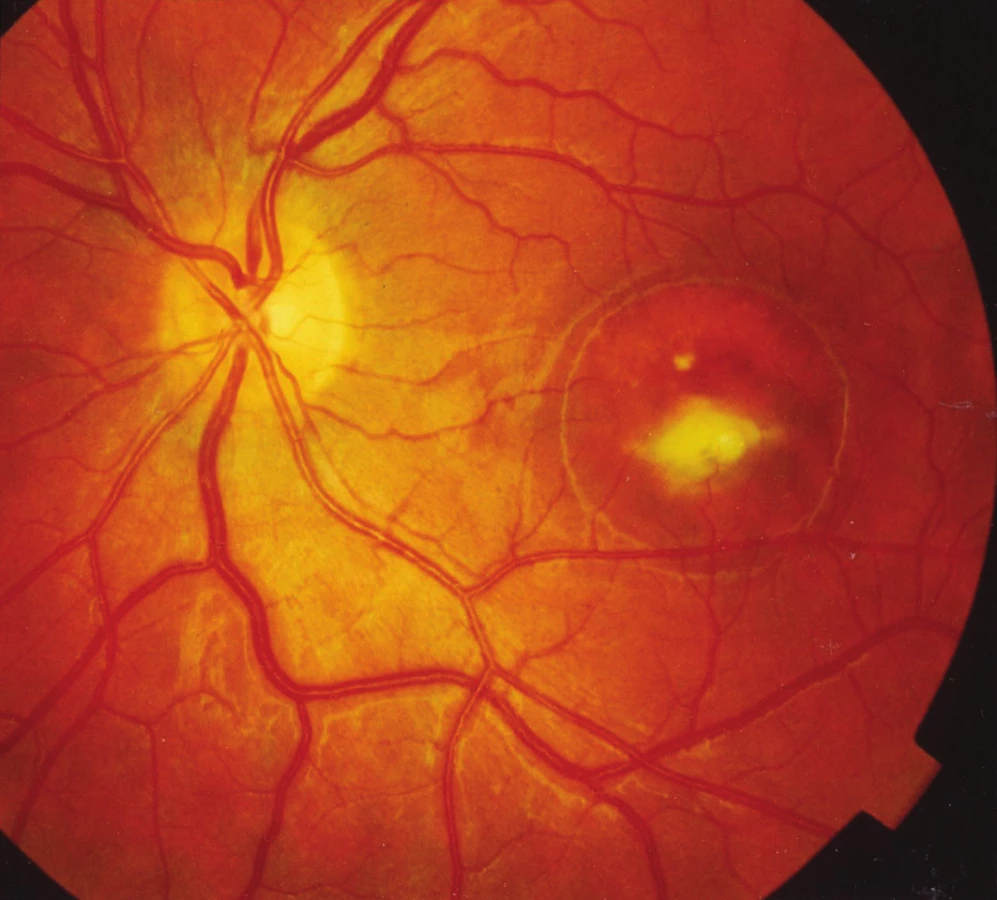
Resekréciu žĺtkového obsahu pseudocysty sme zaznamenali 2-krát. V jednom prípade išlo o samostatný nález u dievčaťa v prvom decéniu, v druhom prípade o dospelého muža z pôvodného rodokmeňa č. 1 z roku 1967 (27) II/4 v 43 roku veku so zmenou hladiny obsahu žĺtkovej hmoty polohou hlavy.
DISKUSIA
Bestova viteliformná makulárna dystrofia (BVMD) je relatívne častá dedičná choroba sietnicového stredu. Má niekoľko štandardných morfologických, elektrofyziologických, angiografických, genetických a funkčných prejavov. Medzi najcharakteristickejšie znaky choroby patria:
- autozomálne dominantný modus dedičnosti,
- patologický gén VMD2 na 11. chromozóme zodpovedný za chorobu a prenos,
- viteliformný solitárny disk makuly,
- patologický EOG,
- nízka alterácia centrálneho vizu v počiatočných štádiách,
- veľká pleomorfia expresivity počas vývoja medzi ostatnými hereditárnymi chorobami makuly,
- žiadna iná okulárna alebo systémová porucha,
- variabilný vek iniciálnej manifestácie choroby,
- veľká variabilita prejavu v intra a interfamiliárnych príbuzenstvách,
- častá hypermetropia bez alebo s astigmatizmom.
Autozomálne dominantný typ dedičnosti s inkompletnou penetranciou a variabilnou expresivitou je všeobecne akceptovaný. Ak však pripustíme, že inkompletnú penetranciu môže ovplyvniť aj patologický EOG, potom doteraz zaužívaný pojem inkompletná penetrancia sa môže meniť. Patologický gén bol odhalený v roku 1992 (8, 26) a označovaný ako VMD2, bol mapovaný na dlhom ramene 11. chromozómu v oblasti 11q12-q13.3. V roku 1998 boli prvýkrát zaznamenané mutácie vo VMD2 géne u pacientov s morbus Best. Tento gén kóduje transmembránový proteín bestrofín, ktorý je exprimovaný v RPE a nachádza sa špecificky v jeho bazolaterálnej plazmatickej membráne a čiastočne v apikálnych výbežkoch. Tu vytvára komplex chloridových kanálikov, ktoré sú zodpovedné za reguláciu transportu tekutín cez RPE. Predpokladá sa, že dysfunkcia bestrofínu vyústi do abnormálneho transportu tekutín a iónov cez RPE, čo následne spôsobí akumuláciu tekutín a debris v oslabenom priestore medzi RPE a fotoreceptormi, prípadne medzi RPE a Bruchovou membránou. Doteraz bolo opísaných viac ako 100 rôznych mutácií s morbus Best. Vo veľkom množstve ide o „missense mutácie“, ktoré vedú k definovaným výmenám aminokyselín v proteínovej sekvencii. Pri pozitívnej rodinnej anamnéze s patologickým EOG boli prakticky vo všetkých prípadoch dokázané mutácie vo VMD2 géne. Ďalší pokrok k objasneniu patogenézy sa očakáva od genetických výskumov a v poznaní zložitých pomerov štruktúr na úrovni bunkovej patológie (12, 33).
Štádiá vývoja viteliformného disku
Bestova choroba je progredujúce ochorenie a nikdy sa nezastaví na úrovni viteliformného disku. V tomto štádiu zostáva vo variabilnom stave, ktorý sa nedá časovo ohraničiť, pretože jeho začiatok nepoznáme. Bolo by potrebné rizikové prípady vyšetrovať v rámci postihnutého príbuzenstva v krátkych časových intervaloch, čo je prakticky nemožné. Z aspektu makromorfológie vývoja sa prijali určité charakteristické štádiá (2, 11, 13, 18, 28).
Štádium I – previteliformné. Má len veľmi necharakteristické alebo žiadne oftalmoskopické prejavy a bez poznania EOG a výskytu choroby v príbuzenstve sotva by sme niekoho zaradili medzi potenciálneho adepta na viteliformný disk.
Štádium II – viteliformné. Solitárny viteliformný disk, do ktorého je pojatá makula, zostáva vedúcim prejavom Bestovej choroby. Farba žĺtka je podstatnou veličinou, či už s inými prímesami v kolorácii disku, vždy je však základná. Obsahom žltej hmoty je nahromadenie žlto-oranžového pigmentu lipofuscínu v bunkách RPE v rozsahu centrálnej fovey. Lipofuscín sa všeobecne považuje za pigment starnutia. V RPE je odvodený od tukov vonkajších segmentov neurosenzorického epitelu pri procese natravovania a obnovovania vonkajších segmentov, vrátane poškodzovania absorpciou svetla a oxidáciou. Lipofuscín sa nachádza už vo včasnom veku v bunkách RPE celého fundu, ale klinicky najvýznačnejšie pri dystrofických chorobách makuly, akou je morbus Best. Mechanizmus, ktorým sa táto substancia akumuluje, je zatiaľ neznámy. K jeho špecifickým vlastnostiam patrí účasť na autofluorescencii makulárnych a extramakulárnych ložísk a blokáda fluorescencie pri FAG viteliformného disku makuly (20, 25). Každý nález takéhoto prejavu na makule nás musí zaväzovať k rutinnému screeningu ako genealogickému vyšetreniu všetkých možných a dostupných členov príbuzenstva. To rozhoduje, či je choroba dedičná alebo ide o samostatný prípad, o prvú mutáciu tejto choroby. V štádiu viteliformného disku je centrálny vizus prekvapujúco len málo alebo vôbec alterovaný. Dokazujú to vyšetrenia funkcií viazaných na foveolu (vizus do diaľky a blízka, farbocit, Haidingerove zväzky, makulárne ERG). To nepriamo naznačuje aj primárne miesto postihnutia a primárne nepoškodené fotoreceptory makuly. Často je nápadná prítomnosť hypermetropie bez alebo s astigmatizmom, čo môžeme potvrdiť aj u našich pacientov. V ďalších štádiách vývoja disku sú už hodnoty zrakovej ostrosti veľmi diferentné a deficit funkcií v každom prípade prítomný.
BMVD nie je spojená so žiadnou očnou alebo systémovou chorobou alebo nejakou abnormalitou s poruchami v biochemickom alebo hematologickém spektre. Výskyt viteliformného disku sa radí do obdobia prvého alebo druhého decénia približne medzi 5. až 15. rokom života, ale ojedinele aj v ďalších decéniách. O tom, či viteliformný disk je kongenitálne prítomný, sa uvažovalo už od prvých publikácií (3, 23). Takúto prítomnosť je ťažko potvrdiť a vyžadovalo by to vyšetriť každého novorodenca z postihnutého príbuzenstva.
Štádium III – pseudohypopyon. Obsah žĺtkovej masy po ukončení produkcie podlieha resorpčnému procesu cez skvapalnenie, pri ktorom sa v závislosti od gravitácie usadzuje s vodorovnou hladinou v potenciálnom priestore, ktorý označujeme ako pseudocysta. Podľa stupňa skvapalnenia sa obsah môže prelievať so zmenou polohy hlavy v rôznych časových reláciách. V našom prípade to bolo po 30 minútach. Súčasne s resorpciou obsahu sa odhaľujú lézie RPE a na FAG zóny hyperfluorescencie z transmisie farbiva. V zriedkavých prípadoch sa obsah pseudocysty môže naplniť znovu žĺtkovou hmotou, ako to pozorovali niektorí autori (11, 17, 23). Tento vzácny fenomén vo vývoji sme pozorovali aj my v dvoch prípadoch.
Štádium IV – (scrambled egg). Tu sa zvyšky masy jazvovite organizujú a pri FAG sa zvyšuje ich sfarbovanie imbibíciou do novotvoreného tkaniva v neskorej fáze. RPE atrofuje spolu s tkanivom, reaktívnou hyperpláziou a akumuláciou pigmentu.
Štádium V – atrofia a neovaskularizácia. V tomto poslednom štádiu sa vývoj uberá prevažne dvomi spôsobmi. Častejšie ide o úplnú atrofiu retinálnych a choroidálnych štruktúr s reaktívnou hyperpláziou RPE. Zriedkavejšie dochádza ku krvácaniu z defektov v Bruchovej membráne a následnej neovaskularizácie s choriokapilaris a tvorbou submakulárnej neovaskulárnej membrány a disciformnej jazvy (19, 30).
Elektrofyziologické vyšetrenia
Variabilná expresivita Bestovej choroby, zvlášť v neskorších štádiách a v neskoršom veku, predstavuje veľké diagnostické problémy a zaradenie tejto choroby. Pokiaľ nezaznamenáme viteliformný disk u jedného člena postihnutého príbuzenstva, zostáva nám nepostrádateľné elektrofyziologické vyšetrenie. Vo výsledkoch elektroretinografie (ERG) sa nezistili okrem makulárneho (pattern) ERG podstatné zmeny. Najčastejšie patologické nálezy sa ale zaznamenali pri EOG vyšetreniach (5). Vyjadrujú sa v pomere L/D (light/dark ratio) alebo Ardenov kvocient a predstavujú index funkčnej kapacity RPE. Deutman (6) stanovil, že L/D ratio nad 185 % je normálne, do 165 % pravdepodobne normálne, do 145 % pravdepodobne subnormálne, menej ako 145 % subnormálne a okolo 100 % jednoznačne patologické. EOG ale nie je jediným špecifickým testom pri BMVD a nedá sa hodnotiť na báze jedného sporadického prípadu.
Fluoroangiografické vyšetrenie
FAG u našich pacientov bolo rutinným vyšetrením každého postihnutého. Jednotlivé nálezy zodpovedali štádiám, v akých sme ich zachytili v čase prvého vyšetrenia buď náhodne pri zrakových problémoch pacienta, alebo v rámci genealogického vyšetrenia príbuzenstva. Charakteristický obraz disku naplnený žĺtkovým materiálom dáva pri FAG obraz blokády backgroud fluorescencie (7, 11). Takýto obraz sa dá zachytiť len zriedkavo. Prekvapil nás ale nález FAG u 6-ročnej pacientky ľavého oka IV/5 z rodokmeňa č. 1 s homogénnym diskom obojstranne (obr. 6a, b), keď sa od začiatku venóznej fázy sfarboval hypefluorescenciou celý disk. Túto oblasť lemovala úzka zóna hypofluorescencie zhusteného RPE. Sfarbovanie disku vo vnútri pigmentového prstenca môže mať rôzne vysvetlenia bez nároku na vysvetlenie: 1. buď je obsah disku riedky a RPE alterovaný tak, že neblokuje, ale odhaľuje choroidálnu fluorescenciu, pritom ešte nie je v stave úplného pseudohypopyonu s gravitáciou v dolných partiách. 2. disk sa sfarbuje akumuláciou farbiva v jeho obsahu cez disrupciu Bruchovej membrány s choriokapilaris. Identifikácia hustého pigmentového lemu okolo hyperfluorescentného terča, cez ktorý prestupujú perifoveolárne kapiláry, bol evidentný na farebných aj ortochromatických snímkach v preinjekčnej fáze. Našli sme ho v literatúre u iných autorov (2, 7, 10, 24) bez zvláštnej pozornosti a vysvetlenia. V ďalších fázach vývoja disku sa tento fenomén vytráca a nezaznamenali sme ho ani u troch ďalších pacientov.


Histopatológia
V roku 1982 vyšli paralelne dve histopatologické štúdie o BVMD, kde oči boli vyšetrené svetelnou a elektrónovou mikroskopiou. Obidva nálezy sa týkali člena príbuzenstva evidentne patriaceho k BVMD. Práca Weingeista a spol. (29) je zatiaľ podľa našich vedomostí demonštráciou najmladšieho postihnutého touto chorobou. Objektom histopatologickej štúdie bol 28-ročný muž, prvýkrát klinicky vyšetrený ako 15-ročný. Pochádza z rodokmeňa II. generácie ako najmladší zo štyroch členov mužského pohlavia. Počas 12-ročného sledovania nemal väčšie zrakové poruchy. Bol vyšetrený angiograficky, EOG mal patologické, získaný defekt farbocitu v ose červeno-zelenej mierneho stupňa a v oftalmoskopickom obraze zmeny vo vývoji viteliformnej lézie. V čase histologického spracovania boli oči v štádiu resorpcie žĺtkovej masy s odlúčením neurosenzorickej sietnice. Výsledky vyšetrení autorov tejto štúdie naznačovali difúznu poruchu RPE s hromadením lipofuscínových granúl, najviac v makule ako zodpovedné za materiál žĺtkovej lézie. Záver štúdie vyznel pre primárnu poruchu v RPE a pre úlohu akumulácie lipofuscínu v potenciálnom priestore medzi senzorickou retinou a RPE.
K iným výsledkom histopatologického vyšetrenia sa dopracovala skupina autorov okolo Frangieho a spol. (9). Objektom ich vyšetrení boli oči 80 ročnej ženy s dobre dokumentovanou BVMD z rodokmeňa so 6 postihnutými v III generáciách. Počas života sa prvé ťažkosti objavili okolo 50. roku života, kedy bola aj prvýkrát oftalmoskopicky vyšetrená. Nález odhalil obojstranne jazvovité zmeny a FAG neovaskularizáciu v obidvoch makulách. Nálezy dvoch príbuzných vo veku 8 a 14 rokov z III. generácie dokumentovali skoršie štádiá vývoja lézie a abnormálny EOG. Pri histologickom vyšetrení autori zistili difúzne ukladanie abnormálneho lipofuscínu v RPE v rozsahu makulárnej oblasti, ale extramakulárne difúzne zmeny neboli evidentné. Prominentným nálezom bol aj PAS pozitívny elektróndenzný granulárny materiál vo vnútorných segmentoch degenerovaných fotoreceptorov a Muellerových buniek. Autori usúdili, že primárnym miestom poruchy môže byť senzorická retina.
Obidva kontroverzné nálezy a názory na miesto primárnej poruchy komentoval Cavender (4) a dal do pozornosti ďalšieho výskumu v súvislosti s BVMD tri otázky: 1. Čo je žltý materiál? 2. Kde sa nachádza tento materiál vo vzťahu k RPE počas viteliformného štádia BVMD? 3. Kde tento žltý materiál vzniká?
Tretia histopatologická štúdia pochádza od OęGormana a spol. (22) z roku 1988. Objektom vyšetrenia boli oči 69-ročného muža s BVMD a klinickým obrazom včasného štádia na jednom a neskorým štádiom na druhom oku. Autori potvrdili nálezy predchádzajúcich dvoch prác, že bunky RPE akumulovali excesívne množstvo lipofuscínu a melanolipofuscínu a stratu buniek vo foveálnej a parafoveálnej oblasti pod RPE. Na podklade získaných nálezov naznačili hypotézu blízku práci Weigeista a spol., že miesto tvorby žĺtkovej masy je RPE a jeho uloženie v ňom a medzi ním a Bruchovou membránou. Disrupciu značne stenčeného RPE naplneného lipofuscínom a neschopnosťou degradovať materiál vonkajších segmentov fotoreceptorov v oblasti makuly vnikne masa do subretinálneho priestoru a začne ďalšiu deštrukciu senzorickej retiny.
Klinická skúsenosť potvrdená viacerými autormi, ako aj vyšetrením 4 našich očí v štádiu viteliformného disku jednoznačne hovorí o minimálnom deficite zrakových funkcií viazaných na foveolu. Z toho vyplýva, že v tomto štádiu nebudú primárnym miestom patológie BVMD fotoreceptory, ale ich poškodenie bude sekundárne.
ZÁVER
Veľká polymorfia vývojových štádií viteliformnej dystrofie makuly vyžaduje štandardný postup v diferenciálnej diagnóze tejto makulárnej choroby. Najspoľahlivejší dôkaz je nález viteliformného disku a jeho posúdenie s ohľadom na vek. Nasleduje vyšetrenie všetkých dostupných členov príbuzenstva pre stanovenie, či choroba je prvou mutáciou, alebo má definitívnu hereditárnu povahu. Nasledujú funkčné a zobrazovacie vyšetrenia s aspektom na určenie morfologického štádia na dôkaz nepostihnutých a nosičov patologického génu, ako aj na predpokladaný prirodzený priebeh choroby. Po získaní jednoznačného záveru je potrebné hovoriť v rámci poradenstva o všetkých podstatných aspektoch choroby. K tomu patrí vysvetlenie povahy choroby, jej priebehu, vplyvu na povolanie a okolnosti bežného života, možnosť dedičnosti a jej riziká a čo všetko je možné poskytnúť v aktuálnom čase choroby pre zlepšenie kvality zraku.
Kauzálna liečba viteliformnej dystrofie makuly zatiaľ neexistuje. Naše úsilie sa zameriava na optimálnu korekciu refrakčných chýb, na zachovanie čírosti optických médií a na prispôsobenie sa podmienkam osvetlenia okolia. Len v niektorých situáciách a celkom výnimočne pri sub a parafoveolárnych neovaskularizáciách je metódou voľby laserová liečba.
Do redakce doručeno dne 24. 9. 2012
Do tisku přijato dne 16. 11. 2012
MUDr. Teodor Streicher
Hornoulická 9
972 01 Bojnice
Slovenská republika
Zdroje
1. Benson, W.E., Kolker, A.E., Enoch, J.M. et al.: Best`s vitelliform makular dystrophy. Am J Ophthalmol, 79; 1975, 1 : 59–66.
2. Blodi, CH.F., Stone, E.M.: Best`s vitelliform dystrophy. Opthalmic Paediatrics and Genetics, 11; 1990, 1 : 49–59.
3. Braley, A.E., Spivey, B.E.: Hereditary vitelline macular degeneration. Arch Ophthalmol, 72; 1964 : 743–762.
4. Cavender, J.C.: Best`s macular dystrophy. Arch Ophthalmol, 100; 1982, 7 : 1067.
5. Cross, H.E., Bard, L.: Electro-oculography in Best`s macular dystrophy. Am J Ophthalmol, 77, 1974, 1 : 46–50.
6. Deutman, A.F.: Electro-oculography in families with vitelliform dystrophy of the fovea. Arch Ophthalmol, 81, 1969, 3 : 305–316.
7. Deutman, A.F.: The hereditary dystrophies of the posterior pole of the eye. Van Gorcum, Assen, 1971, s. 198–299.
8. Forsman, K., Graff, C., Nordström, S., et al.: The gene for Best`s macular dystrophy is located at 11q13 in a Swedish famili. Clin Genet, 42;1992 : 156–159.
9. Frangieh, G.T., Green, R.W., Fine, S.L.: Histopathologic study of Best`s macular dystrophy. Arch Ophthalmol, 100; 1982, 7 : 1115–1121.
10. Friedenwald, J.S., Maumenee, E.A.: Peculiar macular lesions with unaccountably good vision. Arch Ophthalmol, 45; 1951 : 567–569.
11. Godel, V., Chaine, G., Regenbogen, L., et al.: Best`s vitelliform macular dystrophy. Acta Ophthalmol, Supplement 175; 64, 1986 : 5–31.
12. Hartzell, C.H., Qu,Z., Yu, K., et al.: Molecular physiology of bestrophins: multifunctional membrane proteins linked to Best disease and other retinopathies. Physiol Rev, 88; 2008 : 639–672.
13. Huismans, H.: Cysta vitelliformis – Bericht über ein seltenes heredodegeneratives Makulaleiden. Klin Mbl Augenheilk, 166; 1975, 2 : 252–254.
14. Jaeger, W., Bischoff, E.: Vitelliforme Makuladegeneration und Bestsche Makuladgeneration sind dasselbe Krankheitsbild. Klin Mbl Augenheilk, 170; 1977, 6 : 890–899.
15. Kingham, J.D., Lochen, G.P.: Vitelliform macular degeneration. Am J Ophthalmol, 84; 1977, 4 : 526–531.
16. Krämer, F.,White, K., Pauleikhoff, D. et al.: Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur J Hum Genet, 8; 2000, 4 : 286–292.
17. Kraushar, M.F., Margolis, S., Morse, P.H. et al.: Pseudohypopyon in Best`s vitelliform macular dystrophy. Am J Ophthalmol, 94; 1982, 1 : 30–37.
18. Lisch, W.: Die verschiedenen Stadien der vitelliformen Makuladegeneration. Klin Mbl Augenheilk, 176; 1980, 2 : 214–221.
19. Miller, S.A., Bresnick, G.H., Chandra, S.R.: Choroidal neovascular membrane in Best`s vitelliform macular dystrophy. Am J Ophthalmol, 82; 1976, 2 : 252–255.
20. Miller, S.A.: Fluorescence in Best`s vitelliform dystrophy, lipofuscin, and fundus flavimaculatus. Brit J Ophthalmol, 62; 1978 : 256–260.
21. Morse, P.H., MacLean, A.I.: Fluorescein fundus studies in hereditary vitelliruptive macular degeneration. Am J Ophthalmol, 66; 1968, 9 : 485–494.
22. O`Gorman, S., Flaherty, W.A., Fishman, G.A. et al.: Histopathologic findings in Best`s vitelliform macular dystrophy. Arch Ophthalmol, 106; 1988, 9 : 1261–1268.
23. Remky, H., Rix, J., Klier, K.F.: Dominant – autosomale Maculadegeneration (Best, Sorsby) mit zystischen und vitelliformen Stadien (Huysmans, Zanen). Klin Mbl Augenheilk, 146; 1965, 4 : 473–497.
24. Schum, U.: Fluorescenzangiographie bei vitelliformer Maculadegeneration. Bericht 70, Zukunft DOG, Heidelberg, 1969 : 252–257.
25. Spaide,R.: Autofluorescence from the outer retina and subretinal space: Hypothesis and review. Retina, 28; 2008, 1 : 5–35.
26. Stone, E.M., Nichols, B.E., Streb, L.M. et al.: Genetic linkage of vitelliform macular degeneration (Best`s disease) to chromosome 11q13. Nat Genet, 1; 1992 : 246–250.
27. Streicher, T.: Viteliformná heredodegenerácia makuly. Čs Oftal, 23; 1967, 6 : 423–428.
28. Thiel,H.J., Behnke,H.: Klinik und Vererbung der vitelliformen Maculadegeneration. Klin.Mbl.Augenheilk., 158, 1971, 2 : 235–246.
29. Weingeist, T.A., Kobrin, J.I., Watzke, R.C.: Histopathology of Bestęs makular dystrophy. Arch Ophthalm, 100; 1982 : 1108–1114.
30. Záhlava, J., Karel, I., Lešták, J.: Bestova viteliformní dystrofie komplikovaná neovaskulární membránou a krvácením. Čes a Slov Oftal, 58; 2002, 3 : 158–164.
31. Zanen, J., Rausin, G.: Kyste vitelliforme congénital de la macula. Bull Soc belge Ophthal, 96; 1950 : 1–5.
32. Zanen, J., Rausin, G.: Kyste vitelliforme congénital de la macula. Bull Soc belge Ophthal, 98, 1951 : 1–2.
33. Yu, K., Qu, Z., Cui, Y. et al.: Chloride channel activity of bestrophin mutants associate with mild or late-onset macular degeneration. Invest Ophthalmol Vis Sci, 48; 2007 : 4694-4705.
Štítky
OftalmologieČlánek vyšel v časopise
Česká a slovenská oftalmologie

2012 Číslo 5
- Selektivní laserová trabekuloplastika nesnižuje nitroční tlak více než argonová laserová trabekuloplastika
- Progresi glaukomu je třeba hodnotit strukturálními i funkčními parametry
- Ztráta centrálního vidění po filtrujících operacích glaukomu
- Od PGF-2 alfa-isopropyl esteru k latanoprostu: přehled vývoje Xalatanu
- Compliance u pacientů s glaukomem
Nejčtenější v tomto čísle
- Klinická rozmanitosť Bestovej choroby
- Aplikace Dysportu® – možnost doprovodného vedlejšího efektu na postavení víček (klinicko-histologická studie)
- Pars plana vitrektómia a kombinovaná liečba pars plana vitrektómia, triamcinolon acetonid intravitreálne a laserkoagulácia makuly pri diabetickom edéme makuly – ročné výsledky
- Srovnání metody nastavitelných versus fixních stehů při operaci strabismu u pacientů s endokrinní orbitopatií
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy