
Sine syndrómy v klinickom obraze dermatomyozitídy
Sine syndromes in clinical picture of dermatomyositis
This work deals with the issue of sine syndromes in the clinical picture of dermatomyositis. It provides a literature overview of the aforementioned sine syndromes, which is characterized by not fulfilling the diagnostic criteria for dermatomyositis; however, this disease may have a severe course. In the presented case report, we have described the development of acute interstitial pneumonia in systemic polymyositis without clinical signs of muscular involvement, with anti-Jo 1 positivity and elevated levels of myoglobin and troponin. EMG examination did not confirm any disorder in terms of myositis or neuromuscular transmission. This case report points out the necessity of examination of rheumatologic, immunological and interstitial lung involvement in cases of sine syndrome. The efficacy of glucocorticoid therapy is pointed out as well.
Key words:
dermatomyositis, sine syndromes, interstitial lung involvement, rheumatologic and immunological examination
Autoři:
J. Rovenský 1; M. Vašáková 2
Působiště autorů:
Národný ústav reumatických chorôb Piešťany
1; Pneumologická klinika 1. LF UK, Fakultní Thomayerova nemocnice s poliklinikou, Praha
2
Vyšlo v časopise:
Čes. Revmatol., 19, 2011, No. 1, p. 11-16.
Kategorie:
Přehledné články
Souhrn
Práca sa zaoberá problematikou sine syndrómov v klinickom obraze dermatomyozitídy. Uvádza literárny prehľad vyššie uvedeného sine syndrómu, ktorého charakteristika je v tom, že nespĺňa diagnostické kritéria dermatomyozitídy, ale choroba môže mať závažný priebeh. V kazuistickom zdelení sme popísali vývoj pľúcnej akútnej intersticiálnej pneumónie pri systémovej polymyozitíde bez klinických príznakov svalového postihnutia s pozitivitou anti-Jo 1 a eleváciou myoglobínu a troponínu. EMG vyšetrenie nepreukázalo poruchu v zmysle myozitídy alebo neurosvalového prevodu. Kazuistika poukazuje na nutnosť vyšetrenia reumatologického a imunologického aj intersticialneho pľúcneho postihnutia v rámci sine syndrómu. Poukazuje sa na úspešnosť liečby glukokortikoidmi.
Kľúčové slová:
dermatomyozitída, sine syndrómy, postihnutie pľúcného interstícia, reumatologické a imunologické vyšetrenie
Reumatické choroby sú charakteristické tým, že ich diagnóza je stanovená podľa súboru kritérií definovaných skupinami na základe klinického priebehu a laboratórnych parametrov, ktoré umožňujú nozografické ohraničenie danej choroby. Na druhej strane sú však ochorenia hlavne zo skupiny difúznych chorôb spojiva, ktoré nespĺňajú diagnostické kritériá, a preto nie je možné podľa medzinárodných kritérií stanoviť diagnózu ochorenia, pritom však priebeh choroby môže byť závažný.
Jednou z takýchto nozologických jednotiek môže byť amyopatická dermatomyozitída, ktorá má kožné prejavy, ale bez postihnutia svalov, čím sa diagnóza dá predpokladať, ale nedá sa podľa diagnostických kritérií dokázať.
Sontheimer (1) zverejnil históriu klinického popisu dermatomyozitídy, všeobecne je prijatý názor, že Wagner (2) v roku 1863 a Unverricht (3) v roku 1887 popísali ochorenie, ktoré sa nazývalo ako Wagner-Unverrichtova choroba. Heinrich Göttron (4) v roku 1931 bol jedným z prvých, ktorý urobil úplný popis kožnej manifestácie dermatomyozitídy, vrátane úplného popisu atrofických fialových papulóznych vyrážok charakteristických ako vyvýšenie dorzálnej strany MCP kĺbov. Na druhej strane nepopísal sa vzťah medzi kožnou a systémovou manifestáciou dermatomyozitídy.
Pri ich retrospektívnej analýze 40 pacientov s dermatomyozitídou, ktorí sa diagnostikovali na Mayo Clinic, OęLeary a Waisman (5) zistili, že u 14 pacientov bol prítomný edém a kožné lézie. Ochorenie charakterizovali tak, že začiatok choroby bol spojený s dermatologickým postihnutím nasledovaným subklinickými zápalovými svalovými zmenami, v priebehu niekoľkých týždňov alebo mesiacov.
V roku 1942 Harvey Keil (6) podrobne popísal kožnú formu dermatomyozitídy, ktorá bola označovaná ako Keilova varianta dermatomyozitídy, jednalo sa o amyopatickú formu dermatomyozitídy, ktorá bola charakterizovaná ako kožná forma dermatomyozitídy s klasickými kožnými zmenami bez svalového postihnutia po mnohé roky.
História amyopatickej dermatomyozitídy pokračuje pozorovaním Pearsona (7) u 6-ročného dievčaťa s florídnou formou kožných prejavov dermatomyozitídy, pričom v priebehu dvoch rokov nebola prítomná klinická syndromológia zo strany postihnutia svalstva, ani nebola prítomná enzymatická odozva. Pearson (7) ďalej uvádza kazuistiky piatich žien, ktoré mali typický heliotropný exantém s prítomnosťou erytematóznych plátov v okolí lakťových kĺbov, pričom sa však nezistilo žiadne poškodenie svalstva. U jednej pacientky choroba trvala 13 rokov. U ďalších 6 pacientov /štyria muži a dve ženy/, raš bol florídny, na druhej strane svalová slabosť a EMG zmeny boli minimálne. Takýto variant by sa mohol nazvať amyopatická dermatomyozitída. Pearson prvý použil termín amyopatická dermatomyozitída. Problematika sine syndrómu pri amyopatickej dermatomyozitíde však pokračovala ďalej, Krain (8) popísal u 6 pacientov kožné zmeny pri dermatomyozitíde bez svalového postihnutia, neskoršie sa však u pacientov vyvinula polymyozitída. Prvou pacientkou bolo 10-ročné dievča, u ktorého kožné zmeny predchádzali svalovú slabosť 4-mesiace. U druhého pacienta mužského pohlavia kožné zmeny predchádzali svalovej chorobe 6 rokov. U 6 pacientok 3 a 4 kožné ochorenie predchádzalo 5 rokov, pokiaľ sa vyvinula svalová choroba. U 5. pacienta kožné zmeny progredovali a svalová slabosť začala byť klinicky preukazná až za 6 rokov. U 6. pacienta sa zistila tiež pľúcna fibróza. Krain (8) zo svojich pozorovaní vyvodil záver, že zlyhanie rozpoznania dermatomyozitídy pri neprítomnosti svalovej slabosti napriek charakteristickej kožnej erupcii má za následok oneskorenie diagnózy, všeobecne bola horšia prognóza choroby tam, kde bola rezistencia na liečbu glukokortikoidmi.
V roku 1977 Bohan a spol. (9) publikovali prácu s analýzou 153 pacientov s polymyozitídou a dermatomyozitídou. V tejto skupine autori popísali troch pacientov, u ktorých sa nevyvinula svalová slabosť, hoci iné kritéria boli splnené.
Všeobecne sa predpokladá na základe ďalších zistení, že interval medzi kožnými zmenami a vývojom myozitického syndrómu je menej ako dva roky, často menej ako 6 mesiacov.
V priebehu nasledujúcich rokov ďalší autori poukázali na možnosť sine syndrómu pri dermatomyozitíde (10–16), poukázali na skutočnosť, že kožná forma dermatomyozitídy je možná bez prítomnosti svalového syndrómu, pričom sa môže vyskytnúť aj fibrotizujúca alveolitída (10) a nemusí sa dostaviť terapeutický efekt glukokortikoidov samotných, ale až v kombinácii s antimalarikami (11). U jedincov mladších ako 25 rokov boli kožné zmeny pozorované 4 mesiace pred objavením sa svalových ťažkostí a u 9 pacientov nad 25 rokov 8 mesiacov.
Sontheimer (1) uvádza svoje osobné skúsenosti s amyopatickou dermatomyozitídou u 6 pacientov – piatich dospelých a jedného detského pacienta, ktorý mal jednoznačne kožné manifestácie dermatomyozitídy, ale nie svalovú slabosť, neboli zmeny v aktivite svalových enzýmov počas prvých dvoch rokov choroby. Na základe klinických skúseností, keď vyššie uvedené príznaky pretrvávali počas 6 alebo viac mesiacov, ale nie viac ako 24 mesiacov, jednalo sa o predbežnú formu amyopatickej dermatomyozitídy, ale v prípade, že to bolo 24 a viac mesiacov, môže sa so spoľahlivosťou hovoriť o potvrdenej amyopatickej dermatomyozitíde (1, 22). Od amyopatickej dermatomyozitídy treba však odlíšiť hypomyotickú dermatomyozitídu, u ktorej je subklinicky prebiehajúca myopatia dokázaná paraklinickými vyšetreniami /EMG, svalovou biopsiou, magnetickou rezonanciou (17) a ďalšími technikami vrátane imunologických – porušená mikrovaskulatúra kapilárneho riečišťa z polotenkých rezov s využitím lektínu Ulex europeus (18) a dôkaz depozitov C5b9 komplexu atakujúceho membrány (19)/. Lam a spol. (17), uviedli vlastné skúsenosti u 40 pacientov s dermatomyozitídou, u ktorých 10 pacienti boli diagnostikovaní ako amyopatická forma dermatomyozitídy. Magnetická rezonancia môže hrať úlohu pri určení lokalizácie pre biopsiu a prípadne pre jej opakovanie v časovom odstupe, taktiež pre odhalenie zápalových zmien pri klinickom podozrení na amyopatickú dermatomyozitídu. Sontheimer (1) vo svojom prehľadnom článku uviedol, že v roku 2001 bolo v databáze PubMed 49 citácii so slovom amyopatický, pričom len dve citácie uvádzajú termín dermatomyositis bez myozitídy pred rokom 1991. Dôležitým faktom je, že amyopatická dermatomyozitída sa nemusí vyvíjať len u dospelých, ale môže sa vyvíjať aj u detí. Plamondon a Dent (20) uviedli, že u 27 diagnostikovaných pacientov so začiatkom v detskom veku /juvenile-onset amyopathic dermatomyositis/ 10 pacientov bolo liečených celkovo a u 5 sa dosiahla remisia. U ďalších pacientov, ktorí neboli liečení, došlo k remisii spontánne.
Filo (19) definuje amyopatickú dermatomyozitídu vtedy, keď kožné zmeny predchádzajú myozitídu v rôznom časovom intervale. Amyopatická dermatomyozitída sa teda považuje za zriedkavú, ale dobre definovanú formu dermatomyozitídy. Vyskytuje sa u 2–18 % chorých (7, 19, 21, 22). Sontheimer (1) považuje za istú amyopatickú dermatomyozitídu, ktorá pretrváva najmenej dva roky, pri neprítomnosti svalovej slabosti, bez zvýšenej aktivity svalových enzýmov a taktiež nie je dokázaná myopatia modernými diagnostickými prístupmi.
Čo sa týka imunologického obrazu, popisuje sa, že u podskupiny pacientov s amyopatickou dermatomyozitídou sa môžu zistiť protilátky proti antip155 a anti-Se. V tomto smere je však potrebné naďalej bádať (1). Autor vyzýva v článku na možnosť analýz týchto protilátok v jeho laboratóriu v prípade, že je podozrenie na amyopatickú dermatomyozitídu.
Na problematiku amyopatickej dermatomyozitídy je potrebné sa sústrediť aj z aspektu orgánových prejavov, napríklad vývoja intersticiálneho pľúcneho postihnutia /IPP/ (23). Autori upozornili na výskyt u 10 publikovaných prípadov dermatomyozitídy s kožnými príznakmi bez svalového postihnutia /slabosti/, pričom u týchto pacientov sa vyskytol IPP v časovom ohraničení menej ako 6 mesiacov po objavení sa kožných príznakov /autori použili termín pre-myopatická forma dermatomyozitídy/. Ochorenia mali závažný priebeh, pretože siedmi pacienti zomreli, krátko po objavení sa kožných prejavov.
Cao a spol. (24) u skupiny 16 pacientov, ktorí spĺňali kritéria amyopatickej dermatomyozitídy, zistili IPP spojené s respiračnými problémami u 3 pacientov. To znamená, že amyopatická dermatomyozitída môže mať fatálnu komplikáciu z dôvodu vyššie uvedeného IPP. Klinicky sa jednalo o prítomnosť neproduktívneho kašľa, spojeného s rtg obrazom s bazálnymi rachotami na obidvoch pľúcnych bázach, u jedného pacienta došlo k progresívnej dyspnoe a ťažkej hypoxémii, ktorá bola rezistentná na liečbu a pacient zomrel za tri týždne od objavenia sa respiračných symptómov. Funkčné testy pľúc poukázali u 6 pacientov na redukovanú difúznu kapacitu. Okrem problematiky vývoja respiračnej insuficiencie na podklade vývoja IPP pri amyopatickej dermatomyozitíde je možný aj vývoj nádorovej choroby v rámci paraneoplastických syndrómov. Cao a spol. (24) poukázali aj na vývoj karcinómu pankreasu, metastatického adenokarcinómu a nasofaryngeálneho karcinómu. Ďalšia možnosť je vývoj napríklad chronického kožného lupus erythematosus. Čo sa týka imunologického obrazu amyopatickej dermatomyozitídy, 5/16 pacientov mali prítomnosť zrnitého obrazu ANA, ani jeden pacient nemal prítomnosť protilátok Ro/SS-A, La/SS-B alebo U1 RNP v priebehu dvoch rokov od objavenia kožných zmien. 1 pacient mal prítomnosť protilátok proti ds-DNA. U pacienta s chronickým kožným lupus erythematosus sa objavili protilátky antiRo/SSA a antiLa/SS-B, ale až po dvoch rokoch od stanovenia kožných zmien. Čo sa týka terapeutického prístupu, doporučuje sa v začiatku liečba prednizónom /15-40 mg/deň/. Dávku glukokortikoidov je možné znížiť kombináciou s metotrexátom alebo antimalarikami. U štyroch pacientov liečených pre amyopatickú dermatomyozitídu sa objavila malignita, ktorá bola riešená chemoterapiou a rádioterapiou. Traja pacienti, u ktorých sa vyvinula IPF, boli liečení metylprednizolónom (80–120 mg/deň) v kombinácii s metotrexátom. U dvoch pacientov sa zlepšili ako kožný nález, tak aj respiračné problémy. U jedného pacienta vývoj LPF mal fatálny priebeh napriek liečbe. Ďalšou z možností je tiež vývoj klasickej dermatomyozitídy, ako tomu bolo u jedného pacienta /Cao a spol. (24)/.
Pľúcne postihnutie sa vyskytuje u PM/DM relatívne často (5–30 %), väčšinou v rámci tzv. antisyntetázového syndrómu. Najčastejším typom postihnutia pľúc je nešpecifická intersticiálna pneumónia (NSIP), niekedy organizujúca sa pneumónia (OP), u akútnych foriem môže byť aj obraz akútnej intersticiálnej pneumónie so syndrómom dychovej tiesne s histopatologickým obrazom difúzneho alveolárneho poškodenia (AIP/ARDS/DAD). Obraz IPP môže byť modifikovaný svalovým postihnutím dychových svalov a svalov hrtanu (opakované aspirácie).
IPP u PM/DM môže prebiehať buď pod obrazom akútneho, subakútneho, alebo chronického postihnutia interstícia. Ako akútne a subakútne označujeme priebehy, kedy trvanie symptómov, t.j. progredujúcej dušnosti, je kratšie ako 3 mesiace. V prípade akútneho priebehu je väčšina pacientov prijímaných k hospitalizácii pre dušnosť, naopak chronické IPP v rámci PM/DM môžu prebiehať aj klinicky bez prejavov. Väčšinou je prítomné postihnutie kože a svalov, niekedy sa ale môžeme stretnúť s amyopatickou formou PM/DM, niekedy môže chýbať aj kožné postihnutie (25–28). Na skiagrame hrudníka je popísaný u viac než 90 % chorých normálny nález, vzácne sa objavuje retikulácia a retikulonodulácia, zvlášť v dolných pľúcnych poliach. Pri postihnutí bránice dochádza k jej elevácii a v pľúcnych bázach sa objavujú platničkovité atelektázy. Pri počítačovej tomografii hrudníka s vysokou rozlišovacou schopnosťou (HRCT) pozorujeme obraz opacít mliečneho skla, retikulácie a peribronchiálne kondenzácie. V bronchoalveolárnej lavážnej tekutine býva zvýšený celkový počet buniek a v rozpočte je zvýšené zastúpenie lymfocytov, a to hlavne u akútnych a subakútnych foriem. Liečebne podávame kortikoidy v dávke 0,75–1 mg/kg/deň, u akútnych foriem volíme pulzné podanie v dávke 1000 mg Solu-Medrolu denne v úvode liečby, väčšinou pridávame aj imunosupresíva: cyklofosfamid v pulznom režime 500–700 mg mesačne, alternatívou môže byť azathioprín 1–2 mg/kg/deň alebo cyklosporín 2–3 mg/kg/deň. U akútnych fulminantných priebehov môžeme skúsiť intravenózne podanie gamaglobulínov.
AIP u PM/DM môže mať aj fulminantný priebeh, navzdory liečbe, až polovica týchto pacientov zomrie na respiračné zlyhanie do 1–2 mesiacov (5-ročné prežitie 35 %). Chronické IPP majú prognózu jednoznačne lepšiu, 5 rokov prežíva 100 % pacientov. Vzhľadom k závažnosti akútnych a subakútnych foriem pľúcneho postihnutia pri PM/DM musíme po IPP aktívnym screeningom pátrať.
V našom príspevku poukazujeme na možnosť výskytu amyopatickej a adermopatickej PM/DM, s pľúcnym postihnutím ako jediným klinickým prejavom ochorenia.
Kazuistika
V našej kazuistike popisujeme prípad pacientky, ktorá bola prijatá na Pneumologickú kliniku 1. LF UK Fakultnej Thomayerovej nemocnice prekladom z interného oddelenia okresnej nemocnice pre podozrenie na intersticiálny pľúcny proces k dovyšetreniu. Rodinná anamnéza pacientky neobsahovala záznamy o pľúcnych a autoimunitných ochoreniach. T. č. 69-ročná pacientka pracovala ako predavačka, teraz je už ale na starobnom dôchodku. Doma chovala sliepky, teraz už zvieratá nechová a pred 3 rokmi mali v rodinnom dome, kde býva, plesne, teraz ale už nemajú. Alergie na lieky, potraviny, či akékoľvek inhalačné alergény negovala. Fajčila od 25 rokov maximálne 10 cigariet denne, posledných 25 rokov ale nefajčí. Alkohol nepije. Čo sa týka gynekologickej anamnézy, pacientka je po opakovaných kyretážach pre polypy, nikdy nebola tehotná. Lieči sa pre glaukóm posledných 14 rokov, pre vysoký tlak 13 rokov. Má chronickú žilovú insuficienciu posledných 10 rokov. Pred 2 rokmi prekonala úraz s luxáciou hlavice humeru. Má cholecystolitiázu, náhodne zistenú korovú cystu pravej obličky a hyperurikémiu. Trvalo užívala tieto lieky: Micardis 80 mg 1-0-0, Furon 40 mg 2-0-0, Verospiron 25 mg 1-0-1, Citalec 20 mg 1-0-0 a Lusopress 0-0-1.
Od apríla 2010 pacientka pozorovala neproduktívny kašeľ a námahovú dušnosť, ktorej pozvoľna predchádzala aj dušnosť pokojová, zvýraznili sa občasné bolesti za prsnou kosťou, netolerovala polohu ležmo a začal jej opuch nôh. Navštívila pľúcneho lekára a vzhľadom k rádiologickému nálezu bola vyšetrovaná s podozrením na vnútrohrudnú sarkoidózu. Pre zhoršenie ťažkostí musela byť 24. 7. 2010 hospitalizovaná na internom oddelení nemocnice v Benešove. Bola prijímaná s diagnózou akcelerovaná hypertenzia a známky pravostrannej kardiálnej dekompenzácie. Bolo vykonané echokardiografické vyšetrenie s nálezom perikardiálneho výpotku, dilatácie ľavej komory a známok pľúcnej hypertenzie. CT angiografia nepreukázala pľúcnu embóliu, podľa CT boli prítomné známky kardiálnej dekompenzácie a pľúcneho edému, okrsky opacít mliečneho skla nevylučovali aj intersticiálny pľúcny proces (obr. 1, 2). S týmto podozrením potom bola na základe pľúcneho konzília pacientka prijatá na našu kliniku. Pri prijatí bola afebrilná, nesťažovala si na bolesti svalov ani kĺbov. V popredí ťažkostí bol suchý kašeľ, únava a pokojová dušnosť. Schudla približne 15 kg od apríla 2010. Pri fyzikálnom vyšetrení bola konštatovaná obezita, pokojovo nebola pozorovaná manifestná dušnosť, nebola prítomná cyanóza ani ikterus. Pri počúvaní boli zaznamenané chrapoty pod oboma lopatkami, srdečné akcie boli pravidelné a pacientka bola normotenzná. Koža bola bez eflorescencií, dolné končatiny boli už bez opuchov.

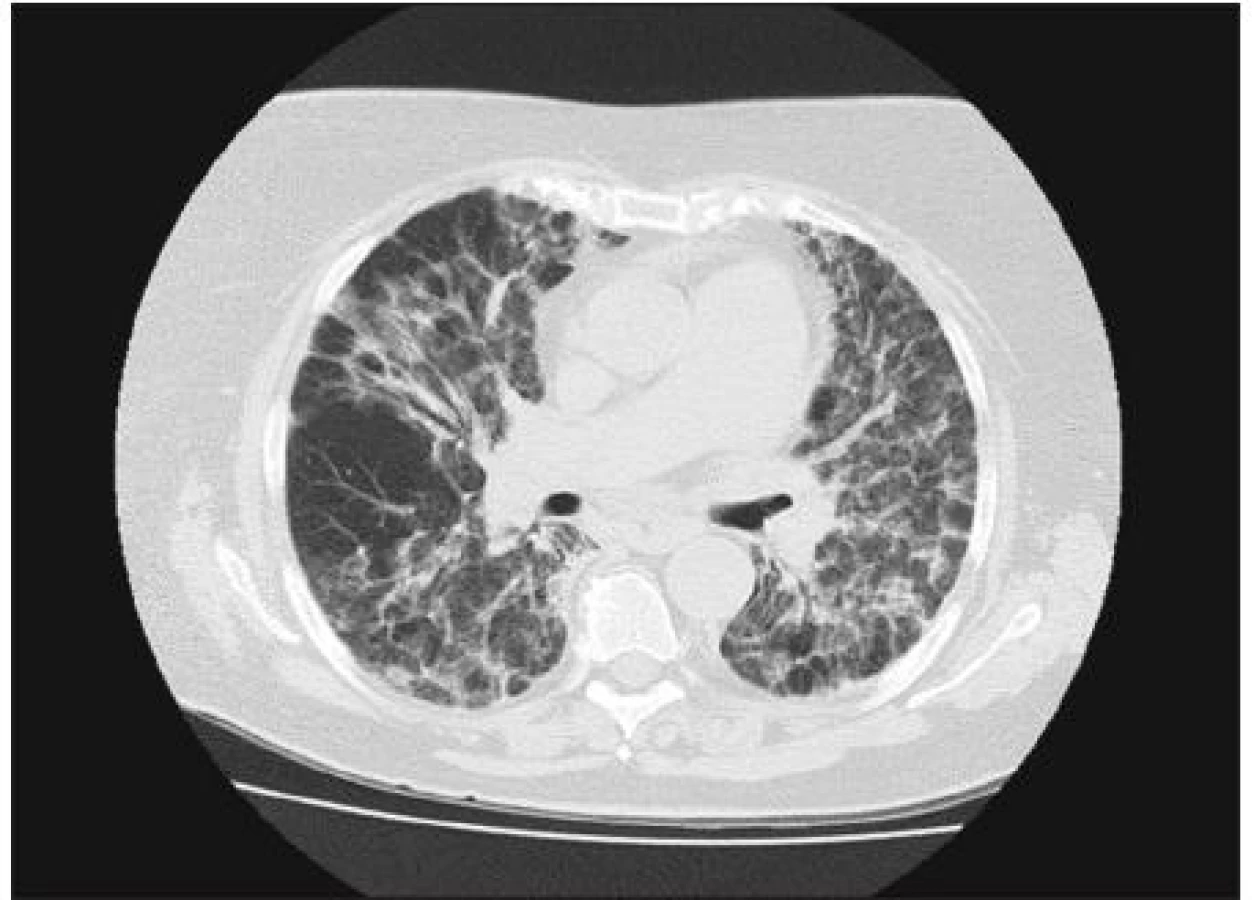
Z laboratórnych nálezov boli minerály v norme, zo začiatku mierne zvýšený kreatinín a transaminázy postupne poklesli k norme, CRP bolo 23,6 mg/l, NT proBNP 175,6 ng/l. Zaujímavým nálezom bola elevácia myoglobínu na 71,3 μg/l a troponínu (0,1 μg/l). V moči bola krv orientačne na +, bielkovina nebola preukázaná. V krvnom obraze bola prechodne neveľká leukocytóza (13,7 x 109) s relatívnou lymfopéniou (7,3 %), červený krvný obraz bol v norme. Bolo doplnené imunologické vyšetrenie vrátane autoprotilátok, kde bola preukázaná izolovaná pozitivita anti-Jo-1. V cytometrickom obraze bolo zrejmé iba klinicky nevýznamné zníženie T lymfocytov, CD4 aj CD8 pozitívnych a NK buniek. Doplnené špecifické IgG, ktoré sa vyšetrilo vzhľadom k udávanej expozícii plesniam v domácom prostredí, nepreukázalo významné zvýšenie vyšetrovaných protilátok. Na ultrazvuku brucha bola popísaná steatóza pečene, mnohopočetná cholecystolitiáza a drobné cysty ľavej obličky. Rádiologický nález na skiagrame hrudníka vykazoval retikuláciu difúzne s ojedinelými nodulami, CT nález, ktorý bol vyššie popísaný, sme potom hodnotili ako obraz akútneho alveolárneho poškodenia. Funkčné vyšetrenie 5. 8. 2010 ukázalo stredne ťažko zníženú vitálnu kapacitu a ťažko zníženú difúznu kapacitu (FVC 55 % NH;FEV1 54 % NH, FEV1/FVC: 108 %; VC 1,42 (55 %); TLco 14 %, Kco44 %; RV 91%,TLC 52%). V krvných plynoch bola zjavná závažná kľudová hypoxémia (pH 7,453; pCO2 5,04 kPa; pO2 7,12 kPa, Sat. 89,2 %).
Kardiologické vyšetrenie s opakovanou echokardiografiou na IKEM nepreukázalo kardiálny podklad ťažkostí pacientky, napriek tomu bola následne 6. 8. vzhľadom k elevácii troponínu a myoglobínu vykonaná aj koronarografia s normálnym nálezom na vencovitých tepnách. Bronchoalveolárna laváž nebola vzhľadom k závažnej pokojovej hypoxémii vykonaná. Nakoniec sme teda z vyššie uvedených vyšetrení stanovili diagnózu pľúcneho postihnutia typu akútnej intersticiálnej pneumónie pri PM/DM bez klinicky zjavného postihnutia svalov a kože. Opreli sme sa hlavne o rádiologické a funkčné vyšetrenie, pozitivitu anti-Jo protilátok a eleváciu myoglobínu a troponínu pri normálnom kardiologickom náleze. Nasadili sme pacientke preto liečbu kortikoidmi, najprv v intravenóznej forme v dávke 160 mg Solu-Medrolu denne. Po nasadení liečby došlo následne k promptnému zlepšeniu stavu pacientky, začali sme preto postupne od tretieho dňa dávku kortikoidov znižovať. Kontrolná spirometria 16. 8. ukázala výrazné zlepšenie funkčných parametrov (FVC 80 %; FEV1 : 59 %, FEV1/FVC 81 %; VC 2,06..80 %; TLco 37 %, Kco 63 %, RV 84 %, TLC 68 %). Kontrolné krvné plyny preukázali normálnu respiráciu (pH 7,395; pCO2 5,02 kPa;pO2 9,38 kPa, Sat. 93,7 %). Na kontrolnom skiagrame hrudníka 16. 8. 2010 bola oproti predchádzajúcemu snímku zrejmá regresia retikulonodulácií a taktiež zmenšenie srdečného tieňa, zrejme pri regresii predtým popisovaného fluidoperikardu.
Nález sme definitívne klinicky uzavreli ako pľúcne postihnutie typu akútnej intersticiálnej pneumónie pri systémovej polymyozitíde bez klinických príznakov svalového postihnutia s pozitivitou anti-Jo1 a eleváciou myoglobínu a troponínu. Pacientku sme na redukovanej dávke kortikoidov (Prednizón 20 mg) prepustili do domáceho ošetrovania. Pri kontrolnom vyšetrení v októbri 2010 je nález na skiagrame a HRCT hrudníku prakticky v norme (obr. 3, 4). EMG vyšetrenie nepreukázalo poruchu v zmysle myozitídy alebo nervovosvalového prevodu. Uvedená kazuistika ukazuje na nutnosť imunologického a prípadne reumatologického vyšetrenia aj u IPP, kde nie je klinicky zrejmé postihnutie v zmysle systémového ochorenia spojiva. I tak sa totiž môže jednať o frustné formy systémových ochorení, čo môže prípadne modifikovať následnú liečbu a sledovanie pacienta.


Záver
Sine syndrómy predstavujú reumatologické ochorenia, ktoré nespĺňajú diagnostické kritéria. V našom prípade sa jednalo o vývoj polymyozitídy bez svalového postihnutia, spojenú v klinickom obraze s intersticiálnou pľúcnou chorobou s prítomnosťou anti-Jo-1 protilátok. Včasná diagnostika a pohotová liečba môže potlačiť prejavy aktivity intersticiálnej pľúcnej choroby v rámci amyopatickej a adermopatickej PM/DM.
Prof. MUDr. Jozef Rovenský, DrSc., FRCP
Národný ústav reumatických chorôb
92101 Piešťany
Slovensko
rovensky.jozef@nurch.sk
Zdroje
1. Sontheimer RD. Would a new name hasten the acceptance of amyopathic dermatomyositis (dermatomyositis siné myositis) as a distinctive subset within the idiopathic inflammatory dermatomyopathies spectrum of clinical illness? J Am Acad Dermatol 2002; 46 : 626-636.
2. Wagner E. Fall einer seltuen Muskelkrankheit. Arch Heilkd 1863; 4 : 282.
3. Unverricht H. Polymyositis acuta progressiva. Zeit Klin Med 1887; 12 : 533.
4. Göttron H. Haut veranderungen bei dermatomyositis. In: Lomholt S, editor. VIII Congress International de Dermatologie; 1930; Copenhagen, Copenhagen: Engelsen and Schroder; 1931.
5. OęLeary PA, Waisman M. Dermatomyositis. A study of forty cases. Arch Dermat Syph 1940; 41 : 1001-1019.
6. Keil H. Manifestations in the skin and mucous membranes in dermatomyositis with special reference to the differential diagnosis from systemic lupus erythematosus. Ann Intern med 1942; 16 : 828-871.
7. Pearson CM. Polymyositis and dermatomyositis. Chapter 52. In: McCardy DJ, editor. Arthritis (and allied conditions). Philadelphia: Lea & Febiger; 1979. p.742-761.
8. Krain L. Dermatomyositis in six patients without initial muscle involvement. Arch Dermatol 1975; 111 : 241-245.
9. Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine 1977; 56 : 255-286.
10. Fernandes L, Goodwill CJ. Dermatomyositis without apparent myositis, complicated by fibrosing alveolitis. J R Soc Med 1979; 72 : 777-779.
11. Braverman I. Connective tissue (rheumatic) diseases. Cutaneous signs of systemic disease. Philadelphia: WB Saunders Co; 1981. p. 299-314.
12. Woo TY, Callen JP, Voorhees JJ, Bickers DR, Hanno R, Hawkins C. Cutaneous lesions of dermatomyositis are improved by hydroxychloroquine. J Am Acad Dermatol 1984; 10 : 592-600.
13. Taieb A, Guichard C, Salamon R, Maleville J. Prognosis in juvenile dermatopolymyositis: a cooperative retrospective study of 70 cases. Pediatr Dermatol 1985; 2(4): 275-281.
14. Gertner E, Urowitz MB. Discordance of skin and muscle involvement in dermatomyositis. Int J Dermatol 1985; 24(8): 518-9.
15. Caro I. A dermatologistęs view of polymyositis/dermatomyositis. Clin Dermatol 1988; 6 : 9-14, 47-48.
16. Rockerbie NR, Woo TY, Callen JP, Giustina T. Cutaneous changes of dermatomyositis precede muscle weakness. J Am Acad Dermatol 1989; 20 : 629-632.
17. Lam WW, Chan H, Chan YL, et al. MR imaging in amyopathic dermatomyositis. Acta Radiologica 1999; 40 : 69-72.
18. Emslie-Smith A, De Visser M, Engel AG. The erliest pathological change in dermatomyositis. Ann Neurol 1989; 26 : 123.
19. Filo V. K problematike amyopatickej dermatomyozitídy. Čas lék čes 2003; 11 : 142.
20. Plamondon S., Dent P.B. Juvenile amyopathic dermatomyositis: Results of a case finding descriptive survey. J Rheumatol 2000; 27 : 2031-2034.
21. Dawkins MA, Jorizzo JL, Walker FO, et al. Dermatomyositis: a dermatology-based case series. J Am Acad Dermatol 1998; 38 : 397-404.
22. Euwer RL, Sontheimer RD. Amyopatic dermatomyositis (dermatitis sine myositis). Presentation of six new cases and review of the literature. J Am Acad Dermatol 1991; 24 : 959-966.
23. Gerami S, Jahromi KT, Ashouri A, Rasoulian G, Heidari A. Sublethal effects of imidacloprid and pymetrozine on the life table parameters of Aphis gossypii Glover (Homoptera: Aphididae). Commun Agric Appl Biol Sci 2005; 70(4): 779-785.
24. Cao H, Parikh TN, Zheng J. Amyopathic dermatomyositis or dermatomyositis-like skin disease: retrospective review of 16 cases with amyopathic dermatomyositis. Clin Rheumatol 2009; 28(10): 1245-1246.
25. Sauty A, Rochat T, Schoch OD, Hamacher J, Kurt AM, Dayer JM, Nicod LP. Pulmonary fibrosis with predominant CD8 lymphocytic alveolitis and anti-Jo-1 antibodies Eur Respir J 1997; 10 : 2907–2912.
26. Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47 : 614-622.
27. Schwarz MI. The lung in polymyositis. Clin Chest Med 1998; 19 : 701-712.
28. Schnabel A, Reuter M, Biederer J, et al. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum 2003; 32 : 273-284.
Štítky
Dermatologie Dětská revmatologie RevmatologieČlánek vyšel v časopise
Česká revmatologie

2011 Číslo 1
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Riziko roztroušené sklerózy u pacientů s psoriázou
- Diagnostika osteoporózy v kontextu současných doporučení
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
Nejčtenější v tomto čísle
- Sine syndrómy v klinickom obraze dermatomyozitídy
- Výskyt autoimunitních onemocnění po aplikaci biologických léků
- Průběh systémového lupus erytematodes v graviditě
- Třeboňské revmatologické dny 5.–7. 1. 2011
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy