
Revised Phylogeny and Novel Horizontally Acquired Virulence Determinants of the Model Soft Rot Phytopathogen SCC3193
Soft rot disease is economically one of the most devastating bacterial diseases affecting plants worldwide. In this study, we present novel insights into the phylogeny and virulence of the soft rot model Pectobacterium sp. SCC3193, which was isolated from a diseased potato stem in Finland in the early 1980s. Genomic approaches, including proteome and genome comparisons of all sequenced soft rot bacteria, revealed that SCC3193, previously included in the species Pectobacterium carotovorum, can now be more accurately classified as Pectobacterium wasabiae. Together with the recently revised phylogeny of a few P. carotovorum strains and an increasing number of studies on P. wasabiae, our work indicates that P. wasabiae has been unnoticed but present in potato fields worldwide. A combination of genomic approaches and in planta experiments identified features that separate SCC3193 and other P. wasabiae strains from the rest of soft rot bacteria, such as the absence of a type III secretion system that contributes to virulence of other soft rot species. Experimentally established virulence determinants include the putative transcriptional regulator SirB, two partially redundant type VI secretion systems and two horizontally acquired clusters (Vic1 and Vic2), which contain predicted virulence genes. Genome comparison also revealed other interesting traits that may be related to life in planta or other specific environmental conditions. These traits include a predicted benzoic acid/salicylic acid carboxyl methyltransferase of eukaryotic origin. The novelties found in this work indicate that soft rot bacteria have a reservoir of unknown traits that may be utilized in the poorly understood latent stage in planta. The genomic approaches and the comparison of the model strain SCC3193 to other sequenced Pectobacterium strains, including the type strain of P. wasabiae, provides a solid basis for further investigation of the virulence, distribution and phylogeny of soft rot bacteria and, potentially, other bacteria as well.
Published in the journal:
. PLoS Pathog 8(11): e32767. doi:10.1371/journal.ppat.1003013
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003013
Summary
Soft rot disease is economically one of the most devastating bacterial diseases affecting plants worldwide. In this study, we present novel insights into the phylogeny and virulence of the soft rot model Pectobacterium sp. SCC3193, which was isolated from a diseased potato stem in Finland in the early 1980s. Genomic approaches, including proteome and genome comparisons of all sequenced soft rot bacteria, revealed that SCC3193, previously included in the species Pectobacterium carotovorum, can now be more accurately classified as Pectobacterium wasabiae. Together with the recently revised phylogeny of a few P. carotovorum strains and an increasing number of studies on P. wasabiae, our work indicates that P. wasabiae has been unnoticed but present in potato fields worldwide. A combination of genomic approaches and in planta experiments identified features that separate SCC3193 and other P. wasabiae strains from the rest of soft rot bacteria, such as the absence of a type III secretion system that contributes to virulence of other soft rot species. Experimentally established virulence determinants include the putative transcriptional regulator SirB, two partially redundant type VI secretion systems and two horizontally acquired clusters (Vic1 and Vic2), which contain predicted virulence genes. Genome comparison also revealed other interesting traits that may be related to life in planta or other specific environmental conditions. These traits include a predicted benzoic acid/salicylic acid carboxyl methyltransferase of eukaryotic origin. The novelties found in this work indicate that soft rot bacteria have a reservoir of unknown traits that may be utilized in the poorly understood latent stage in planta. The genomic approaches and the comparison of the model strain SCC3193 to other sequenced Pectobacterium strains, including the type strain of P. wasabiae, provides a solid basis for further investigation of the virulence, distribution and phylogeny of soft rot bacteria and, potentially, other bacteria as well.
Introduction
The soft rot bacteria Pectobacterium and Dickeya of the family Enterobacteriaceae are responsible for significant, global economic losses of crops and ornamental plants, both in the field and in storage. Although soft rot enterobacteria can infect a wide variety of plants, the main crop affected is potato (Solanum tuberosum L.). Potato is number five among food crops in the world and is a staple food in a number of countries (FAOSTAT 2009, http://faostat.fao.org/). Previously, the Pectobacterium and Dickeya species were classified into the Erwinia genus, and relatively recent phylogenetic analyses have elevated them to novel genera [1], [2]. In addition, some of the subspecies have been raised to the species level [3]. For historical reasons, three distinct potato diseases caused by soft rot enterobacteria are described: common soft rot (tuber symptoms), blackleg (tuber-born stem disease) and aerial stem rot (spread mechanically or via insects) [4]. Pectobacterium and Dickeya are characterized as opportunistic pathogens that switch from an asymptomatic latent phase into a virulent phase in suitable environmental conditions [5]. The virulent phase is thought to begin under anoxic conditions when oxygen radical-dependent plant defense mechanisms decline, allowing bacterial multiplication and the induction of plant cell wall-degrading enzymes (PCWDEs). The induction of PCWDEs occurs when the bacterial cell density reaches a quorum of 107 cfu/g of plant tissue [5].
The soft rot enterobacteria are necrotrophs and are generally considered to be brute-force pathogens relying on PCWDEs for pathogenicity. In fact, several regulatory mutants affecting enzyme production are essentially avirulent [6]–[9]. Studies of more fine-tuned virulence mechanisms may have been hampered by this massive production of PCWDEs [6]. Therefore, the latent stage preceding necrotrophy remains poorly understood. However, during the last 25 years, a number of other virulent lifestyle promoting determinants have been identified from Pectobacterium. These determinants include the following: flagella-based motility, cell membrane structures, such as enterobacterial common antigen (ECA) and lipopolysaccharide (LPS), type III secretion systems (T3SS), type IV secretion systems (T4SS), type VI secretion systems (T6SS), necrosis-inducing protein (Nip), a protein similar to an avirulence protein in Xanthomonas (Svx), coronafacic acid synthesis pathways (cfa genes), plant ferredoxin-like protein (FerE) and citrate uptake and 3-hydroxy-2-butanone pathways (bud) [10]–[19]. The virulence strategies of necrotrophic bacteria differ from hemibiotrophic and biotrophic phytopathogens, such as Erwinia amylovora, Pantoea sp. and Agrobacterium tumefaciens, which rely mainly on T3SS or T4SS for pathogenicity [20], [21]. The genomic era has provided novel tools for the identification of previously unknown virulence determinants without large-scale biological experiments. Several genomic studies of plant and animal pathogenic enterobacteria have been conducted, but only two have been published on soft rot bacteria. The first study compared P. atrosepticum and Salmonella, and the second study characterized P. atrosepticum, P. carotovorum and P. carotovorum subsp. brasiliensis [11], [22].
The soft rot pathogen Pectobacterium sp. SCC3193 was originally isolated from a diseased potato stem from a Finnish field in the early 1980s and was characterized as P. carotovorum (previously called Erwinia carotovora subsp. carotovora) [23]. Since its discovery, SCC3193 has been a model strain in soft rot research, and much is known about its virulence and molecular biology [7], [15], [17], [23]–[33]. In this work, we present a revised phylogeny, an analysis of virulence determinants and a characterization of genomic islands; we also discuss novel virulence strategies utilized throughout the lifestyle of Pectobacterium sp. SCC3193. We show that SCC3193 can be taxonomically classified as Pectobacterium wasabiae and that P. wasabiae has unique features when compared with other Pectobacterium strains; these features were most likely acquired via horizontal gene transfer. This work indicates that P. wasabiae has been present, though unnoticed, in European and maybe in American potato fields for a long time. Genome analysis is supplemented with experimental results to suggest novel virulence determinants of Pectobacterium; many of these determinants could be important during the poorly characterized latent stage of infection.
Results/Discussion
Phylogenetic analysis identifies SCC3193 as Pectobacterium wasabiae
The species status of Pectobacterium sp. SCC3193 was questioned after an initial review of the recently sequenced genome of SCC3193 by our Pectobacterium sequencing consortium in Helsinki, Finland (CP003415, Koskinen et al., in press). We discovered distinctive sequence similarity of SCC3193 to the strain WPP163 (NC_013421.1), which was sequenced in 2009 by another group (Nicole Perna and coworkers, and US DOE Joint Genome Institute; unpublished). WPP163 was isolated from potato stem and classified as P. wasabiae prior to the genome sequencing [34]. Originally, Pectobacterium sp. SCC3193 was identified as Pectobacterium carotovorum based on disease symptoms in potato, the ability to produce PCWDEs, fatty acid composition and other biochemical properties. Subsequent studies suggested that SCC3193 may not be a typical P. carotovorum strain due to its LPS composition, sensitivity to T4 phage, decreased ability to macerate plant tissues and inability to grow at +37°C [23]. However, P. carotovorum has been recognized as a highly variable species that is composed of several subspecies. Thus, differences in phenotype have been accepted [3]. At the time of the isolation and characterization of SCC3193, the species Pectobacterium wasabiae (previously called Erwinia carotovora subsp. wasabiae) had not yet been described. The type strain (CFBP 3304T) of P. wasabiae was isolated from wasabi (Japanese horseradish) in 1987 [3], [35]. Reports of P. wasabiae isolates are limited when compared to the number of P. carotovorum and P. atrosepticum reports [35]–[40]. To evaluate the species status of SCC3193, we conducted a thorough phylogenetic analysis of SCC3193, which included biochemical and genomic methods. The genome of P. wasabiae CFPB 3304T was sequenced by our consortium to be used as a reference in this phylogenetic analysis.
A phylogenetic tree indicates that SCC3193 is Pectobacterium wasabiae
We first performed a brief set of standard biochemical tests, which are commonly used to distinguish P. wasabiae from P. carotovorum, to determine the species of SCC3193. These tests could not unambiguously place SCC3193 into any of the known species underlining previous difficulties to determine the taxon. SCC3193, as well as the P. wasabiae type strain (CFBP 3304T), differed from the P. carotovorum type strain (CFBP 2046T) with respect to growth in 5% NaCl and the ability to grow at +37°C and from the P. atrosepticum type strain (HAMBI 1429T) with respect to growth in 5% NaCl, production of reducing sugars and fermentation of α-methyl-glucoside. However, SCC3193 also differed from the P. wasabiae type strain (CFBP 3304T) in utilization of melibiose and raffinose. Subsequently, we utilized the genomic information for the taxonomic characterization of SCC3193. Pectobacterium sp. SCC3193 and P. wasabiae CFBP 3304T, which was sequenced for this purpose (AKVS00000000), and 52 additional reference species were compared in an extended multilocus sequence analysis (51 loci) and used for phylogenetic analysis. For the selection of the 51 orthologous groups (Dataset S1), reciprocal best hits were determined based on similarities detected using the fast protein sequence database search tool SANS [41]. For the 51 orthologous groups, we created multiple alignments using Muscle and bootstrapped them using RAxML. All the bootstrapped trees were merged to generate a phylogenetic tree using the Consense program in the Phylip package. In this analysis, SCC3193 was grouped with P. wasabiae WPP163 and P. wasabiae CFBP 3304T. The P. wasabiae clade including SCC3193 was also close to P. atrosepticum SCRI1043. Notably, SCC3193 was placed in a separate clade from P. carotovorum strains (Figure 1). This finding indicates that SCC3193, WPP163 and the P. wasabiae type strain form an evolutionary group distinct from other Pectobacterium species. The phylogenetic tree is in agreement with previous reports regarding the taxonomy of the soft rot enterobacteria Pectobacterium and Dickeya [2], [3], [36], [42], further supporting our results. In conclusion, our phylogenetic analysis of SCC3193 resulted in the novel finding that SCC3193 does not belong to the species P. carotovorum but to a completely different species, P. wasabiae.
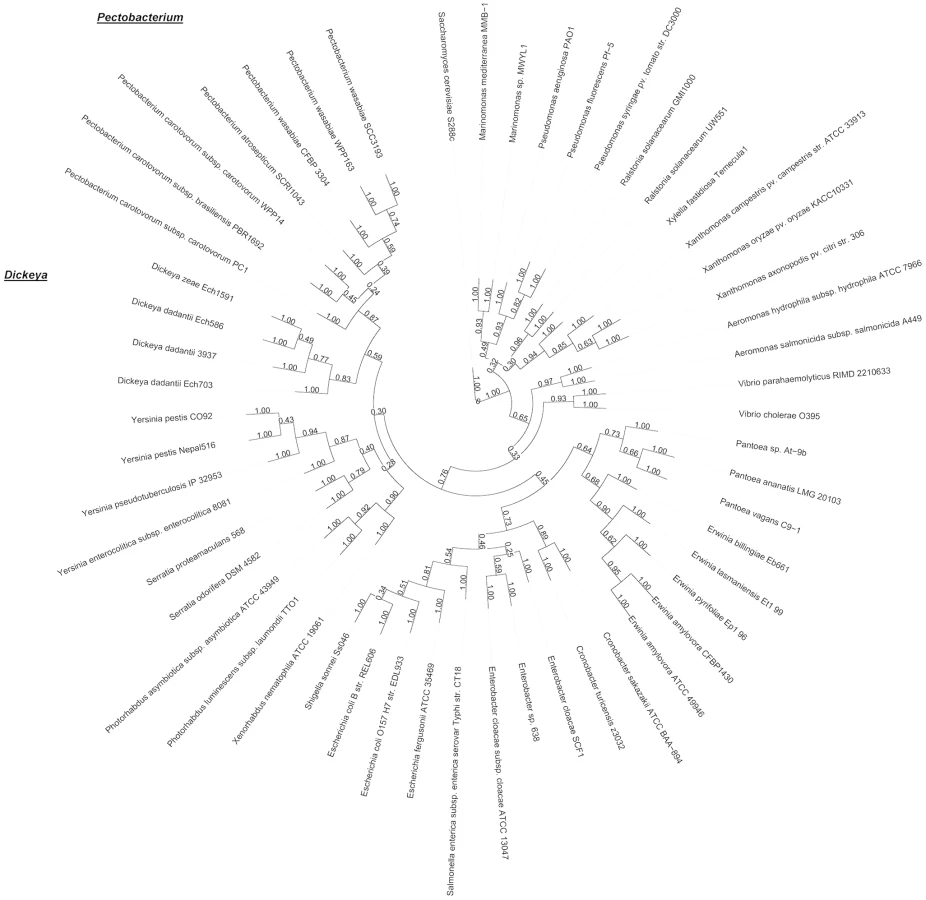
The proteome comparison of soft rot bacteria confirmed the species of SCC3193
We applied a novel approach to find evolutionarily relevant groups within the soft rot bacteria and confirm the species of SCC3193. We compared proteomes using clustering with OrthoMCL and visualized them on a heat map. In the heat map, core genome, strain, species and genus-specific protein clusters are clearly visible; the heat map also groups phylogenetic clades together (Figure 2A). The correlations of all strains were calculated based on proteomes (Figure 2B), and they were in agreement with the phylogenetic clades of Pectobacterium and Dickeya (Figure 1). The comparison of all the sequenced Pectobacterium and Dickeya species with Yersinia pestis CO92 as an outgroup showed that the correlation between SCC3193 and P. wasabiae WPP163 was approximately 0.9 (bright red squares in Figure 2B). The correlation of SCC3193 and WPP163 with the type strain of P. wasabiae was also high. The high correlation of proteomes may indicate a close evolutionary relationship, suggesting that SCC3193 and WPP163 belong to the same species, which is most likely P. wasabiae.

The proteome comparison approach is a novel and simple tool for the characterization of new isolates and the revision of previous phylogenetic taxa. Clustering may also work as a tool for detecting species or group-specific proteins and investigating functional differences between strains. However, we found that the use of different ORF predictions for different strains may result in a relatively high error rate; therefore, we propose that ORF predictions need to be unified before the clustering of proteomes.
SCC3193 has nearly complete synteny with Pectobacterium wasabiae WPP163
To investigate the relationships among all sequenced P. wasabiae strains isolated from different continents and host crops (SCC3193 from potato in Europe, WPP163 from potato in North-America and CFBP 3304T from Japanese horseradish in Asia), we compared their genome sequences to each other and to the closest neighbor in the clade (P. atrosepticum SCRI1043) using Mauve, which is a multiple genome alignment tool. SCC3193 shows almost complete synteny with P. wasabiae WPP163 and differs by only 7.4% in the pairwise alignment (pairwise genome content distance) (Figure 3). Because the genome of the type strain is in contigs, it was aligned according to SCC3193. The type strain differs by 19.4% and by 19.1% from SCC3193 and WPP163, respectively. The differences between P. wasabiae strains in the pairwise alignments are much smaller than the differences between P. wasabiae strains and P. atrosepticum; SCC3193 differs 28.7%, WPP163 differs 27.8% and CFBP 3304T differs 30.4% from P. atrosepticum SCRI1043. Taken together, the results of the pairwise alignment support the notion that SCC3193, WPP163 and CFBP 3304T belong to the same species. This is most evident for SCC3193 and WPP163, due to their almost complete synteny. However, we cannot rule out the possibility that SCC3193 and WPP163 could represent a novel potato-related subspecies of P. wasabiae, a species that has previously been found mainly from Japanese horseradish. This possibility is especially notable when we consider the small number of P. wasabiae isolates characterized and the relatively large difference between the type strain and SCC3193/WPP163 compared with the small difference between SCC3193 and WPP163. Based on the current knowledge, we suggest that these three strains are best classified as P. wasabiae.

Pectobacterium wasabiae may have been present but unnoticed on potato fields for a long time
Our work and the very recent work of Nabhan and colleagues at the end of 2011 [40] suggest that some P. wasabiae strains have been misidentified as P. carotovorum in the past. Nabhan et al. [40] showed that three P. carotovorum strains isolated from Europe can now be classified as P. wasabiae based on multilocus sequence alignment. At this point, it is unclear whether P. wasabiae is an emerging pathogen in potato fields or if the rising number of P. wasabiae reports [36], [37], [39] is a consequence of improved DNA sequence-based characterization methods. Our genomic approach and comparison of SCC3193 to the type strain of P. wasabiae could provide a solid basis for further investigation of the distribution and phylogeny of P. wasabiae.
Pectobacterium wasabiae SCC3193 has an arsenal of established virulence determinants
To obtain an overall view of SCC3193 virulence factors, we mined the genome for known virulence determinants of Pectobacterium. Because we are working with an established model strain, many virulence-associated determinants of SCC3193 have already been identified in genetic studies, and some of these were even originally described in SCC3193. This may create a bias that is reflected in the number of published virulence-associated genes found in the genome of SCC3193.
Plant cell wall-degrading enzymes are essential for pathogenesis of Pectobacterium
The production of PCWDEs is the hallmark of soft rot pectobacteria. We identified a total of 39 known or putative pectinases, cellulases and proteinases (Table S1). Most of the PCWDE genes are shared by all sequenced Pectobacterium strains, with the exception of several genes for putative proteinases, of which two may be specific to P. wasabiae, and one is present only in SCC3193 and the type strain. The number of strain-specific proteinases is similar in all the compared strains. PehK, HrpW and a putative pectate lyase are not present in P. wasabiae strains, although they are present in the other compared Pectobacterium strains. P. carotovorum subsp. carotovorum PC1 has one additional putative polygalacturonase. Our GO term based proteinase analysis is supplemented with previously described putative proteinases that are not present in E. coli strains; therefore, they may represent a novel proteinase class associated with plant pathogens [22]. One previous report indicates that potato isolates of P. wasabiae are less virulent than P. carotovorum strains [34]. In our observation, we have also found that SCC3193 has a lower capacity to macerate potato tubers than P. atrosepticum SCRI1043 and P. carotovorum SCC1. It remains unclear whether the lack of PehK, HrpW and the pectate lyase present in other Pectobacterium has an effect on the virulence of P. wasabiae.
Previously characterized virulence determinants of Pectobacterium present in SCC3193 and other P. wasabiae strains with sequenced genome
The flagella-encoding cluster (W5S_1760–W5S_1810) is most likely elementary to the virulent lifestyle [24], [43], [44] and can be found in the genomes of all P. wasabiae strains. The enterobacterial common antigen (ECA) encoding cluster (W5S_4355–W5S_4365) and the LPS cluster (W5S_4520–W5S_4538) are present in all P. wasabiae strains as well. Mutagenesis of the rffG (ECA and LPS core) and waaJ (LPS) genes reduces the virulence of P. atrosepticum in potato [12], [45]. All P. wasabiae strains also harbor the 3-hydroxy-2-butanone pathway (budRAB; W5S_0740–W5S_0742 and budC; W5S_0317) that was recently shown to have an effect on the alkalization of the environment, which is hypothesized to be a partial reason for the decreased virulence of the budB mutant in potato compared with the wild-type P. carotovorum [19]. P. wasabiae and other Pectobacterium species contain a few virulence factors encoded by single genes. One is the necrosis-inducing virulence protein (Nip; W5S_1316), which has been experimentally demonstrated in P. wasabiae SCC3193, P. atrosepticum SCRI1043 and P. carotovorum ATTn10 to be a virulence determinant [15], [46]. Another single gene encoding a putative virulence factor (svx; W5S_0937) is highly similar to a secreted avirulence factor gene in Xanthomonas, and it has been shown to contribute to the virulence of P. atrosepticum [16]. The citrate transporter, which is proposed to enhance the colonization of P. atrosepticum in potato tubers by decreasing citrate concentration [18], was also found in P. wasabiae strains SCC3193 (W5S_4105), WPP163 and CFBP 3304T.
Virulence is regulated via a complex network
The regulation of virulence in Pectobacterium has been extensively studied, and a large number of regulators controlling pathogenicity have been characterized both genetically and through molecular studies [11]. SCC3193 has been one of the main models utilized to elucidate virulence regulation in pectobacteria, and as a result, a number of regulatory mutants affecting virulence and PCWDE production have been characterized. The complex regulatory network in SCC3193 is relatively well described and includes the following: a cell density-dependent quorum sensing system, first described in SCC3193 (ExpI; W5S_4607, ExpR; W5S_4606/W5S_1749, LuxS; W5S_1019); several two-component systems involved in sensing the environment (ExpAS; W5S_1457/W5S_3687, PehRS; W5S_2096/W5S_2095, PmrAB; W5S_4173/W5S_4174); the Rcs phosphorelay system (RcsC; W5S_3208, RcsD; W5S_3206, RcsB; W5S_3207); a few global regulators (RsmA; W5S_1009, KdgR; W5S_2118, ExpM; W5S_2224, Hor; W5S_2637); and a regulatory RNA (rsmB; 3645019–3644673) [7]–[9], [17], [27], [30], [32], [33], [47]–[50]. All the genes encoding the above mentioned components of the regulatory network are also present in the genomes of P. wasabiae WPP163 and CFBP 3304T. In addition to these previously characterized regulators of virulence, we identified a number of additional putative regulators in the genome of SCC3193 (Broberg et al., in preparation) both from the core genome, often conserved among enterobacteria, and from the genomic islands, which may represent novel regulators or regulators co-opted for species - or niche-specific interactions.
Secretion systems pass virulence determinants across the bacterial cell wall from cytosol to environment
The delivery of virulence determinants into the host is typically a central feature in pathogenesis. In pectobacteria, pectinases and cellulases are secreted through a type II secretion system (T2SS, W5S_1291–W5S_1305) that is also called the Out-system, and its inactivation renders Pectobacterium avirulent [24], [51]. Proteinases are usually secreted through a type I secretion system (T1SS), and it has been shown that PrtW (W5S_2894) contributes to virulence in SCC3193 [28]. In SCC3193, a virB-type IV secretion system (T4SS, W5S_1616–W5S_1624), which is best known as the Ti-plasmid transferring system of A. tumefaciens, is similar to that of P. atrosepticum [13]. A similar T4SS is also present in P. wasabiae CFBP 3304T, but we were not able to find it from P. wasabiae WPP163 on protein or nucleotide level. Interestingly, the similar T4SS cluster is also present in P. carotovorum subsp. brasiliensis but not in P. carotovorum subsp. carotovorum WPP14 or PC1. Furthermore, P. wasabiae has a T6SS that was previously characterized in P. atrosepticum as a virulence determinant [14]. Intriguingly, SCC3193 harbors two T6SS clusters (W5S_0962–W5S_0978 and W5S_2418–W5S_2441) and this is also the case for strains WPP163 and CFBP 3304T. Additionally, SCC3193 has genes for 26 haemolysin co-regulated proteins (Hcp) or valine-glycine repeat protein G (VgrG) proteins that may be related to the function of T6SS. The number of Hcp and VgrG encoding genes varies among bacterial species and even strains: P. wasabiae WPP163 carries 18 and P. wasabiae CFBP 3304T eight hcp or vgrG genes. The actual function of these proteins is not fully understood; they may have a role in the construction of the syringe-like secretion machinery of T6SS, or they may act as effectors [52]. The roles of the T6SS among Gram-negative bacteria appear to be diverse. Depending on the type of effectors delivered and possibly on the structure of the syringe, T6SS targets several organisms, including humans, other animals, plants and bacteria [52]–[54].
Pectobacterium wasabiae lacks type III secretion system
Only a few previously described virulence-related genes in Pectobacterium were not found in SCC3193: namely, the genes for coronafacic acid synthesis (specific to P. atrosepticum) and the type III secretion system (T3SS) present in many P. atrosepticum and P. carotovorum strains [11], [13]. The T3SS is composed of the injection machinery encoded by the conserved hrp/hrc gene cluster and of a species/strain-specific collection of effectors required to suppress the basal defenses of the host [55]. Analysis of the SCC3193, P. wasabiae WPP163 and the type strain (CFBP 3304T) genomes failed to identify any traces of T3SS. It appears typical of P. wasabiae that it lacks this widespread and important virulence determinant. These results are in agreement with previous failed efforts to detect T3SS in SCC3193 or other P. wasabiae strains; for example, no signs of this injection machinery or the associated effectors (encoded by hrpN, dspE and hecB genes) have been found [34], [36], [56]. Although absent from P. wasabiae, T3SS contributes to the virulence of other soft rot species [57]–[59].
Contrary to hemibiotrophic pathogens such as Pseudomonas syringae, where T3SS is essential for pathogenicity [21], the role of T3SS in necrotrophic soft rot bacteria appears quite complex. T3SS contributes to the virulence of P. carotovorum, P. atrosepticum and Dickeya strains, but even strains naturally lacking T3SS, such as P. wasabiae, are still able to infect potatoes. A recent study showed no clear correlation between virulence and the presence of T3SS in P. carotovorum [34]. The relatively modest significance of T3SS to pathogenicity in pectobacteria is also reflected in the small number of T3 effectors found in these species compared, for example, to the dozens of known effectors of Pseudomonas syringae [60]. Thus, Pectobacterium may have alternative ways of modifying the host plant at the initiation of infection.
The lack of T3SS and numerous effectors may also benefit Pectobacterium by widening its host range. Soft rot bacteria, excluding the potato-specific P. atrosepticum, are often found to colonize a wide range of food crops and ornamental plants, while many T3SS-dependent plant pathogens, such as Erwinia, Pseudomonas, Xanthomonas and Xylella, have a very narrow host range. It is well documented that some T3 effectors may act as colonization-inhibiting avirulence proteins that are recognized by the plant [55]. Virulence assays to investigate the role of T3SS in soft rot bacteria in planta thus far indicate that in a complex natural niche, T3SS may have an important role under certain conditions but not in others.
Genomic islands have a Pectobacterium wasabiae twist
Horizontal gene transfer plays an important role in the evolution of bacteria. It enables the rapid acquisition of beneficial traits in a single event. These gene clusters of probable horizontal origin are termed genomic islands (GIs/GEIs) or horizontally acquired islands (HAIs) [11], [61], [62]. To characterize the genome composition of SCC3193 and identify horizontally acquired virulence determinants or other adaptive traits, we determined putative GIs in the genome of SCC3193. We used two sequence composition-based GI prediction methods, SIGI-HMM and IslandPath-DIMOB [63], [64], which were found to have the highest overall accuracy of the six methods tested in a recent bioinformatic study [65]. In addition, we used one comparative genomic-based GI prediction method, IslandPick [65]. The three methods predicted a different number of islands and smaller islets: six for IslandPath-DIMOB, ten for IslandPick and over hundred for SIGI-HMM (63 consisted of five or more successive ORFs) (Figure 4). Automated predictions were subsequently manually curated, resulting in a total of 56 genomic islands (Table S2). The GIs comprise ∼0.86 Mb, which is 16.7% of the size of the genome and encompasses 21.6% of all ORFs (1040 of the 4804 ORFs). In comparison, 17.6% of the ORFs of Escherichia coli MG1655 were estimated to have been acquired horizontally [66]. In general, GIs are estimated to comprise between 1.6% and 32.6% of the ORFs in prokaryotic genomes [67].

A majority of the SCC3193 islands can be found identically or with slight permutations in P. wasabiae WPP163, whereas less than half of the islands are present in the genome of P. wasabiae CFBP 3304T (Table S2). Approximately 15 islands were specific to P. wasabiae among the soft rot group. Some Pectobacterium-specific islands (for example, GI_44) or islands partially present in other Pectobacterium strains but completely lacking from Dickeya (for example GI_13 and GI_36) were also discovered. A number of islands were specific to SCC3193 and could not be found in any other strains of soft rot bacteria (for example, GI_20, GI_40, GI_43 and GI_54). Furthermore, a few islands showed a significant similarity to the genomes of bacteria outside the soft rot species (GI_9, GI_10, GI_30, GI_40, GI_44 and GI_50). Functional predictions for the genes on the islands in SCC3193 suggested the presence of a high number of mobile element genes and genes with unknown functions. The islands were also found to contain several genes for known virulence determinants, such as Nip (GI_17), which is necessary for the full virulence of SCC3193 [15], and DsbA (GI_56), which is required for the correct conformation of many secreted virulence proteins in P. atrosepticum and P. carotovorum [68], [69]. Most of the islands were shown to carry genes with predicted functions that could often be potentially associated with plant colonization or virulence (Table 1, Table S2, Figure 4).

Plant ferredoxin-like protein and benzoic acid/salicylic acid carboxyl methyltransferase may have a eukaryotic origin
Two genes of possible eukaryotic origin were identified in the genome of SCC3193, and they were also present in the genome of P. wasabiae WPP163. These genes appear to be unique to potato isolates of P. wasabiae, as similar genes are not present in the genome of the P. wasabiae type strain CFBP 3304T isolated from Japanese horseradish or in the genomes of any other bacterial species characterized, and the deduced proteins are most similar to plant proteins. One of these genes encodes the plant ferredoxin-like protein FerE (W5S_4691, GI_55), which has been found to enhance oxidative stress tolerance and have an effect on the fitness of SCC3193 in planta [17]. The other gene encodes a putative S-adenosyl-L-methionine∶benzoic acid/salicylic acid carboxyl methyltransferase (W5S_2852, GI_35). The corresponding enzymes in plants, which methylate benzoic acid and salicylic acid, participate in plant defense responses and the biosynthesis of floral scents [70], [71]. Methyl salicylate, produced by methylation of salicylic acid in response to pathogen attack, is thought to act as a mobile signal for systemic acquired resistance, which provides broad-spectrum resistance against further pathogen attacks throughout the plant [71]. The overexpression of plant salicylic acid methyltransferase has been shown to affect plant defense responses and pathogen resistance [72], [73]. This finding suggests the intriguing possibility that the P. wasabiae protein may be used to modify salicylic acid-dependent defenses and could represent a novel mechanism for a plant pathogen to manipulate its host. The ability to manipulate the induction of defense responses could be of great advantage to the T3SS-deficient P. wasabiae at the beginning of the infection, enabling it to colonize the host without being confronted with an arsenal of defenses triggered by plant recognition of the conserved microbe-associated molecular patterns (MAMPs).
A novel bacterial microcompartment is present in Pectobacterium wasabiae
We found a novel bacterial microcompartment (BMC) cluster (GI_50) present only in P. wasabiae strains (SCC3193, WPP163 and CFBP 3304T) and a handful of non-soft rot bacterial species. Bacterial microcompartments are enclosed protein complexes that encapsulate the sequential reaction steps for selected metabolic pathways, and they are thought to be horizontally transferring genomic elements [74]–[76]. Thus far, the best-known BMCs are CO2-fixing carboxysome, propanediol utilization (Pdu) BMC and ethanolamine utilization (Eut) BMC. Additional findings include the pyruvate-to-ethanol pathway in Vibrio furnissii and the ethanol-to-acetate pathway, together with the separate BMC for glycerol metabolism in Clostridium kluyveri [76], although these results have not yet been fully investigated. The BMC of SCC3193 is similar to the pyruvate-to-ethanol BMC of V. furnissii; this is based on the core enzyme pyruvate formate-lyase. However, the SCC3193 cluster includes an additional acetate kinase that is not present in any of the described microcompartment clusters. In Salmonella, it is thought that a household acetate kinase participates in the Eut pathway [77]. The rest of the genes in the cluster are typical components of Eut/Pdu BMCs, which mainly encodes the microcompartment multiprotein complex.
The BMC cluster in P. wasabiae, with a small variation in synteny, is present in the following strains with complete genomes: Escherichia coli CFT073 (c4524–c4538 and c4545–c4548), Rhodospirillum rubrum ATCC11170 (Rru_A0902–Rru_A0919), Rhodopseudomonas palustris BisB18 (RPC_1163–RPC_1179), Rhodobacter capsulatus SB1003 (RCAP_rcc02196–RCAP_rcc02214), Clostridium beijerinckii NCIMB 8052 (Cbei_4050–Cbei_4065) and Shewanella sp. W3-18-1 (SputW3181_0417–SputW3181_0429). These bacteria are anaerobes or facultative anaerobes with extremely versatile metabolic systems [78]–[86]. It is not clear what this microcompartment does in P. wasabiae strains, but we speculate that it enhances the anaerobic utilization of different carbon sources. It is well known that Pectobacterium can grow in anaerobic conditions, and it is possible that this BMC provides an advantage in the utilization of carbon for P. wasabiae when compared to other soft rot bacteria.
Liposaccharide-encoding clusters differ from Pectobacterium carotovorum
Previously, it was shown that P. wasabiae SCC3193 has a different LPS composition than P. carotovorum strains [23]. Two novel putative liposaccharide clusters were found in SCC3193. These clusters localized to GI_38 and GI_53; the first cluster is found only in potato isolates of P. wasabiae (SCC3193, WPP163), with the exception of the first three genes (rfbA, rfbC and rfbD), which are commonly present in Pectobacterium. This previously unknown cluster contains features of the clusters that encode LPS, capsular polysaccharides and lipo-oligosaccharide (LOS). The second cluster is present in potato isolates of P. wasabiae and in P. atrosepticum (waa cluster), and it is partially present in other Pectobacterium strains including the P. wasabiae type strain CFBP 3304T. These findings likely explain the differences found between the LPS composition of SCC3193 and that of P. carotovorum. Lipopolysaccharides and similar structures on the surface of bacteria have several roles in plant-microbe interactions. They may enhance the attachment of bacteria to the plant tissue, protect the bacteria from plant-derived toxins and be recognized by the plant, which may lead to a defense response or to a mutualistic lifestyle [87]. The variation observed in the LPS composition within Pectobacterium may indicate an adaptation to different environmental conditions.
Arsenic resistance may have a recent origin
SCC3193 was shown to harbor an arsenic resistance cluster. Arsenic resistance genes (ars) are widespread among bacteria, and they can be plasmid-borne or chromosomal [88]. SCC3193 GI_40 harbors an arsenic resistance cluster (6303 bp). The ars cluster was originally located adjacent to an SIGI-HMM predicted small (3133 bp) island, but we manually expanded the island to cover the cluster as well. The novel island (13686 bp) showed significant similarity to the genome of Enterobacter cloacae subsp. cloacae ATCC 13047 (blastn query coverage 87%, E 0.0). This region is missing from the genomes of other Pectobacterium strains. However, less similar Ars proteins are present in P. atrosepticum SCRI1043 (ECA1603–ECA1606, HAI7) and P. carotovorum WPP14 (GI:227326266, GI:227326267, GI:227326268). Evolutionary studies suggest that the exchange of ars clusters between plasmids and chromosomes and the horizontal transfer of the cluster are frequent [89]. Therefore, it appears likely that the ars clusters of SCC3193 and E. cloacae share a relatively recent common origin that is different from the clusters of P. atrosepticum SCRI1043 and P. carotovorum WPP14. Other Pectobacterium strains, including P. wasabiae WPP163 and CFBP 3304T, contain only putative arsenate reductase (arsC) and lack other components. SCC3193 also has two additional copies of arsC (W5S_3181 and W5S_3448 on GI_39). SCC3193 was isolated from southern Finland, where the highest arsenic concentrations of the cultivated soils in Finland are found [90]. The arsenic resistance cluster may have been an advantage under these environmental conditions. It is likely that arsenic resistance genes benefit all Pectobacterium species in the environment and inside plants that grow in soils containing arsenic.
Rhs elements are often related to outer membrane structures
A number of Rhs (recombination hot-spot) element genes were found in the genomic islands of SCC3193 (GI_2, GI_4, GI_8, GI_18, GI_29 and GI_45) and were partially or completely missing from other soft rot strains, except GI_8 which is present with 100% query coverage and GI_29 which is present with 93% query coverage in P. wasabiae WPP163 (Table S2). Rhs elements comprise a variable number of core rhs genes from different families and extensions to the core. In addition, they contain adjacent vgrG and hcp genes and an H-rpt insertion sequence [91]. Based on early studies, Rhs elements were first thought to be mediators of chromosomal rearrangements. With an increasing number of genomic sequences available for comparative studies, the view of Rhs elements is changing, and the loci are observed as a diverse and ancient family of protein-encoding genes [92].
Rhs element genes are reported to have several roles, depending on the species. Rhs elements have been associated with T6SS, encoding its functional core proteins, namely, Hcp and VgrG [93], [94]. To identify putative T6SS-related Hcp and VgrG proteins in SCC3193, we aligned all known Hcp and VgrG proteins in SCC3193 and P. atrosepticum, and a crude phylogenetic analysis was performed using ClustalW2 [95], [96]. Based on this analysis, only some of the vgrG and hcp genes present in SCC3193 are related to T6SS (data not shown). vgrG genes have also been associated with a social behavior called self-recognition. In Proteus mirabilis, Hcp and VgrG protein-encoding genes form the IdsA-IdsB locus. IdsB (VgrG) is required for self-recognition [97]. Many of the Rhs elements of SCC3193 contain several YD repeat-encoding genes. Previously, Rhs-YD repeat elements have been associated with social motility in Myxococcus xanthus [98]. Recently, Rhs elements have also been related to a toxin/immunity system, where they play a role in the contact-dependent intercellular competition of bacteria. This system, referred to as contact-dependent growth inhibition (Cdi), is also present in P. wasabiae WPP163; in D. dadantii 3937, it has an effect on the growth of E. coli [99]. A similar Cdi locus is present in SCC3193 on GI_5, but it is missing from the P. wasabiae type strain (CFBP 3304T). The function of Rhs elements is not clear; based on the current evidence, Rhs, Hcp and VgrG proteins likely form bacterial cell membrane-associated complexes, where additional domains have an effect on other organisms.
Several fimbria/pilus systems and flagella are on genomic islands
One genomic area in particular appeared to have accumulated a number of predicted horizontally transferred genes and gene clusters (GI_18–GI_22). Of these genomic islands, GI_18–GI_21 can be found partially (>50% query coverage) also from other P. wasabiae strains CFPB 3304T and WPP163. However, GI_22 can be found only from P. wasabiae WPP163 and SCC3193 but not from CFPB 3304T. In addition to several predicted plasmid or phage origin clusters, this region contains putative fimbria or pilus encoding clusters, including the virB-T4SS that lies between GI_20 and GI_21. Interestingly, similar T4SS is present in P. wasabiae SCC3193 and CFPB 3304T but not in P. wasabiae WPP163 pointing toward possibility that the corresponding T4SS clusters have horizontal origin or that it is deleted from WPP163. GI_18 also carries one of the putative T6SS effector encoding clusters, which is highly similar to corresponding clusters in P. atrosepticum (ECA2866-ECA2869 and ECA4275–ECA4278) [14], [100]. Additionally, the single copy flagellar system, which is related to the virulent lifestyle, was found on a genomic island (GI_24). The type IV pilus cluster (pil genes) found on GI_42 is similar to a genomic island (HAI2) in P. atrosepticum [11]. Similar type IV pilus is present, among soft rot bacteria, only in P. wasabiae SCC3193, P. wasabiae WPP163 and P. atrosepticum SCRI1043. Type IV pili mediate attachment to surfaces and twitching motility, and they are important in the pathogenesis of many animal pathogenic species in the Enterobacteriaceae family but are not known to influence virulence in Pectobacterium [101], [102]. Overall, very little is known about the attachment and biofilm formation of the Pectobacterium species. In P. atrosepticum, LPS is needed for full attachment on artificial surfaces [12]. In addition, Pectobacterium species contain several gene clusters encoding putative attachment structures, suggesting that the lack of experimental data on attachment could be due to the functional redundancy of these systems.
Aerobactin siderophore locus is present in potato isolates of Pectobacterium wasabiae
Siderophores facilitate iron acquisition from the environment. Aerobactin is a siderophore present in several enterobacterial strains. It has been characterized best from human pathogens, such as E. coli, Shigella flexneri, Klebsiella pneumonia and Salmonella sp., and the cluster is thought to be on a horizontally transferred element [103], [104]. In P. wasabiae SCC3193, an aerobactin synthesis cluster (W5S_0838–W5S_0842) was identified but not predicted to be located on GIs. Nevertheless, we speculate that it is a horizontally transferred element in the SCC3193 genome. An aerobactin synthesis cluster is not present in any other sequenced Pectobacterium except for P. wasabiae WPP163 (Pecwa_0947–Pecwa_0951). Previously, only one Pectobacterium strain (P. carotovorum W3C105) producing aerobactin has been identified [103]. The role of siderophores in the virulence of the Pectobacterium species has not been determined, but the capability of iron acquisition is an important virulence determinant for Dickeya [105]. Therefore, it is likely that the presence of aerobactin would contribute to the virulence of certain P. wasabiae strains and/or benefit the bacteria in other iron-deficient environments.
Novel virulence determinants
The characterization of the SCC3193 genome revealed novel genes that could contribute to the virulent lifestyle. We selected a group of interesting potential virulence determinants for experimental verification. The corresponding genes were inactivated by targeted mutagenesis, and their contribution to virulence was tested on axenic tobacco seedlings and potato tuber slices. A previously characterized phytase gene in P. wasabiae CFBP 3304T (Y17_1078) [106] is also present in P. wasabiae SCC3193 (W5S_4347) and P. wasabiae WPP163 (Pecwa_4189). In our virulence assays the phenotype of an SCC3193 phytase gene knock-out mutant was inconclusive and its potential role in virulence would require additional studies. However, the phytase gene is not unique to P. wasabiae as it can be found from all sequenced Pectobacterium strains and many other plant associated bacteria. The mutants that had phenotypes in planta were tested for their ability to grow in vitro and produce polygalacturonases and cellulases. No differences were observed between the mutants and the wild - type strain (data not shown), indicating that the phenotypes in planta are due to plant-microbe interactions and not general growth defects or a major reduction in PCWDE production.
SirB1 contributes to virulence of Pectobacterium wasabiae SCC3193
The sirB locus and its surroundings are conserved within Enterobacteriaceae. SirB is suggested to regulate virulence in Salmonella, but very little is known about its function or contribution to virulence [107], [108]. The SCC3193 sirB− mutant was considerably impaired in its capacity to infect axenic tobacco and macerate potato tubers (p<0.001, Figure 5AC), which suggests that sirB could play an important role in the virulence of P. wasabiae SCC3193. The delayed development of symptoms in tobacco seedlings could be complemented by the addition of the wild-type sirB locus (W5S_2384–W5S_2385) in trans (p<0.001). Interestingly, full complementation was also achieved when only the second gene of the locus, sirB1 (W5S_2384), was used (p<0.001). The addition of the first gene of the locus, sirB2 (W5S_2385), had no effect when it was added alone. This result suggests that sirB1, not sirB2, is required for the virulence of SCC3193 and that the phenotype is not due to the polarity of the mutation. We also measured the growth of the sirB− mutant in planta and found that the mutant showed significantly reduced growth when compared to the wild-type strain (p<0.01, Figure 5B). The addition of the sirB locus in trans restored the growth to levels approximating wild-type growth.

In Salmonella, the overexpression of the sirB locus in trans can suppress effects caused by mutations in sirA, which encodes a two-component system response-regulator similar to ExpA (GacA) in Pectobacterium [8], [107]. SirA is needed for the transcriptional activation of Salmonella invasion genes within the pathogenicity island 1 (SPI1). The Salmonella SirB is also necessary for the full expression of SirC (not present in SCC3193), a transcription factor encoded within SPI1 [108]. Although SirC is essential for the invasion of the intestinal epithelium, the sirB locus is not needed for the invasive phenotype [108]. Due to its effect on SirC expression and the ability to suppress sirA mutant phenotypes, SirB is considered to be a putative transcriptional regulator, although it does not belong to any known class of transcription factors. In E. coli, the sirB locus is nonessential [109], and no function has been reported. However, the putative operon in which the locus is situated is well characterized and contains genes involved in basic cellular functions, such as protein synthesis and the biosynthesis of LPS [109]–[111]. To our knowledge, SirB has not been reported to regulate the other genes in the operon. An online program for transmembrane helix prediction (TMHMM Server v. 2.0 [112]) predicts the SCC3193 sirB2 to encode a 14.9-kDa (131 aa) membrane protein with four hydrophobic transmembrane helices; no transmembrane helices are predicted for the 30.7-kDa (269 aa) protein encoded by sirB1, indicating that the protein is soluble. Therefore, if the locus encodes for a transcriptional regulator, SirB1 is a strong candidate.
Our results highlight the possibility that SirB is an important regulator of virulence in P. wasabiae and possibly in other soft rot bacteria. However, further experimental evidence is needed to confirm that SirB functions as a regulator. In contrast to the reduced maceration levels and reduced growth in planta, the sirB mutant appears to exhibit similar in vitro PCWDE production and growth as the wild-type, indicating that SirB could be important for bacterial fitness specifically during infection.
Type VI secretion system clusters have overlapping functions in Pectobacterium wasabiae SCC3193 during potato infection
The mutagenesis of a type VI secretion system machinery encoding cluster, either T6SS-1 (W5S_0962–W5S_0978) or T6SS-2 (W5S_2418–W5S_2441), did not affect maceration capacity in the potato tuber slice assay (data not shown). However, the T6SS-double mutant showed a reduced level of maceration (p<0.05, Figure 5C), suggesting that the two separate T6SS machineries occupy at least partially overlapping functions in planta. The phenotype of the SCC3193 T6SS-double mutant is similar to that of P. atrosepticum, in which mutations of its single T6SS cluster genes decrease its virulence in potato [14]. However, there is a contradictory report regarding the virulence of single-gene T6SS mutants of P. atrosepticum [100], which may be partially explained by the different virulence assays used in these studies.
Based on the comparison of the T6SS loci with those in other sequenced bacteria in GenBank using blastn, WGS-tblastn and blastp, it is obvious that the first locus (T6SS-1) is conserved among Pectobacterium and many other bacteria. The second locus (T6SS-2) provides the best hit for P. wasabiae WPP163, P. wasabiae type strain and Pantoea and Erwinia species; its synteny differs from the first locus. This clearly explains why our island prediction picked T6SS-2, not T6SS-1, as a genomic island of probable horizontal origin (GI_30). T6SS-2 was found to be similar, apart from a few additional ORFs, to a corresponding cluster in Erwinia amylovora CFBP1430 (EAMY_3000–EAMY_3028). The same cluster is also present in E. pyrifoliae and E. tasmaniensis [113]. We were not able to find a typical Pectobacterium style T6SS in the available Erwinia strains.
Our results support the hypothesis that T6SS contributes to virulence in Pectobacterium and that the function of the two T6SS clusters of possibly different evolutionary origins in P. wasabiae strains is at least partially redundant. However, we cannot rule out that T6SS in Pectobacterium may have another target in addition to plants. The process by which Pectobacterium utilizes T6SS during potato colonization and for its potential other targets remains to be elucidated.
Virulence cluster 2 contains a putative lipoprotein transport system and a hopL1-like gene
The virulence cluster 2 (Vic2) deletion mutant exhibited a reduced level of maceration (p<0.01, Figure 5C) in the potato tuber slice assay. The respective cluster (W5S_0498–W5S_0477) contains 22 ORFs, has a total size of ∼22.3 kb and has unique features when compared to other bacterial sequences available (Figure 6). The first 12 ORFs (W5S_0498–W5S_0487) form a cluster found in a handful of bacterial species, in addition to P. wasabiae strains (SCC3193, WPP163, CFPB 3304T), with completed genome sequences, such as Enterobacter sp. 638 (Ent638_4231–Ent638_4241), Xenorhabdus bovienii (XBJ1_1142–XBJ1_1152), Azotobacter vinelandii (Avin_51980–Avin_52100), Pasteurella multocida subsp. multocida (PM1818–PM1828), P. syringae (Psyr_2623–Psyr_2633, PSPTO_2870–PSTO_2880), Pseudomonas putida S16 (PPS_0194–PPS_0182) and Haemophilus parainfluenzae (PARA_16340–PARA_16450). Notably, this cluster is not found in any soft rot species other than P. wasabiae, and it was predicted to be horizontally acquired in SCC3193 (GI_7).

The cluster containing the first 12 ORFs encodes a protein that is similar to HopL1 (query coverage 99%, identity 42–43%, E 0.0), which has been characterized as T3SS secreted in P. syringae [114], and a putative lipoprotein transport system (Figure 6). As P. wasabiae lacks T3SS, it seems reasonable that HopL1 may be secreted through the putative lipoprotein transport system or some other machinery in SCC3193. We investigated how common it is that T3SS is missing from strains harboring the cluster that carries hopL1. Therefore, we compared two core proteins of T3SS derived from P. syringae DC3000 (HrpN/HrpU, HrcC) against the genomes of P. wasabiae CFPB 3304T, P. wasabiae WPP163, Enterobacter sp. 638, X. bovienii, A. vinelandii, P. multocida subsp. multocida, P. putida and H. parainfluenzae using tblastn. We were unable to find T3SS-related genes in these strains. The lack of T3SS in P. wasabiae WPP163 and other P. wasabiae strains tested previously, Enterobacter sp. 638, X. bovienii and P. putida are also noted in the literature [34], [115], [116]. We suggest that HopL1 could be functional without T3SS. In conclusion, we hypothesize that this putative lipoprotein transport system and/or HopL1-like protein may be responsible for the Vic2 deletion mutant phenotype in potato.
Virulence cluster 1 contains phage-related genes and a putative type I site-specific restriction-modification system
The Virulence cluster 1 (Vic1) deletion mutant exhibited a reduced capacity to macerate potato tuber slices when compared with the wild-type SCC3193 (p<0.01, Figure 5C), suggesting that one or several of the deleted genes could have an effect on SCC3193 virulence. The ∼14.4-kb genomic region contains ten ORFs (W5S_0467–W5S_0476) and is partially overlapping with GI_6 (W5S_0459–W5S_0472). The region is largely lacking from the genomes of other Pectobacterium strains, including P. wasabiae WPP163 and CFBP 3304T. ORFs W5S_0467 to W5S_0472 are part of a putative phage. The last ORF of Vic1 (W5S_0476) is annotated to encode a toxin component of a toxin-antitoxin system (Fic family) and is classified into COG3943 as a virulence protein, but no specific function for this class of proteins is known.
ORFs W5S_0473–W5S_0475 form a three-gene operon encoding a putative type I restriction modification (R-M) system that is thought to protect bacteria against invading foreign DNA [117]. Restriction-modification systems are common among bacteria, and there are implications of an extensive lateral transfer of the R-M genes [118], [119]. The position of the SCC3193 R-M system between GI_6 and GI_7, and the absence of this system in other Pectobacterium strains, including P. wasabiae WPP163 and CFBP 3304T, implies that the system may have been horizontally acquired in SCC3193. Furthermore, mutation of the type I R-M methyltransferase of Yersinia pseudotuberculosis was shown to lead to decreased virulence in a mouse model by an unknown mechanism [120]. Whether the SCC3193 methyltransferase has a role in virulence is a matter of speculation. Further studies are necessary to identify the exact virulence determinant(s) responsible for the observed reduced virulence phenotype of the Vic1 deletion mutant.
Concluding remarks
The model strain Pectobacterium sp. SCC3193 has been one of the most intensively studied soft rot strains for over two decades. Therefore, the molecular-level information on its virulence has had a significant impact on the theory of virulence in Pectobacterium. However, SCC3193 was originally incorrectly identified as P. carotovorum at the time of isolation, and our extensive phylogenetic analysis in this report reveals that it belongs to the species P. wasabiae, which is a less characterized species in the soft rot group. It is likely that other Pectobacterium isolates could also be incorrectly classified, and P. wasabiae may be more common than previously thought. Our report will facilitate further studies of the distribution and molecular biology of P. wasabiae, as a well-studied model strain has been added to the species, and three genome sequences are now publicly available, including the Japanese type strain sequenced in this study.
The absence of the T3SS and T3 effectors is the most distinctive feature separating Pectobacterium wasabiae SCC3193 and other P. wasabiae strains from other soft rot bacteria, other plant pathogens and other animal pathogenic enterobacteria. However, the number of confirmed T3 effectors is limited in soft rot bacteria overall, compared with the dozens in hemibiotrophs, and the effect of T3SS on virulence is not central in Pectobacterium. Our genomic approach, which was supplemented with in planta experiments, revealed putative ways for Pectobacterium wasabiae and other pectobacteria to establish infection. Promising candidates for novel virulence determinants having an effect at the early stage of infection are Virulence cluster 2, which carries genes for a putative novel lipoprotein transport system; HopL1, which is an adjacent T3SS effector-like protein; and T6SS. The latter has similar features to T3SS, which injects effectors into the host cell. T6SS is found in a variety of bacterial species and is not confined to pathogens. On the nitrogen-fixing plant symbiont Rhizobium leguminosarum, T6SS is related to host specificity [121]. The P. wasabiae benzoic acid/salicylic acid methyltransferase may also represent a novel way to manipulate the host in the latent stage, but we have not yet found experimental evidence to support this hypothesis.
In the future, to learn more about the latent stage of Pectobacterium infection, we should also consider bacteria outside the pathogens relying on T3SS. Many endophytic bacteria lack T3SS, T4SS and/or the production of pectinolytic enzymes common in phytopathogens [122]–[125]. They are known to use a diverse set of attachment structures and flagella-based motility to colonize plants and to produce plant hormones and other compounds for the modification of plant metabolism. Altogether, endophytic bacteria and symbionts, which efficiently establish interactions with plants, could be viewed as an opportunity to learn more about colonization mechanisms, which may also be important for the pathogenic lifestyle of soft rot bacteria during the latent stage of infection.
Materials and Methods
Bacterial strains and growth conditions
In this work, we used Pectobacterium wasabiae SCC3193, its derivatives and Pectobacterium wasabiae CFBP 3304T for the sequencing and/or biological experiments (Table S3). Standard growth conditions included culturing bacteria on Luria Broth (L3522, Sigma-Aldrich, US) for 1 d at +28°C. The antibiotics chloramphenicol (20 µg/mL) or ampicillin (100 or 150 µg/mL) were added when appropriate.
Genome sequencing
Genomic DNA of Pectobacterium wasabiae CFBP 3304T was extracted from an overnight culture using phenol-ether purification and ethanol precipitation. The quality and quantity of the DNA was assessed using spectrophotometry and agarose gel electrophoresis. The DNA concentration used for the hybridization step was 8 pM. Template amplification/cluster generation was performed using the Cluster station and the Single-Read Cluster generation kit v. 4 (Catalog # GD-103-4001). The sequencing was performed with an Illumina Genome Analyzer IIx using a v. 5 sequencing kit (FC-104-5001). All operations were performed following the manufacturer's protocols (Cluster Station User Guide, Part # 15005236 Rev. B, November 2009, Sequencing Kit v5 Reagent Preparation Guide, Part # 15013595 Rev. A, May 2010). Data generated by Illumina Solexa GAIIx were analyzed with SCS-RTA v. 2.9. Matrix and phasing parameters that were estimated from the PhiX control were used for base-calling. The demultiplexing and conversion of bcl-files to FASTQ-files were performed using OLB v. 1.9.0 and CASAVA GERALD v. 1.7.0. The instrument vendor provided all the software. Adapter sequences were clipped from the reads using cutadapt-tool. Additionally, if an adapter was removed from one read, the other read was shortened to reflect the change when necessary. The read data were assembled with ABySS using the following parameters: −j = 2 k = 60 n = 10 q = 15 ABYSS_OPTIONS = ‘–illumina-quality -c 25 -e 25’.
Accession number of Pectobacterium wasabiae CFBP 3304T
This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AKVS00000000. The version described in this paper is the first version, AKVS01000000.
ORF annotation
The ORF prediction was completed for CFBP 3304T using the Prodigal gene prediction program [126]. Systematic errors made by Prodigal were corrected, and the intergenic areas were double checked for missed gene predictions with the GenePRIMP program [127] and manual correction. Predicted and corrected protein sequences were then functionally annotated with descriptions (DE), Gene Ontologies (GO) and Enzyme Commission Numbers (EC) using the PANNZER tool (Koskinen et al., in preparation; method is unpublished). The COGnitor tool [128] was used to link sequences into COG database clusters. Finally, functional elements (for example, domains) were searched using the InterProScan tool [129].
Operon prediction
The annotated ORFs of SCC3193 (Koskinen et al., submitted) were grouped into hypothetical operons using the ofs1.2 software [130]. The grouping was based on link probabilities, which represent the probability of a given ORF to be co-expressed with a downstream gene (see Dataset S2). Based on an E. coli benchmark (RegulonDB 6.7 release [131]), our estimates suggest that thresholding these link probabilities at 0.54 will recall approximately 83% of true operon links with 81% specificity. The OFS operon prediction is based on intergenic distances, similarities between annotations and the conservative clustering of similar genes in other species. The set of “other species” (referred to as informant species) used in this analysis was compiled using a single representative of each bacterial order enlisted in the NCBI bacteria taxonomy. At the time of compilation, there were 119 orders with at least one full genome sequenced; therefore, the list contains 119 entries (Dataset S3).
Phylogenetic tree
In addition to the predicted protein sequences of Pectobacterium wasabiae SCC3193 and Pectobacterium wasabiae CFBP 3304T, full RefSeq proteomes were fetched from NCBI for 52 additional species. One-to-one orthologs of SCC3193 proteins were determined using the RBH (reciprocal best hit) criterion. Protein X.G1 from proteome G1 and protein X.G2 from proteome G2 are reciprocal best hits, if there X.G2 is the best match of X.G1 in proteome G2 and X.G1 is the best match of X.G2 in proteome G1. The full proteomes of SCC3193 and 53 target species were compared using SANS [41] with window size of 100. For phylogenetic analysis, we selected 51 groups of orthologs of SCC3193 proteins present in each of the 53 other species. The ortholog groups were then aligned using Muscle v. 3.8.31 [132]. For all multiple alignments, 1000 bootstrap trees were created using RAxML v. 7.0.4 [133]. The settings used for RAxML were “raxmlHPC -m PROTGAMMAJTT -c 4 -f d -n %s -s %s -x 137 -N 1000”, where %s was replaced with the correct input and corresponding output file names. All the bootstrapped trees were merged using the Consense program v. 3.68 from the Phylip package [134]. The merged trees were visualized using iTOL webtools [135].
Biochemical tests
Biochemical tests for the ability to reduce sugars, phosphatase activity, indole production, growth on sorbitol, growth on melibiose, growth on raffinose, growth on lactose, utilization of keto-methyl glucoside, growth in 5% NaCl and growth at +37°C were conducted for P. wasabiae SCC3193 and the type strains P. wasabiae CFBP 3304T, P. carotovorum CFBP 2046T and P. atrosepticum HAMBI 1429T according to the previously described protocols [136], [137].
Proteome comparison
In the proteome comparison, we re-identified all the ORFs for the sequenced Pectobacterium and Dickeya species and for the outgroup Yersinia pestis CO92. This was performed using Prodigal for gene prediction. The proteomes were then aligned using the blastp program and clustered into orthologous groups with OrthoMCL program. OrthoMCL clusters were converted into an orthologs vs. species (OvsS) matrix. The obtained OvsS matrix can be altered for ease of interpretation by ordering similar columns and rows next to each other, which was accomplished by creating a hierarchical cluster tree from the rows and columns. The internal nodes of the tree were flipped to place more similar neighboring clusters next to each other [138]. The similarity of the columns was based on the Pearson correlation, while the similarity of the rows was based on the cosine similarity. These measures were selected by reviewing the obtained visualization from the different similarity measures. The ordered matrix was then visualized on a heat map. A distance matrix was created by calculating the Pearson correlation similarity scores between species in the OvsS matrix and then visualized as a heat map.
Multiple genome alignment
Pectobacterium wasabiae CFBP 3304T contigs were ordered by aligning them against a reference genome, Pectobacterium wasabiae SCC3193. This alignment was created with Mauve v.2.3.1 [139]. The reordered Pectobacterium wasabiae CFBP 3304T contigs were then aligned against the genomes of Pectobacterium wasabiae WPP163 and Pectobacterium atrosepticum SCRI1043. Mauve produced a genome content distance matrix as an output from the pairwise alignments, which was used to quantify the differences in the aligned sequences.
Genomic islands
Genomic island predictions were performed using three computational tools: two that utilize sequence composition-based GI prediction methods, SIGI-HMM from the Colombo package [63] and IslandPath-DIMOB [64], and one that is based on a comparative genomic-based GI prediction, IslandPick [65]. Automated predictions were manually curated. The borders were adjusted such that genes known to be frequently associated with GIs or mobile genetic elements, such as integrase and phage genes, were added when these genes were located adjacent to automatically predicted islands. In addition, when necessary, the proximity of tRNA genes, similar functions encoded adjacent to the predicted island and the absence of the region in closely related strains based on blastn were used as criteria to modify the islands. Finally, we filtered out all islands consisting of less than five ORFs. To determine whether the SCC3193 genomic islands are also present in the genomes of other soft rot bacteria, blastn searches were performed with the nucleotide sequences of the islands against all available Pectobacterium and Dickeya genomes in GenBank. For P. carotovorum WPP14 and P. carotovorum subsp. brasiliensis PBR1692, whose genomes have not been completed, the combined query coverage of all contigs for each island was estimated based on a WGS-blastn search against these genomes. For P. wasabiae CFBP 3304T, the estimation was based on blastn pairwise alignment.
Genome comparisons
The comparison of orthologous proteins of Pectobacterium wasabiae SCC3193 was conducted against 41 selected soft rot bacteria, plant pathogens, animal pathogens and insect pathogens (Dataset S4) to identify novel virulence genes in SCC3193, in P. wasabiae or in soft rot bacteria. The comparison was performed manually, based on clusters created with OrthoMCL [140]. The genome and specific proteins identified were analyzed using the genome viewer Argo v. 1.0.31 [141], operon predictions, tblastn via Embster 2.0 beta launched from the CSC Chipster platform (http://chipster.csc.fi/embster/), annotation, blastn and blastp in GenBank NCBI [142], [143]. These analyses were conducted to identify known virulence determinants, detect missing virulence determinants and identify novel putative virulence determinants. The analysis of PCWDEs was performed by utilizing publications, sequence similarity and selected GO terms (Table S4) known to be associated with the enzymes of interest. The enzymes with the selected GO terms were mined using PANNZER (Koskinen et al., in preparation; method is unpublished), InterProScan [129] or BioMart [144]. The proteinases present in E. coli were discarded according to Glasner et al. [22], and they were not considered as putative enzymes targeting plants.
Targeted mutagenesis
The inactivation of individual genes and gene clusters was performed by deleting target sequences and replacing them with an antibiotic cassette, according to Datsenko and Wanner [145]. The antibiotic cassette was amplified from a pKD3 template plasmid with primers carrying 50 bp of similar sequence to the genomic DNA of Pectobacterium wasabiae SCC3193 (Table S5). The cloning was conducted using the proofreading PCR enzyme Phusion according to the manufacturer's 3-step protocol (Finnzymes). The insert was gel purified. For the electrocompetent cells, bacteria were grown overnight, diluted 1∶50 and grown to OD600 0.4. The cells were cooled down and washed twice with sterile ice-cold ddH2O and once with sterile ice-cold 10% glycerol. The cells were resuspended in 1.5–2x volume of sterile ice-cold 10% glycerol. Electroporation was performed with the following settings: 2.5 kV, 25 µF and 200 Ω in 0.2 cm cuvettes (Bio-Rad Laboratories). The recovery times for pKD46 and the antibiotic cassette insertion were 15 min and 3.5 h, respectively. The mutations and the position of the insert were confirmed with two PCR reactions according to Datsenko and Wanner [145] in addition to sequencing (Table S5). For the double mutant of T6SS, the first antibiotic cassette was digested using flipase produced in trans from the pFLP2 plasmid [146].
Plasmid constructs
For the complementation experiments, the genes of the sirB locus were amplified by PCR from wild-type SCC3193 genomic DNA using the proofreading PCR enzyme Phusion (Finnzymes). The following primers were utilized: SirB1_compl_F and SirB1_compl_R for sirB1; SirB2_compl_F and SirB2_compl_R for sirB2; and SirB2_compl_F and SirB1_compl_R for the complete sirB locus (Table S5). The PCR products were gel purified, digested with HindIII and SacI and ligated into pMW119 (Nippon Gene), which was digested with the corresponding enzymes. The constructs were confirmed via PCR and sequencing.
Tobacco seedling virulence assay
The bacterial virulence was tested on axenic tobacco seedlings (Nicotiana tabacum cv. ‘Samsun’) according to Pirhonen et al. [24]. The seedlings were propagated in 24-well tissue culture plates on ½ MS medium supplemented with vitamins (Duchefa) and 2% sucrose and solidified with 0.8% agar. The seedlings were grown for 16 days with a 16/8 h day/night cycle at 26/22°C. The bacterial strains were grown overnight, washed and diluted in 10 mM MgSO4. A single leaf from each of 48 plants was wounded with a needle and inoculated with 1.5 µl of bacterial solution (OD600 0.05). The inoculated plants were kept in the dark at room temperature, and the development of disease symptoms (leaf maceration) was scored visually at 24 and 48 h after inoculation on a scale of 0 to 6 according to the severity of symptoms. To determine bacterial growth in planta (cfu/plant), the inoculated plants were kept in the dark at 26°C and homogenized into 10 mM MgSO4 after 24 and 44 h; serial dilutions of bacteria were plated. All experiments were repeated three times, and the data of each replicate were analyzed statistically utilizing a Mann-Whitney significance test for the pairwise comparison of two independent samples using PASW Statistics 18.
Potato tuber slice virulence assay
For the tuber slice assay [17], bacterial strains were grown overnight, washed and resuspended into 10 mM MgSO4. Tubers (cv. Van Gogh, H&H Tuominen, Finland) were washed with tap water and surface sterilized with 10% Na-hypochlorite for 5 min. The tubers were sliced 0.6 cm thick, surface sterilized again with flaming and placed on wet paper tissue in a Petri dish. Ten to 12 tuber slices were inoculated with 10 µl of bacterial solution containing 106 cfu/ml and incubated at room temperature in a shady place for three days. The macerated area was assessed, and the results were classified into seven groups: 0%, ∼5%, ∼20%, ∼50%, ∼75%, ∼90% and 100% rotten tissue per tuber slice surface area. The experiments were repeated five times, and data from all the replicates were combined for statistical analyses utilizing the Mann-Whitney significance test for the pairwise comparison of two independent samples using PASW Statistics 18.
Enzyme assays and growth curve
Cellulase (Cel) and polygalacturonase (PehA) activities were assayed from 10 µl of supernatant of liquid cultures grown overnight and from appropriate dilutions of the supernatant on corresponding enzyme indicator plates [7]. The growth of the sirB, Vic1, Vic2 and T6SS-double mutants were compared with the SCC3193 wild-type strain in an hrp-inducing minimal medium [147] using 0.4% polygalacturonic acid (Sigma P3850) as a sole carbon source. The bacteria were washed once with 1x minimal medium, diluted to OD600 0.1 and then grown for 20 h at +28°C with shaking (200 rpm).
Supporting Information
Zdroje
1. HaubenL, MooreER, VauterinL, SteenackersM, MergaertJ, et al. (1998) Phylogenetic position of phytopathogens within the Enterobacteriaceae. Syst Appl Microbiol 21 : 384–397.
2. SamsonR, LegendreJB, ChristenR, Fischer-Le SauxM, AchouakW, et al. (2005) Transfer of Pectobacterium chrysanthemi (Burkholder et al. 1953) Brenner et al. 1973 and Brenneria paradisiaca to the genus Dickeya gen. nov. as Dickeya chrysanthemi comb. nov. and Dickeya paradisiaca comb. nov. and delineation of four novel species, Dickeya dadantii sp. nov., Dickeya dianthicola sp. nov., Dickeya dieffenbachiae sp. nov. and Dickeya zeae sp. nov. Int J Syst Evol Microbiol 55 : 1415–1427 doi:10.1099/ijs.0.02791-0
3. GardanL, GouyC, ChristenR, SamsonR (2003) Elevation of three subspecies of Pectobacterium carotovorum to species level: Pectobacterium atrosepticum sp. nov., Pectobacterium betavasculorum sp. nov. and Pectobacterium wasabiae sp. nov. Int J Syst Evol Microbiol 53 : 381–391.
4. CzajkowskiR, PérombelonMCM, van VeenJA, van der WolfJM (2011) Control of blackleg and tuber soft rot of potato caused by Pectobacterium and Dickeya species: a review. Plant Pathol 60 : 999–1013 doi:10.1111/j.1365-3059.2011.02470.x
5. PérombelonMCM (2002) Potato diseases caused by soft rot erwinias: an overview of pathogenesis. Plant Pathol 51 : 1–12 doi:10.1046/j.0032-0862.2001.Shorttitle.doc.x
6. TothIK, BirchPRJ (2005) Rotting softly and stealthily. Curr Opin Plant Biol 8 : 424–429 doi:10.1016/j.pbi.2005.04.001
7. PirhonenM, FlegoD, HeikinheimoR, PalvaET (1993) A small diffusible signal molecule is responsible for the global control of virulence and exoenzyme production in the plant pathogen Erwinia carotovora. EMBO J 12 : 2467–2476.
8. ErikssonAR, AnderssonRA, PirhonenM, PalvaET (1998) Two-component regulators involved in the global control of virulence in Erwinia carotovora subsp. carotovora. Mol Plant Microbe Interact 11 : 743–752 doi:10.1094/MPMI.1998.11.8.743
9. HyytiäinenH, MontesanoM, PalvaET (2001) Global regulators ExpA (GacA) and KdgR modulate extracellular enzyme gene expression through the RsmA-rsmB system in Erwinia carotovora subsp. carotovora. Mol Plant Microbe Interact 14 : 931–938 doi:10.1094/MPMI.2001.14.8.931
10. TothIK, BellKS, HolevaMC, BirchPRJ (2003) Soft rot erwiniae: from genes to genomes. Mol Plant Pathol 4 : 17–30 doi:10.1046/j.1364-3703.2003.00149.x
11. TothIK, PritchardL, BirchPRJ (2006) Comparative genomics reveals what makes an enterobacterial plant pathogen. Annu Rev Phytopathol 44 : 305–336 doi:10.1146/annurev.phyto.44.070505.143444
12. EvansTJ, IndA, KomitopoulouE, SalmondGPC (2010) Phage-selected lipopolysaccharide mutants of Pectobacterium atrosepticum exhibit different impacts on virulence. J Appl Microbiol 109 : 505–514 doi:10.1111/j.1365-2672.2010.04669.x
13. BellKS, SebaihiaM, PritchardL, HoldenMTG, HymanLJ, et al. (2004) Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp. atroseptica and characterization of virulence factors. Proc Natl Acad Sci U S A 101 : 11105–11110 doi:10.1073/pnas.0402424101
14. LiuH, CoulthurstSJ, PritchardL, HedleyPE, RavensdaleM, et al. (2008) Quorum sensing coordinates brute force and stealth modes of infection in the plant pathogen Pectobacterium atrosepticum. PLoS Pathog 4: e1000093 doi:10.1371/journal.ppat.1000093
15. MattinenL, TshuikinaM, MäeA, PirhonenM (2004) Identification and characterization of Nip, necrosis-inducing virulence protein of Erwinia carotovora subsp. carotovora. Mol Plant Microbe Interact 17 : 1366–1375 doi:10.1094/MPMI.2004.17.12.1366
16. CorbettM, VirtueS, BellK, BirchP, BurrT, et al. (2005) Identification of a new quorum-sensing-controlled virulence factor in Erwinia carotovora subsp. atroseptica secreted via the type II targeting pathway. Mol Plant Microbe Interact 18 : 334–342 doi:10.1094/MPMI-18-0334
17. SjöblomS, HarjunpääH, BraderG, PalvaET (2008) A novel plant ferredoxin-like protein and the regulator Hor are quorum-sensing targets in the plant pathogen Erwinia carotovora. Mol Plant Microbe Interact 21 : 967–978 doi:10.1094/MPMI-21-7-0967
18. UrbanyC, NeuhausHE (2008) Citrate uptake into Pectobacterium atrosepticum is critical for bacterial virulence. Mol Plant Microbe Interact 21 : 547–554 doi:10.1094/MPMI-21-5-0547
19. Marquez-VillavicencioMDP, WeberB, WitherellRA, WillisDK, CharkowskiAO (2011) The 3-Hydroxy-2-Butanone Pathway Is Required for Pectobacterium carotovorum Pathogenesis. PLoS ONE 6: e22974 doi:10.1371/journal.pone.0022974
20. PoueymiroM, GeninS (2009) Secreted proteins from Ralstonia solanacearum: a hundred tricks to kill a plant. Curr Opin Microbiol 12 : 44–52 doi:10.1016/j.mib.2008.11.008
21. CollmerA, SchneiderDJ, LindebergM (2009) Lifestyles of the effector rich: genome-enabled characterization of bacterial plant pathogens. Plant Physiol 150 : 1623–1630 doi:10.1104/pp.109.140327
22. GlasnerJD, Marquez-VillavicencioM, KimH-S, JahnCE, MaB, et al. (2008) Niche-specificity and the variable fraction of the Pectobacterium pan-genome. Mol Plant Microbe Interact 21 : 1549–1560 doi:10.1094/MPMI-21-12-1549
23. PirhonenM, HeinoP, HelanderI, HarjuP, PalvaET (1988) Bacteriophage T4 resistant mutants of the plant pathogen Erwinia carotovora. Microb Pathog 4 : 359–367.
24. PirhonenM (1991) Identification of Pathogenicity Determinants of Erwinia carotovora subsp. carotovora by Transposon Mutagenesis. Mol Plant Microbe Interact 4 : 276 doi:10.1094/MPMI-4-276
25. SaarilahtiHT, PirhonenM, KarlssonMB, FlegoD, PalvaET (1992) Expression of pehA-bla gene fusions in Erwinia carotovora subsp. carotovora and isolation of regulatory mutants affecting polygalacturonase production. Mol Gen Genet 234 : 81–88.
26. MäeA, HeikinheimoR, PalvaET (1995) Structure and regulation of the Erwinia carotovora subspecies carotovora SCC3193 cellulase gene celV1 and the role of cellulase in phytopathogenicity. Mol Gen Genet 247 : 17–26.
27. AnderssonRA, PalvaET, PirhonenM (1999) The response regulator expM is essential for the virulence of Erwinia carotovora subsp. carotovora and acts negatively on the sigma factor RpoS (sigma s). Mol Plant Microbe Interact 12 : 575–584 doi:10.1094/MPMI.1999.12.7.575
28. MaritsR, KõivV, LaasikE, MäeA (1999) Isolation of an extracellular protease gene of Erwinia carotovora subsp. carotovora strain SCC3193 by transposon mutagenesis and the role of protease in phytopathogenicity. Microbiology (Reading, Engl) 145 (Pt 8) 1959–1966.
29. AnderssonRA, KõivV, Norman-SetterbladC, PirhonenM (1999) Role of RpoS in virulence and stress tolerance of the plant pathogen Erwinia carotovora subsp. carotovora. Microbiology (Reading, Engl) 145 (Pt 12) 3547–3556.
30. KõivV, MäeA (2001) Quorum sensing controls the synthesis of virulence factors by modulating rsmA gene expression in Erwinia carotovora subsp. carotovora. Mol Genet Genomics 265 : 287–292.
31. MontesanoM, BraderG, PonceDE, LeónI, PalvaET (2005) Multiple defence signals induced by Erwinia carotovora ssp. carotovora elicitors in potato. Mol Plant Pathol 6 : 541–549 doi:10.1111/j.1364-3703.2005.00305.x
32. SjöblomS, BraderG, KochG, PalvaET (2006) Cooperation of two distinct ExpR regulators controls quorum sensing specificity and virulence in the plant pathogen Erwinia carotovora. Mol Microbiol 60 : 1474–1489 doi:10.1111/j.1365-2958.2006.05210.x
33. AndresenL, KõivV, AlamäeT, MäeA (2007) The Rcs phosphorelay modulates the expression of plant cell wall degrading enzymes and virulence in Pectobacterium carotovorum ssp. carotovorum. FEMS Microbiol Lett 273 : 229–238 doi:10.1111/j.1574-6968.2007.00794.x
34. KimH-S, MaB, PernaNT, CharkowskiAO (2009) Phylogeny and virulence of naturally occurring type III secretion system-deficient Pectobacterium strains. Appl Environ Microbiol 75 : 4539–4549 doi:10.1128/AEM.01336-08
35. GotoM, MatsumotoK (1987) Erwinia carotovora subsp. wasabiae subsp. nov. Isolated from Diseased Rhizomes and Fibrous Roots of Japanese Horseradish (Eutrema wasabi Maxim.). Int J Syst Bacteriol 37 : 130–135 doi:10.1099/00207713-37-2-130
36. MaB, HibbingME, KimH-S, ReedyRM, YedidiaI, et al. (2007) Host range and molecular phylogenies of the soft rot enterobacterial genera pectobacterium and dickeya. Phytopathology 97 : 1150–1163 doi:10.1094/PHYTO-97-9-1150
37. PitmanAR, WrightPJ, GalbraithMD, HarrowSA (2008) Biochemical and genetic diversity of pectolytic enterobacteria causing soft rot disease of potatoes in New Zealand. Australasian Plant Pathol 37 : 559 doi:10.1071/AP08056
38. KimMH, ChoMS, KimBK, ChoiHJ, HahnJH, et al. (2012) Quantitative Real-Time Polymerase Chain Reaction Assay for Detection of Pectobacterium wasabiae Using YD Repeat Protein Gene-Based Primers. Plant Dis 96 : 253–257 doi:10.1094/PDIS-06-11-0511
39. Baghaee-RavariS, RahimianH, Shams-BakhshM, Lopez-SolanillaE, Antúnez-LamasM, et al. (2010) Characterization of Pectobacterium species from Iran using biochemical and molecular methods. Eur J Plant Pathol 129 : 413–425 doi:10.1007/s10658-010-9704-z
40. NabhanS, WydraK, LindeM, DebenerT (2011) The use of two complementary DNA assays, AFLP and MLSA, for epidemic and phylogenetic studies of pectolytic enterobacterial strains with focus on the heterogeneous species Pectobacterium carotovorum. Plant Pathology 61 : 498–508 Available: http://onlinelibrary.wiley.com/doi/10.1111/j.1365-3059.2011.02546.x/abstract. Accessed 24 February 2012.
41. KoskinenJP, HolmL (2012) SANS: high-throughput retrieval of protein sequences allowing 50% mismatches. Bioinformatics 28: i438–i443 doi:10.1093/bioinformatics/bts417
42. YishayM, BurdmanS, ValverdeA, LuzzattoT, OphirR, et al. (2008) Differential pathogenicity and genetic diversity among Pectobacterium carotovorum ssp. carotovorum isolates from monocot and dicot hosts support early genomic divergence within this taxon. Environ Microbiol 10 : 2746–2759 doi:10.1111/j.1462-2920.2008.01694.x
43. MulhollandV, HintonJC, SidebothamJ, TothIK, HymanLJ, et al. (1993) A pleiotropic reduced virulence (Rvi-) mutant of Erwinia carotovora subspecies atroseptica is defective in flagella assembly proteins that are conserved in plant and animal bacterial pathogens. Mol Microbiol 9 : 343–356.
44. HossainMM, ShibataS, AizawaS-I, TsuyumuS (2005) Motility is an important determinant for pathogenesis of Erwinia carotovora subsp. carotovora. Physiol Mol Plant Pathol 66 : 134–143 doi:10.1016/j.pmpp.2005.06.001
45. TothIK, ThorpeCJ, BentleySD, MulhollandV, HymanLJ, et al. (1999) Mutation in a gene required for lipopolysaccharide and enterobacterial common antigen biosynthesis affects virulence in the plant pathogen Erwinia carotovora subsp. atroseptica. Mol Plant Microbe Interact 12 : 499–507 doi:10.1094/MPMI.1999.12.6.499
46. PembertonCL, WhiteheadNA, SebaihiaM, BellKS, HymanLJ, et al. (2005) Novel quorum-sensing-controlled genes in Erwinia carotovora subsp. carotovora: identification of a fungal elicitor homologue in a soft-rotting bacterium. Mol Plant Microbe Interact 18 : 343–353 doi:10.1094/MPMI-18-0343
47. AnderssonRA, ErikssonAR, HeikinheimoR, MäeA, PirhonenM, et al. (2000) Quorum sensing in the plant pathogen Erwinia carotovora subsp. carotovora: the role of expR(Ecc). Mol Plant Microbe Interact 13 : 384–393 doi:10.1094/MPMI.2000.13.4.384
48. LaasikE, AndresenL, MäeA (2006) Type II quorum sensing regulates virulence in Erwinia carotovora ssp. carotovora. FEMS Microbiol Lett 258 : 227–234 doi:10.1111/j.1574-6968.2006.00222.x
49. FlegoD, MaritsR, ErikssonAR, KõivV, KarlssonMB, et al. (2000) A two-component regulatory system, pehR-pehS, controls endopolygalacturonase production and virulence in the plant pathogen Erwinia carotovora subsp. carotovora. Mol Plant Microbe Interact 13 : 447–455 doi:10.1094/MPMI.2000.13.4.447
50. HyytiäinenH, SjöblomS, PalomäkiT, TuikkalaA, Tapio PalvaE (2003) The PmrA-PmrB two-component system responding to acidic pH and iron controls virulence in the plant pathogen Erwinia carotovora ssp. carotovora. Mol Microbiol 50 : 795–807.
51. ReevesPJ, WhitcombeD, WharamS, GibsonM, AllisonG, et al. (1993) Molecular cloning and characterization of 13 out genes from Erwinia carotovora subspecies carotovora: genes encoding members of a general secretion pathway (GSP) widespread in gram-negative bacteria. Mol Microbiol 8 : 443–456.
52. RecordsAR (2011) The type VI secretion system: a multipurpose delivery system with a phage-like machinery. Mol Plant Microbe Interact 24 : 751–757 doi:10.1094/MPMI-11-10-0262
53. SchwarzS, WestTE, BoyerF, ChiangW-C, CarlMA, et al. (2010) Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog 6: e1001068 doi:10.1371/journal.ppat.1001068
54. ZhengJ, HoB, MekalanosJJ (2011) Genetic analysis of anti-amoebae and anti-bacterial activities of the type VI secretion system in Vibrio cholerae. PLoS ONE 6: e23876 doi:10.1371/journal.pone.0023876
55. AlfanoJR, CollmerA (2004) Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu Rev Phytopathol 42 : 385–414 doi:10.1146/annurev.phyto.42.040103.110731
56. PitmanAR, HarrowSA, VisnovskySB (2009) Genetic characterisation of Pectobacterium wasabiae causing soft rot disease of potato in New Zealand. Eur J Plant Pathol 126 : 423–435 doi:10.1007/s10658-009-9551-y
57. KimH-S, ThammaratP, LommelSA, HoganCS, CharkowskiAO (2011) Pectobacterium carotovorum elicits plant cell death with DspE/F but the P. carotovorum DspE does not suppress callose or induce expression of plant genes early in plant-microbe interactions. Mol Plant Microbe Interact 24 : 773–786 doi:10.1094/MPMI-06-10-0143
58. HolevaMC, BellKS, HymanLJ, AvrovaAO, WhissonSC, et al. (2004) Use of a pooled transposon mutation grid to demonstrate roles in disease development for Erwinia carotovora subsp. atroseptica putative type III secreted effector (DspE/A) and helper (HrpN) proteins. Mol Plant Microbe Interact 17 : 943–950 doi:10.1094/MPMI.2004.17.9.943
59. YangC-H, Gavilanes-RuizM, OkinakaY, VedelR, BerthuyI, et al. (2002) hrp genes of Erwinia chrysanthemi 3937 are important virulence factors. Mol Plant Microbe Interact 15 : 472–480 doi:10.1094/MPMI.2002.15.5.472
60. CunnacS, LindebergM, CollmerA (2009) Pseudomonas syringae type III secretion system effectors: repertoires in search of functions. Curr Opin Microbiol 12 : 53–60 doi:10.1016/j.mib.2008.12.003
61. HackerJ, CarnielE (2001) Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep 2 : 376–381 doi:10.1093/embo-reports/kve097
62. DobrindtU, HochhutB, HentschelU, HackerJ (2004) Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol 2 : 414–424 doi:10.1038/nrmicro884
63. WaackS, KellerO, AsperR, BrodagT, DammC, et al. (2006) Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7 : 142 doi:10.1186/1471-2105-7-142
64. HsiaoW, WanI, JonesSJ, BrinkmanFSL (2003) IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19 : 418–420.
65. LangilleMGI, HsiaoWWL, BrinkmanFSL (2008) Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9 : 329 doi:10.1186/1471-2105-9-329
66. LawrenceJG, OchmanH (1998) Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A 95 : 9413–9417.
67. KooninEV, MakarovaKS, AravindL (2001) Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol 55 : 709–742 doi:10.1146/annurev.micro.55.1.709
68. Vincent-SealyLV, ThomasJD, CommanderP, SalmondGP (1999) Erwinia carotovora DsbA mutants: evidence for a periplasmic-stress signal transduction system affecting transcription of genes encoding secreted proteins. Microbiology (Reading, Engl) 145 (Pt 8) 1945–1958.
69. CoulthurstSJ, LilleyKS, HedleyPE, LiuH, TothIK, et al. (2008) DsbA plays a critical and multifaceted role in the production of secreted virulence factors by the phytopathogen Erwinia carotovora subsp. atroseptica. J Biol Chem 283 : 23739–23753 doi:10.1074/jbc.M801829200
70. EffmertU, SaschenbreckerS, RossJ, NegreF, FraserCM, et al. (2005) Floral benzenoid carboxyl methyltransferases: from in vitro to in planta function. Phytochemistry 66 : 1211–1230 doi:10.1016/j.phytochem.2005.03.031
71. ParkS-W, KaimoyoE, KumarD, MosherS, KlessigDF (2007) Methyl salicylate is a critical mobile signal for plant systemic acquired resistance. Science 318 : 113–116 doi:10.1126/science.1147113
72. KooYJ, KimMA, KimEH, SongJT, JungC, et al. (2007) Overexpression of salicylic acid carboxyl methyltransferase reduces salicylic acid-mediated pathogen resistance in Arabidopsis thaliana. Plant Mol Biol 64 : 1–15 doi:10.1007/s11103-006-9123-x
73. LiuP-P, YangY, PicherskyE, KlessigDF (2010) Altering expression of benzoic acid/salicylic acid carboxyl methyltransferase 1 compromises systemic acquired resistance and PAMP-triggered immunity in arabidopsis. Mol Plant Microbe Interact 23 : 82–90 doi:10.1094/MPMI-23-1-0082
74. YeatesTO, KerfeldCA, HeinhorstS, CannonGC, ShivelyJM (2008) Protein-based organelles in bacteria: carboxysomes and related microcompartments. Nat Rev Microbiol 6 : 681–691 doi:10.1038/nrmicro1913
75. KerfeldCA, HeinhorstS, CannonGC (2010) Bacterial microcompartments. Annu Rev Microbiol 64 : 391–408 doi:10.1146/annurev.micro.112408.134211
76. YeatesTO, CrowleyCS, TanakaS (2010) Bacterial microcompartment organelles: protein shell structure and evolution. Annu Rev Biophys 39 : 185–205 doi:10.1146/annurev.biophys.093008.131418
77. GarsinDA (2010) Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol 8 : 290–295 doi:10.1038/nrmicro2334
78. AnforaAT, HalladinDK, HaugenBJ, WelchRA (2008) Uropathogenic Escherichia coli CFT073 is adapted to acetatogenic growth but does not require acetate during murine urinary tract infection. Infect Immun 76 : 5760–5767 doi:10.1128/IAI.00618-08
79. BryantWA, KrabbenP, BaganzF, ZhouY, WardJM (2009) The Analysis of Multiple Genome Comparisons in Genus Escherichia and Its Application to the Discovery of Uncharacterised Metabolic Genes in Uropathogenic Escherichia coli CFT073. Comp Funct Genomics 782924 doi:10.1155/2009/782924
80. MosbergJA, YepA, MeredithTC, SmithS, WangP-F, et al. (2011) A unique arabinose 5-phosphate isomerase found within a genomic island associated with the uropathogenicity of Escherichia coli CFT073. J Bacteriol 193 : 2981–2988 doi:10.1128/JB.00033-11
81. ReslewicS, ZhouS, PlaceM, ZhangY, BriskaA, et al. (2005) Whole-genome shotgun optical mapping of Rhodospirillum rubrum. Appl Environ Microbiol 71 : 5511–5522 doi:10.1128/AEM.71.9.5511-5522.2005
82. MujahidM, SasikalaC, RamanaCV (2010) Aniline-induced tryptophan production and identification of indole derivatives from three purple bacteria. Curr Microbiol 61 : 285–290 doi:10.1007/s00284-010-9609-2
83. LarimerFW, ChainP, HauserL, LamerdinJ, MalfattiS, et al. (2004) Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat Biotechnol 22 : 55–61 doi:10.1038/nbt923
84. ImamS, YilmazS, SohmenU, GorzalskiAS, ReedJL, et al. (2011) iRsp1095: a genome-scale reconstruction of the Rhodobacter sphaeroides metabolic network. BMC Syst Biol 5 : 116 doi:10.1186/1752-0509-5-116
85. MilneCB, EddyJA, RajuR, ArdekaniS, KimP-J, et al. (2011) Metabolic network reconstruction and genome-scale model of butanol-producing strain Clostridium beijerinckii NCIMB 8052. BMC Syst Biol 5 : 130 doi:10.1186/1752-0509-5-130
86. RodriguesJLM, SerresMH, TiedjeJM (2011) Large-scale comparative phenotypic and genomic analyses reveal ecological preferences of shewanella species and identify metabolic pathways conserved at the genus level. Appl Environ Microbiol 77 : 5352–5360 doi:10.1128/AEM.00097-11
87. NewmanM-A, DowJM, MolinaroA, ParrilliM (2007) Priming, induction and modulation of plant defence responses by bacterial lipopolysaccharides. J Endotoxin Res 13 : 69–84 doi:10.1177/0968051907079399
88. StolzJF, BasuP, SantiniJM, OremlandRS (2006) Arsenic and selenium in microbial metabolism. Annu Rev Microbiol 60 : 107–130 doi:10.1146/annurev.micro.60.080805.142053
89. JacksonCR, DugasSL (2003) Phylogenetic analysis of bacterial and archaeal arsC gene sequences suggests an ancient, common origin for arsenate reductase. BMC Evol Biol 3 : 18 doi:10.1186/1471-2148-3-18
90. Mäkelä-Kurtto R, Eurola M, Justen A, Backman B, Luoma S, et al..(Geological Survey of Finland) (2006) Arsenic and other elements in agro-ecosystems in Finland and particularly in the Pirkanmaa region. Final report 2006. Espoo, Finland: Geological Survey of Finland. 121 p. Available from: http://en.gtk.fi/Geoinfo/Publications/Publicationsales.html
91. WangYD, ZhaoS, HillCW (1998) Rhs elements comprise three subfamilies which diverged prior to acquisition by Escherichia coli. J Bacteriol 180 : 4102–4110.
92. JacksonAP, ThomasGH, ParkhillJ, ThomsonNR (2009) Evolutionary diversification of an ancient gene family (rhs) through C-terminal displacement. BMC Genomics 10 : 584 doi:10.1186/1471-2164-10-584
93. PukatzkiS, MaAT, SturtevantD, KrastinsB, SarracinoD, et al. (2006) Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A 103 : 1528–1533 doi:10.1073/pnas.0510322103
94. MougousJD, CuffME, RaunserS, ShenA, ZhouM, et al. (2006) A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312 : 1526–1530 doi:10.1126/science.1128393
95. LarkinMA, BlackshieldsG, BrownNP, ChennaR, McGettiganPA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23 : 2947–2948 doi:10.1093/bioinformatics/btm404
96. GoujonM, McWilliamH, LiW, ValentinF, SquizzatoS, et al. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38: W695–699 doi:10.1093/nar/gkq313
97. GibbsKA, UrbanowskiML, GreenbergEP (2008) Genetic determinants of self identity and social recognition in bacteria. Science 321 : 256–259 doi:10.1126/science.1160033
98. YouderianP, HartzellPL (2007) Triple mutants uncover three new genes required for social motility in Myxococcus xanthus. Genetics 177 : 557–566 doi:10.1534/genetics.107.076182
99. PooleSJ, DinerEJ, AokiSK, BraatenBA, t' Kint de RoodenbekeC, et al. (2011) Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet 7: e1002217 doi:10.1371/journal.pgen.1002217
100. MattinenL, SomervuoP, NykyriJ, NissinenR, KouvonenP, et al. (2008) Microarray profiling of host-extract-induced genes and characterization of the type VI secretion cluster in the potato pathogen Pectobacterium atrosepticum. Microbiology (Reading, Engl) 154 : 2387–2396 doi:10.1099/mic.0.2008/017582-0
101. MattickJS (2002) Type IV pili and twitching motility. Annu Rev Microbiol 56 : 289–314 doi:10.1146/annurev.micro.56.012302.160938
102. CraigL, PiqueME, TainerJA (2004) Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2 : 363–378 doi:10.1038/nrmicro885
103. IshimaruCA, LoperJE (1992) High-affinity iron uptake systems present in Erwinia carotovora subsp. carotovora include the hydroxamate siderophore aerobactin. J Bacteriol 174 : 2993–3003.
104. RatledgeC, DoverLG (2000) Iron metabolism in pathogenic bacteria. Annu Rev Microbiol 54 : 881–941 doi:10.1146/annurev.micro.54.1.881
105. BoughammouraA, FranzaT, DellagiA, RouxC, Matzanke-MarksteinB, et al. (2007) Ferritins, bacterial virulence and plant defence. Biometals 20 : 347–353 doi:10.1007/s10534-006-9069-0
106. ShaoN, HuangH, MengK, LuoH, WangY, et al. (2008) Cloning, expression, and characterization of a new phytase from the phytopathogenic bacterium Pectobacterium wasabiae DSMZ 18074. J Microbiol Biotechnol 18 : 1221–1226.
107. JohnstonC, PeguesDA, HueckCJ, LeeA, MillerSI (1996) Transcriptional activation of Salmonella typhimurium invasion genes by a member of the phosphorylated response-regulator superfamily. Mol Microbiol 22 : 715–727.
108. RakemanJL, BonifieldHR, MillerSI (1999) A HilA-independent pathway to Salmonella typhimurium invasion gene transcription. J Bacteriol 181 : 3096–3104.
109. StrohmaierH, RemlerP, RennerW, HögenauerG (1995) Expression of genes kdsA and kdsB involved in 3-deoxy-D-manno-octulosonic acid metabolism and biosynthesis of enterobacterial lipopolysaccharide is growth phase regulated primarily at the transcriptional level in Escherichia coli K-12. J Bacteriol 177 : 4488–4500.
110. NakahigashiK, KuboN, NaritaS, ShimaokaT, GotoS, et al. (2002) HemK, a class of protein methyl transferase with similarity to DNA methyl transferases, methylates polypeptide chain release factors, and hemK knockout induces defects in translational termination. Proc Natl Acad Sci U S A 99 : 1473–1478 doi:10.1073/pnas.032488499
111. RydenM, MurphyJ, MartinR, IsakssonL, GallantJ (1986) Mapping and complementation studies of the gene for release factor 1. J Bacteriol 168 : 1066–1069.
112. KroghA, LarssonB, von HeijneG, SonnhammerEL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305 : 567–580 doi:10.1006/jmbi.2000.431510.1006/jmbi.2000.4315.
113. SmitsTHM, RezzonicoF, KamberT, BlomJ, GoesmannA, et al. (2010) Complete genome sequence of the fire blight pathogen Erwinia amylovora CFBP 1430 and comparison to other Erwinia spp. Mol Plant Microbe Interact 23 : 384–393 doi:10.1094/MPMI-23-4-0384
114. Petnicki-OcwiejaT, SchneiderDJ, TamVC, ChanceyST, ShanL, et al. (2002) Genomewide identification of proteins secreted by the Hrp type III protein secretion system of Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci U S A 99 : 7652–7657 doi:10.1073/pnas.112183899
115. TaghaviS, van der LelieD, HoffmanA, ZhangY-B, WallaMD, et al. (2010) Genome sequence of the plant growth promoting endophytic bacterium Enterobacter sp. 638. PLoS Genet 6: e1000943 doi:10.1371/journal.pgen.1000943
116. Goodrich-BlairH, ClarkeDJ (2007) Mutualism and pathogenesis in Xenorhabdus and Photorhabdus: two roads to the same destination. Mol Microbiol 64 : 260–268 doi:10.1111/j.1365-2958.2007.05671.x
117. TockMR, DrydenDTF (2005) The biology of restriction and anti-restriction. Curr Opin Microbiol 8 : 466–472 doi:10.1016/j.mib.2005.06.003
118. KobayashiI (2001) Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res 29 : 3742–3756.
119. NobusatoA, UchiyamaI, KobayashiI (2000) Diversity of restriction-modification gene homologues in Helicobacter pylori. Gene 259 : 89–98.
120. PouillotF, FayolleC, CarnielE (2007) A putative DNA adenine methyltransferase is involved in Yersinia pseudotuberculosis pathogenicity. Microbiology (Reading, Engl) 153 : 2426–2434 doi:10.1099/mic.0.2007/005736-0
121. BladergroenMR, BadeltK, SpainkHP (2003) Infection-blocking genes of a symbiotic Rhizobium leguminosarum strain that are involved in temperature-dependent protein secretion. Mol Plant Microbe Interact 16 : 53–64 doi:10.1094/MPMI.2003.16.1.53
122. WuX, MonchyS, TaghaviS, ZhuW, RamosJ, et al. (2011) Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida. FEMS Microbiol Rev 35 : 299–323 doi:10.1111/j.1574-6976.2010.00249.x
123. RosenbluethM, Martínez-RomeroE (2006) Bacterial endophytes and their interactions with hosts. Mol Plant Microbe Interact 19 : 827–837 doi:10.1094/MPMI-19-0827
124. RyanRP, GermaineK, FranksA, RyanDJ, DowlingDN (2008) Bacterial endophytes: recent developments and applications. FEMS Microbiol Lett 278 : 1–9 doi:10.1111/j.1574-6968.2007.00918.x
125. TylerHL, TriplettEW (2008) Plants as a habitat for beneficial and/or human pathogenic bacteria. Annu Rev Phytopathol 46 : 53–73 doi:10.1146/annurev.phyto.011708.103102
126. HyattD, ChenG-L, LocascioPF, LandML, LarimerFW, et al. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11 : 119 doi:10.1186/1471-2105-11-119
127. PatiA, IvanovaNN, MikhailovaN, OvchinnikovaG, HooperSD, et al. (2010) GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 7 : 455–457 doi:10.1038/nmeth.1457
128. TatusovRL, GalperinMY, NataleDA, KooninEV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28 : 33–36.
129. ZdobnovEM, ApweilerR (2001) InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17 : 847–848.
130. WestoverBP, BuhlerJD, SonnenburgJL, GordonJI (2005) Operon prediction without a training set. Bioinformatics 21 : 880–888 doi:10.1093/bioinformatics/bti123
131. Gama-CastroS, Jiménez-JacintoV, Peralta-GilM, Santos-ZavaletaA, Peñaloza-SpinolaMI, et al. (2008) RegulonDB (version 6.0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation. Nucleic Acids Res 36: D120–124 doi:10.1093/nar/gkm994
132. EdgarRC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 : 1792–1797 doi:10.1093/nar/gkh340
133. StamatakisA, LudwigT, MeierH (2005) RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21 : 456–463 doi:10.1093/bioinformatics/bti191
134. FelsensteinJ (1989) PHYLIP – Phylogeny Inference Package (Version 3.2). Cladistics 5 : 164–166 doi:10.1111/j.1096-0031.1989.tb00562.x
135. LetunicI, BorkP (2007) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23 : 127–128 doi:10.1093/bioinformatics/btl529
136. Schaad NW, Jones JB, Chun W (2001) Laboratory Guide for Identification of Plant Pathogenic Bacteria. St Paul, USA: American Phytopathological Society Press. 373 p.
137. Hyman LJ, Toth IK, Pérombelon MCM (2002) Isolation and identification. In: Pérombelon MCM, van der Wolf JM, editors. Methods for the detection and quantification of Erwinia carotovora subsp. atroseptica (Pectobacterium carotovorum subsp. atrosepticum) on potatoes: a laboratory manual. Scotland, UK: Scottish Crop Research Institute. pp. 66–77.
138. ToronenP (2004) Selection of informative clusters from hierarchical cluster tree with gene classes. BMC Bioinformatics 5 : 32 doi:10.1186/1471-2105-5-32
139. DarlingAE, MauB, PernaNT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5: e11147 doi:10.1371/journal.pone.0011147
140. LiL, StoeckertCJJr, RoosDS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13 : 2178–2189 doi:10.1101/gr.1224503
141. EngelsR, YuT, BurgeC, MesirovJP, DeCaprioD, et al. (2006) Combo: a whole genome comparative browser. Bioinformatics 22 : 1782–1783 doi:10.1093/bioinformatics/btl193
142. AltschulSF, MaddenTL, SchäfferAA, ZhangJ, ZhangZ, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 : 3389–3402.
143. AltschulSF, WoottonJC, GertzEM, AgarwalaR, MorgulisA, et al. (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J 272 : 5101–5109 doi:10.1111/j.1742-4658.2005.04945.x
144. HaiderS, BallesterB, SmedleyD, ZhangJ, RiceP, et al. (2009) BioMart Central Portal–unified access to biological data. Nucleic Acids Res 37: W23–27 doi:10.1093/nar/gkp265
145. DatsenkoKA, WannerBL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 : 6640–6645 doi:10.1073/pnas.120163297
146. HoangTT, Karkhoff-SchweizerRR, KutchmaAJ, SchweizerHP (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212 : 77–86.
147. HuynhTV, DahlbeckD, StaskawiczBJ (1989) Bacterial blight of soybean: regulation of a pathogen gene determining host cultivar specificity. Science 245 : 1374–1377.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- A Wolf in Sheep's Clothing: SV40 Co-opts Host Genome Maintenance Proteins to Replicate Viral DNA
- Intracellular Vesicle Acidification Promotes Maturation of Infectious Poliovirus Particles
- Whole Genome Sequencing Reveals Local Transmission Patterns of in Sympatric Cattle and Badger Populations
- The Role of Auxin-Cytokinin Antagonism in Plant-Pathogen Interactions
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy