
Integrative Genomic Analyses Identify as a Novel Lineage-Specific Oncogene in Lung Squamous Cell Carcinoma
Background:
Traditionally, non-small cell lung cancer is treated as a single disease entity in terms of systemic therapy. Emerging evidence suggests the major subtypes—adenocarcinoma (AC) and squamous cell carcinoma (SqCC)—respond differently to therapy. Identification of the molecular differences between these tumor types will have a significant impact in designing novel therapies that can improve the treatment outcome.
Methods and Findings:
We used an integrative genomics approach, combing high-resolution comparative genomic hybridization and gene expression microarray profiles, to compare AC and SqCC tumors in order to uncover alterations at the DNA level, with corresponding gene transcription changes, which are selected for during development of lung cancer subtypes. Through the analysis of multiple independent cohorts of clinical tumor samples (>330), normal lung tissues and bronchial epithelial cells obtained by bronchial brushing in smokers without lung cancer, we identified the overexpression of BRF2, a gene on Chromosome 8p12, which is specific for development of SqCC of lung. Genetic activation of BRF2, which encodes a RNA polymerase III (Pol III) transcription initiation factor, was found to be associated with increased expression of small nuclear RNAs (snRNAs) that are involved in processes essential for cell growth, such as RNA splicing. Ectopic expression of BRF2 in human bronchial epithelial cells induced a transformed phenotype and demonstrates downstream oncogenic effects, whereas RNA interference (RNAi)-mediated knockdown suppressed growth and colony formation of SqCC cells overexpressing BRF2, but not AC cells. Frequent activation of BRF2 in >35% preinvasive bronchial carcinoma in situ, as well as in dysplastic lesions, provides evidence that BRF2 expression is an early event in cancer development of this cell lineage.
Conclusions:
This is the first study, to our knowledge, to show that the focal amplification of a gene in Chromosome 8p12, plays a key role in squamous cell lineage specificity of the disease. Our data suggest that genetic activation of BRF2 represents a unique mechanism of SqCC lung tumorigenesis through the increase of Pol III-mediated transcription. It can serve as a marker for lung SqCC and may provide a novel target for therapy.
: Please see later in the article for the Editors' Summary
Published in the journal:
. PLoS Med 7(7): e32767. doi:10.1371/journal.pmed.1000315
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1000315
Summary
Background:
Traditionally, non-small cell lung cancer is treated as a single disease entity in terms of systemic therapy. Emerging evidence suggests the major subtypes—adenocarcinoma (AC) and squamous cell carcinoma (SqCC)—respond differently to therapy. Identification of the molecular differences between these tumor types will have a significant impact in designing novel therapies that can improve the treatment outcome.
Methods and Findings:
We used an integrative genomics approach, combing high-resolution comparative genomic hybridization and gene expression microarray profiles, to compare AC and SqCC tumors in order to uncover alterations at the DNA level, with corresponding gene transcription changes, which are selected for during development of lung cancer subtypes. Through the analysis of multiple independent cohorts of clinical tumor samples (>330), normal lung tissues and bronchial epithelial cells obtained by bronchial brushing in smokers without lung cancer, we identified the overexpression of BRF2, a gene on Chromosome 8p12, which is specific for development of SqCC of lung. Genetic activation of BRF2, which encodes a RNA polymerase III (Pol III) transcription initiation factor, was found to be associated with increased expression of small nuclear RNAs (snRNAs) that are involved in processes essential for cell growth, such as RNA splicing. Ectopic expression of BRF2 in human bronchial epithelial cells induced a transformed phenotype and demonstrates downstream oncogenic effects, whereas RNA interference (RNAi)-mediated knockdown suppressed growth and colony formation of SqCC cells overexpressing BRF2, but not AC cells. Frequent activation of BRF2 in >35% preinvasive bronchial carcinoma in situ, as well as in dysplastic lesions, provides evidence that BRF2 expression is an early event in cancer development of this cell lineage.
Conclusions:
This is the first study, to our knowledge, to show that the focal amplification of a gene in Chromosome 8p12, plays a key role in squamous cell lineage specificity of the disease. Our data suggest that genetic activation of BRF2 represents a unique mechanism of SqCC lung tumorigenesis through the increase of Pol III-mediated transcription. It can serve as a marker for lung SqCC and may provide a novel target for therapy.
: Please see later in the article for the Editors' Summary
Introduction
Lung cancer is the most common cause of cancer deaths worldwide [1],[2]. It is projected that by 2020, lung cancer will be the fifth most lethal entity among all diseases [3]. Improvement in survival has been very modest. Less than 16% of lung cancer patients survive 5 y or more [2], owing to late diagnosis and a paucity of effective therapies.
Squamous cell carcinoma (SqCC) and adenocarcinoma (AC) are the predominant non-small cell lung cancer (NSCLC) cell types [4]. Currently, they are regarded as a single disease entity in terms of systemic therapy. There is increasing evidence that AC and SqCC respond differently to therapy [5],[6]. The differences in therapeutic response may be related to the specific cell lineages from which they develop. Biological differences that segregate with lineage may also lead to differences in progression and response to therapies [7]. Specific genes and their respective pathways may lead to carcinogenesis only when disrupted in permissive conditions [8]. For example, a gene may have oncogenic properties when overexpressed in basal cells in the central airway compartment because it supports growth under these conditions; however, the same gene may have no effect on Clara cells in the lung periphery. Recent studies using transgeneic mouse models support this theory. For example, in murine models with KRAS mutations [9]–[12], although all airway epithelial cells contained this mutation, only adenomatous hyperplastic lesions—precursors to AC—developed in the peripheral lung in these mice, suggesting that only particular gene alterations in specific cell types in a certain local environment or niche can lead to the development of the individual lung cancer subtypes.
Cell lineage may also have a dramatic effect on the manifestation of genetic alterations during the development of lung cancer subtypes, as only those promoting a specific malignant phenotype will be selected and maintained [7],[13]. DNA amplification and subsequent overexpression is a predominant mechanism of oncogene activation in epithelial cancers, including those of lung origin [14],[15]. The subsistence of a DNA amplicon is thought to result from selection of genes within the amplified region that promote tumor growth [14]. Thus, the specific requirements for tumorigenesis in different cell lineages may therefore be associated with selection of different amplicons. Copy number increase of 8p12-8p11.21 is one of the most frequent focal changes in NSCLC occurring in ∼9%–35% of cases, with amplification present in ∼3%–8% of cases in the literature, a frequency rivaling those of established NSCLC oncogenes such as MYC (∼6%) and EGFR (∼3%) [16],[17]. In this study, we sought to determine the lineage specificity of the 8p amplicon in order to discover novel oncogene(s) restricted to tumorigenesis, in particular NSCLC cell types.
Methods
Ethics Statement
All patient samples were collected under informed patient consent and anonymized as approved by the University of British Columbia - British Columbia Cancer Agency Research Ethics Board (REB number H04-60060).
Tumor Samples
Formalin-fixed, paraffin-embedded, and fresh-frozen tissues were collected from St. Paul's Hospital, Vancouver General Hospital and Princess Margaret Hospital following approval by the Research Ethics Boards. Formalin-fixed paraffin-embedded lung carcinoma in situ (CIS) samples were collected by fluorescence bronchoscopy-directed biopsies at the British Columbia Cancer Agency. Hematoxylin and eosin-stained sections of each sample were examined by a lung pathologist to select regions of interest for microdissection to ensure >70% tumor cell content. DNA was isolated using standard procedure with proteinase K digestion followed by phenol-chloroform extraction as previously described [18].
Tiling-Path Array Comparative Genomic Hybridization
Array hybridization was performed as previously described [19]–[21]. Briefly, equal amounts (200–400 ng) of sample and single male reference genomic DNA were differentially labeled and hybridized to SMRT array v.2 (BCCRC Array Laboratory, Vancouver, BC) previously described to give optimal genome coverage [22],[23].
Hybridized arrays were imaged using a charge-coupled device (CCD) camera system and analyzed using SoftWoRx Tracker Spot Analysis software (Applied Precision). Systematic biases (intensity, spatial, plate, and background biases) were removed from all array data files using a stepwise normalization procedure as previously described [15],[24]. SeeGH software was used to combine replicates and visualize all data as log2 ratio plots [25],[26]. All replicate spots with a standard deviation above 0.075 or signal-to-noise ratios below three were removed from further analysis. The clones were then positioned on the basis of the human March 2006 (hg18) genome assembly. Genomic imbalances (gains and losses) within each sample were identified using aCGH-Smooth [27] with lambda and breakpoint per chromosome settings at 6.75 and 100, respectively, as previously described [21]. The resulting frequency of alteration was then determined for each lung cancer cell type as described previously [21]. High-level amplifications were determined using an algorithm previously described with the log2 threshold set at >0.6 for tumors and >0.8 for CIS cases (because of different levels of cell heterogeneity) [15]. Regions were only scored as amplified if two or more consecutive array elements met these criteria. All array comparative genomic hybridization (CGH) data are available on the System for Integrative Genomic Microarray Analysis (SIGMA) Web site at http://sigma.bccrc.ca/.
Comparison of Cell Type Alteration Frequencies
Regions of differential copy number alteration between AC and SqCC genomes were identified as follows. Each array element was scored as 1, gain/amplification; 0, neutral/retention; or −1, loss/deleted, for each individual sample. Values for elements filtered on the basis of quality control criteria were inferred by using neighbouring clones within 10 Mb. Probes were then aggregated into genomic regions if the similarity in copy number status between adjacent clones was at least 90% across all samples from the same cell type. The occurrence of copy number gain/amplification, loss/deletion, and retention at each locus was then compared between AC and SqCC datasets using the Fisher exact test. Testing was performed using the R statistical computing environment on a 3×2 contingency table as previously described, generating a p-value for each clone [21]. A Benjamini-Hochberg multiple hypothesis testing correction based on the number of distinct regions was applied and resulting p-values ≤0.01 were considered significant. Adjacent regions within 1 Mb that matched both the direction of copy number difference and statistical significance were then merged. Finally, regions had to be altered in >20% of samples in a group and the difference between groups >10% to be considered.
Gene Expression Microarray Analysis of Clinical Tumor Specimens
47 fresh-frozen lung tumors were obtained from Vancouver General Hospital as described above. Microdissection of tumor cells was performed and total RNA was isolated using RNeasy Mini kits (QIAGEN Inc.). Samples along with universal reference RNA were labeled and hybridized to a custom Agilent Whole Genome Oligonucleotide microarray according to the manufacture's protocols. The resulting expression data were processed and normalized using Rosetta Resolver software. Affymetrix U133 Plus 2 expression data for 111 NSCLC tumors were downloaded from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, accession number GSE3141) and normalized using Microarray Suite (MAS) 5.0 [28]. A summary containing the number of samples analyzed and corresponding platform is presented in Table S1.
Gene Expression Microarray Analysis of Normal Bronchial Epithelial Cells
RNA was obtained from exfoliated bronchial cells of 67 lung cancer–free individuals obtained during fluorescence bronchoscopy [29]. All individuals were either current or former smokers without lung cancer. Expression profiles were generated for all cases using the Affymetrix U133 Plus 2 platform and normalized using MAS 5.0.
Statistical Analysis of Gene Expression Data
Gene expression probes were mapped to March 2006 (hg18) genomic coordinates and those within the regions of copy number difference between the cell types on Chromosome arm 8p were determined. Comparisons between expression levels for AC and SqCC tumors as well as SqCC tumors and normal bronchial cells were performed using the Mann-Whitney U test and computed with the ranksum function in MATLAB. As the direction of gene expression difference was predicted to match the direction of copy number difference, one-tailed p-values were calculated. A Benjamini-Hochberg multiple hypothesis testing correction was applied on the basis of the total number of gene expression probes analyzed. Probes with a corrected p-value ≤0.01 were considered significant. If multiple probes mapped to the same gene, the one with the lowest p-value (Agilent data) or with maximum intensity across the data (Affymetrix) was used.
Integration of Genetic and Gene Expression Data
To integrate gene expression with copy number data, two methods were used. First, a 10-kb moving average was generated using the normalized log2 array CGH ratios for each sample with copy number and expression. These values were subsequently standardized using a Z-transformation for each sample throughout the whole genome in order to facilitate better comparisons across the sample set. An average Z-score was then calculated using the values corresponding to the genomic intervals spanning each of the genes of interest on Chromosome 8p. Finally, a nonparametric Spearman correlation coefficient was calculated using the Z-scores for copy number and log10 ratios for gene expression across all samples of interest. The corresponding p-value representing the statistical significance of a positive correlation was calculated and a Benjamini-Hochberg multiple hypothesis testing correction applied as described above. For the second method, the copy number status was determined by aCGH-Smooth as described above and mapped to genes of interest from clones using genomic coordinates from the UCSC Genome Browser (hg18). The gene expression levels for all genes were then compared between samples with copy number gain/amplification against samples that were copy number neutral using the Mann-Whitney U test [15]. An association was deemed significant if the Benjamini-Hochberg corrected p-value ≤0.05 and the median and mean gene expression in the samples with gain/amplification were higher than those samples that were copy number neutral. Again, as the direction of gene expression difference was predicted to match the direction of copy number difference, one-tailed p-values were calculated.
Reverse Transcriptase PCR Analysis of Transcription Levels in Clinical Tumor Samples
Quantitative reverse transcriptase (RT)-PCR was performed on SDS7900HT (Applied Biosystems) using SYBR Green and the ΔΔCt method with RPS13 expression levels used as reference for normalization. Primers used were: BRF2_F: GTGAAGCTCCTGGGACTGGAT, BRF2_R: GTATTTGGCTGGCACAGAAGG, RPS13F: GTTGCTGTTCGAAAGCATCTTG, and RPS13R: AATATCGAGCCAAACGGTGAA. Associations between BRF2 expression and clinicopathological features were evaluated by the Wilcoxon test. Breakdown of samples used are provided in Table S1.
Cell Lines and Culture Conditions
NSCLC cell lines H520, H1395, and H2347 were purchased from American Type Culture Collection (ATCC). Cells were maintained in RPMI-1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen). The HBEC3-KT immortalized normal human bronchial epithelial cell (HBEC) line was established by introducing mouse Cdk4 and hTERT into normal HBECs obtained from a 65-y-old woman without cancer [30]. The HBEC3-KT53 line was established by stably knocking down p53 in the original cell line, HBEC3-KT [31]. These two parental lines were used to overexpress BRF2 via the pMSCV vector (see below). All HBEC3 cell lines were cultured in K-SFM (Invitrogen) medium containing 50 µg/µl bovine pituitary extract (Invitrogen) and 5 ng/µl EGF (Invitrogen).
TaqMan Analysis of Transcript Levels in Cancer Cell Lines
5 µg micrograms of total RNA isolated from cultured cells (H2122, H2347, HCC193, H1395, H2009, H1993, HCC4006, HCC2279, H2087, HCC78, HCC461, HCC1195, H1819, H1648, HCC366, H3255, HCC2450, HCC15, HCC95, and H520) was converted to cDNA using an ABI High Capacity cDNA Archive kit (Applied Biosystems). An aliquot of 100 ng of cDNA was used for each real-time PCR reaction. TaqMan (Applied Biosystems) gene expression assays: BRF2 (Hs00217757_m1) and 18s rRNA (Hs99999901_s1) were performed using standard TaqMan reagents and protocols on a Applied Biosystems 7500 Fast Real-Time PCR system (Applied Biosystems). The ΔΔCt method was used for expression quantification using the average cycle threshold for 18S rRNA to normalize gene expression levels between samples [21]. Cycle thresholds for the primers were then compared between the individual cell lines and a pooled normal lung cDNA reference sample generated from Human Lung Total RNA (AM7968, Ambion) to identify the fold change represented.
Western Blot Analysis of Protein Levels
Cells were washed twice with cold PBS and lysed in the presence of protease inhibitors. Each cleared lysate was diluted and boiled for electrophoresis and transferred to polyvinylidene membrane [31]. Membranes preblocked with 3% bovine serum albumin in PBS with 0.05% Tween-20 (PBST) were incubated with primary antibodies against BRF2 (Abcam, 1∶500 dilution) for 1 h at room temperature. After three washes in PBST, the membranes were incubated with horseradish-peroxidase-conjugated donkey anti-goat polyclonal antibody (Abcam, 1∶2,000 dilution) for 45 min at room temperature. After three PBST washes, antibody binding was visualized by enhanced chemiluminescence (GE Healthcare). Subsequently, the bound antibodies were stripped from the membranes with a buffer containing 62.5 mM Tris-HCl, (pH 6.7), 2% SDS and β-mercaptoethanol and reprobed with monoclonal antibody to beta-actin (Abcam, 1∶6,000) to confirm equal sample loading.
Northern Blot Analysis of Pol III Transcript Levels
Briefly, total RNA was isolated from cell lines using TRIzol reagent (Invitrogen) and 4 µg was resolved on an 8% UreaGel denaturing gradient gel as per manufacturer's instructions (National Diagnostics). The gel was then equilibrated in 0.5× TBE for 15 min and RNA was capillary transferred to positively charged nylon membrane using 0.5× TBE according to the protocol described by Sambrook and Russell [32]. RNA was bound to the membrane by UV-crosslinking with 120 J UV light using a Stratalinker (Stratagene). The blot was then prehybridized at 45°C for 30 min using DIG Easy hybridization buffer (Roche), and 50 pmol of DIG labeled30-mer oligonucleotide probe was then added. Specific probe sequences were as follows: 5S rRNA, 5′ - CCTGCTTAGCTTCCGAGATCAGACGAGATC-3′; U6 small nuclear RNA (snRNA), 5′ - CTTGCGCAGGGGCCATGCTAATCTTCTCTG-3′; 7SK snRNA, 5′-CGTCCTCTTCGACCGAGCGCGCAGCTTCGG-3′. Hybridization proceeded for 16–20 h and blots were washed two times for 15 min at room temperature in 2× SSC, 1% SDS followed by a 15-min wash in the same buffer at 45°C. Further washes and luminescence detection was performed using the DIG Wash and Block Buffer set and the DIG Luminescent Detection kit following the manufacturer's instructions (Roche). Blots were then exposed to Lumi-Film (Roche) and developed to image.
RNAi Knockdown
For RNA interference (RNAi) experiments, two methods were employed. For the first method, lentiviral short hairpin RNAs (shRNA) vectors targeted against BRF2 were purchased from Open Biosystems. Briefly, individual lentiviruses, each containing a single pLKO plasmid construct coding an shRNA targeted for BRF2, were prepared by transfecting 293T cells with the packaging plasmids VSVG and d8.91 and the shRNA plasmids using TransIT-LT1 transfection reagent (Mirus). Virus containing empty pLKO vector served as a control. Virus supernatant was collected from the transfected 293T cells each day for 3 consecutive days post-transfection. H520 cells were infected at 50% confluency, using 1 ml of each respective virus. After 48 h, cells were selected with 2.5 µg/ml puromycin. Selection was continued until all nontransfected cells were dead. Stably transfected cell lines were maintained in growth media supplemented with 2.5 µg/ml of puromycin. Sequence details for BRF2-1 (TRCN0000016128) and BRF2-2 (TRCN0000016129) can be found on the Open Biosystems Web site (www.openbiosystems.com).
For the second method, ON-TARGETplus SMARTpool small interfering RNAs (siRNAs) targeting BRF2 along with a negative control (ON-TARGETplus siCONTROL nontargeting siRNA pool) were obtained from Dharmacon. H520 and H1395 cells were subcultured at a ratio of 1∶3 or 1∶6 using 0.25% tryspin-EDTA (Gibco). Transfection efficiency was optimized using siGLO Green Transfection Indicator (Dharmacon). For the transfection experiments, cells were seeded in 24-well culture plates at 60,000 cells/ml 24 h before transfection. Cells were transfected at a final concentration of 100 nM siRNA using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. The cells were then incubated at 37°C for 24 h before RNA analysis, 48 h for protein, and 72 h for MTT assays. For both shRNA and siRNA experiments, BRF2 expression levels for multiple independent knockdowns were determined by qRT-PCR as described above and scaled relative to the average of the control treated cells (±standard error measure [SEM]).
Cell Proliferation Assays
The 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT) assay (Trevigen) was used to determine the status of cell proliferation in both shRNA and siRNA experiments according to the manufacturer's instructions. For siRNA experiments, exponentially growing cells were diluted to a concentration of 313,000 cells/ml in RPMI-1640 with 10% FBS, seeded in triplicate in 96-well plates and incubated at 37°C for 4 h. The cells were then treated with 10 µl of MTT reagent for 4 h before adding 100 µl of detergent reagent to solubilize the formazan precipitate. The reaction product was then quantified by measuring absorbance at 570 nm with reference to 650 nm using an EMax plate reader (Molecular Devices). The mean ±SEM absorbance values for experiments from three independent transfections were normalized to the average of the respective controls.
For shRNA experiments, 4,000 cells for each condition were seeded in triplicate for each time point in 96-well plates and treated with MTT as described above at 24, 72, and 96 h. The absorbance reading for blank (media) wells was subtracted from the mean absorbance readings for each time point and plotted (±SEM) to quantify cell proliferation. Replicate experiments were performed and a representative experiment is shown.
Soft Agar Anchorage-Independent Growth Assay
The H520 stably transfected pLKO vector control and shRNA BRF2 gene knockdown cell lines were used in the colony formation assay. Single cell suspensions were prepared in growth media supplemented with 20% fetal bovine serum (Invitrogen), 2.5 µg/ml puromycin, 0.1% penicillin-streptomycin (Invitrogen), and 0.3% low-melting point agarose (Invitrogen), resulting in a final concentration of 1,000 cells/ml. 1 ml of cell suspension (1,000 cells) was plated onto an equal volume of supplemented media with a 0.5% low-melting point agarose concentration. Supplemented media lacking cells was plated as a negative control. Each cell line was seeded in triplicate in 12-well plates and cultured for 14 d at 37°C, after which colonies were counted and the mean ±SEM were normalized to the average of the control. Replicate experiments were performed and a representative experiment is shown.
Construction of the BRF2 Expression Vector
The BRF2 sequence from the pBRF2-HORF construct (Invitrogen) was cloned into the retroviral vector pMSCV-hygro (Clontech) and sequenced. This construct was named pMSCV-BRF2. The pMSCV-BRF2 construct and the vector alone (pMSCV) were then transfected into the Pheonix Ampho retroviral packaging cell line (Orbigen) according to manufacturer's protocols. Subsequent infections into HBEC3-KT and HBEC3-KT53 were performed and plasmid-containing cells were selected by treating with 20 µg/µl of hygromycin for 10 d. This step resulted in the generation of four stable cell lines: HBEC3-KT-pMSCV-BRF2, HBEC3-KT-pMSCV (vector control), HBEC3-KT53-pMSCV-BRF2, and HBEC3-KT53-pMSCV (vector control).
In Vitro Cell Growth Assays
Growth curves were determined for each of the six HBEC cell lines by culturing 1,000 cells in triplicate in 12-well plates and counting on the third, sixth, eighth, and tenth day. The average ±SEM for each line is reported. Replicate experiments were performed and a representative experiment is shown.
Statistical Analysis of Functional Assays
For all cell model assays, p-values were calculated using the Student's t test when comparing two conditions and ANOVA when comparing three. All calculations were performed with MATLAB software and one-tailed p-value ≤0.05 was considered significant.
Immunohistochemistry
Slides were deparaffinized using xylene and rehydrated through an ethanol series to water. Antigen retrieval was performed using a decloaking chamber at 15 psi for 20 min in sodium citrate buffer (pH 6.0). Endogenous peroxidase enzyme activity was blocked using 3% H2O2 in methanol for 30 min at room temperature. Slides were washed in 1% PBS and then blocked using 10% skim milk for 6 h at room temperature. Slides were incubated for 16 h at 4°C with a 1∶200 dilution of goat polyclonal anti-BRF2 primary antibody (Abcam), followed by incubation with a donkey anti-goat biotinylated secondary antibody (Santa Cruz Biotechnology). Normal goat IgG was used as negative control (Santa Cruz Biotechnology). Detection was accomplished using DAB (ImmunoCruz staining system, Santa Cruz Biotechnology). Slides were then counterstained using hematoxylin, and the area within the diagnostic area was scored by three independent observers on the basis of the following criteria: 0, no positive staining; 1, 25% positive cells; 2, 50% positive cells; 3, 75% positive cells; and 4, 100% positive cells. Conflicting scores were resolved by choosing the value consistent between two observers or the average of all three varying scores.
Significance Analysis of Microarrays
Using the 111 NSCLC samples in the dataset by Bild et al. (Table S1) [28], samples were sorted by highest to lowest expression for BRF2 on the basis of the probe with the highest average intensity across the dataset [28]. Differential gene expression analysis using significance analysis of microarrays (SAM) [33] was performed using the ten samples with highest BRF2 expression against the ten samples with lowest BRF2 expression. A q-value threshold of ≤0.05 was used to identify differentially expressed genes associated with high BRF2 expression.
Functional Assessment of BRF2-Associated Genes
Functional analyses were generated through the use of Ingenuity Pathways Analysis (Ingenuity Systems) as previously described [15]. Functional Analysis identified the biological functions that were most significant to the dataset. Fisher exact test was used to calculate a p-value determining the probability that each biological function assigned to the dataset is due to chance alone.
Results
A Focal Region on 8p Is Preferentially Amplified in SqCC
We compared the 8p chromosome arm of 161 microdissected NSCLC tumors—103 AC and 58 SqCC (Table S1, sample set 1a)—by tiling resolution array CGH [22]. After hybridization experiments, genomic profiles were normalized and subjected to a smoothing algorithm in order to computationally define regions of copy number gain and loss along the entire length of Chromosome arm 8p [21]. Individual samples were then grouped by their corresponding cell type, and probes were aggregated into regions on the basis of similar copy number status. The resulting frequency of alteration for each region along the arm was compared between cell types using the Fisher exact test to identify regions of copy number disparity, and the resulting p-values were corrected for multiple comparisons with a cut-off of ≤0.01 considered significant (Methods). Although the telomeric portion of 8p was frequently lost in both AC and SqCC cell types, two regions spanning a total of 5.65 Mbp at 8p12–8p11.21 were found to be frequently gained specifically in SqCC (Figure 1a and 1b; Table S2). Copy number increase of focal regions at 8p12–p11.21 was found in up to 40% of SqCC tumors, while DNA loss was the most prevalent event in AC (∼39%). In addition, high-level amplification (log2 ratio >0.6) was present in ∼12% of SqCC samples (seven out of 58) demonstrating the preferential selection for this alteration in tumors of this cell lineage. The increased incidence of 8p amplification in comparison to previous reports is attributed to analyzing the cell types as distinct groups, as opposed to combining all the NSCLC cell types as a single entity. In addition, the small sample sizes of previous studies may also have limited the detection of specific disruptions unique to each cell type to just extremely high frequency events, such as the gain of Chromosome 3q [17],[34],[35]. These results indicate that gain/amplification of 8p12–8p11.21 is restricted to SqCC and occurs far more frequently than previously thought, highlighting the importance of considering cell lineage in genomic studies of malignancies from the same tissue site.

BRF2 Gene Expression Drives Selection of the 8p Amplicon in Lung SqCC
The cell type dependent pattern of 8p amplification raised the possibility that a lineage-specific oncogene may be driving the preferential selection of this amplicon in SqCC. Such a gene should display five fundamental properties each translating into its own testable hypothesis. First, increased expression would be restricted to SqCC tumors mirroring the specificity of DNA amplification (hypothesis 1). Second, as the target of the amplicon, expression would be higher in SqCC tumors with gain/amplification than those without (hypothesis 2). Third, expression should be significantly higher in SqCC tumors than normal bronchial epithelial cells; that is, the gene should be activated in cancerous and not normal tissue (hypothesis 3). Fourth, the gene should have oncogenic potential and provide a growth and/or survival advantage to cells when overexpressed (hypothesis 4). Lastly, if necessary for initiating tumorigenesis, amplification should occur early in tumor development and therefore be present in lung SqCC precursor lesions (hypothesis 5).
To test the first hypothesis, we generated gene expression microarray profiles for a subset of 47 tumors (34 AC, 13 SqCC) with sufficient amounts of material that were also analyzed by array CGH in order to integrate genetic and gene expression information (Table S1, sample set 1b). In total, 62 probes corresponding to 44 unique genes mapped to within the alteration boundaries (Table S2). To identify lineage-restricted genes, we compared the expression levels for all probes between the AC and SqCC samples. Since we predicted candidate genes to be overexpressed in SqCC, a one-tailed Mann-Whitney U test was used with Benjamini-Hochberg corrected p-values ≤0.01 considered significant. Ten unique genes meeting these criteria were uncovered from this analysis that showed a clear distinction in expression levels between the AC and SqCC tumors (Figure 1c; Table S3).
After identifying these SqCC specific genes, we next aimed to ensure that amplification is responsible for their differential expression, as these will be candidate targets driving amplicon selection (hypothesis 2). For this purpose, we utilized two complementary approaches. First, a nonparametric Spearman correlation coefficient was calculated for each gene using Z-transformed copy number ratios and log10 gene expression ratios (Methods). Five of the ten genes (LSM1, BRF2, ASH2L, TM2D2, and WHSC1L1) had a correlation coefficient of >0.75 and a corrected p-value (representing the statistical significance of a positive correlation) of <0.01 and were further considered as candidates (Table S3 for all values). The second approach involved the comparison of expression levels between SqCC tumors with gene dosage increase (gain/amplification) and those with neutral copy number status (Methods). Of the five genes with a positive association between copy number and expression, only three (LSM1, BRF2, and ASH2L) also showed significantly elevated transcript levels specifically in SqCC samples with gain or amplification and were therefore determined to be regulated by copy number (Table S3). qRT-PCR analysis of BRF2 (the most likely target gene, see below) confirmed the microarray results (Figure S1; Table S4). Importantly, none of these genes demonstrated a correlation between copy number and expression in AC, reinforcing the specificity of this alteration to SqCC.
In addition to demonstrating a linkage between expression and amplification, a candidate oncogene should only be expressed at elevated levels in cancerous, and not normal, tissues [36]. Therefore, to test the third hypothesis, we analyzed the RNA levels of these three genes in an independent panel of 53 SqCC lung tumors and 67 samples of exfoliated bronchial cells from cancer-free individuals generated using the Affymetrix U133 Plus 2 platform (Table S1, sample sets 2 and 3). Strikingly, only BRF2 was aberrantly expressed (>2-fold, p<1.0×10−8) in cancerous tissues identifying it as the sole gene passing the three main criteria of a candidate lineage-specific oncogene described above (Figure 2; Table S3). To further confirm these observations, a third, independent sample set consisting of 118 NSCLC tumors and 39 non-neoplastic lung tissues (Table S1, sample set 4) was analyzed for BRF2 expression by qRT-PCR (Methods). Consistent with the microarray results, expression of BRF2 in primary tumors was significantly higher than that in the non-neoplasia tissues (p<0.001) with overexpression more common in SqCC than AC (p = 0.03), supporting our findings.

Taken together, results from testing the first three hypotheses clearly demonstrate that BRF2 is the driver gene of the 8p amplicon and identify it as a candidate lineage-specific oncogene in SqCC. Previous studies investigating this amplified region in NSCLC have proposed FGFR1 and WHSC1L1 as potential oncogenes [17],[37]. However, we ruled out FGFR1 as a possible target as it was not differentially expressed between AC and SqCC, and as such, was excluded from further analysis. This conclusion is in agreement with a study by Tonon et al. that suggested WHSC1L1 as the more likely amplification target in NSCLC [17]. Although we demonstrated that WHSC1L1 expression was restricted to SqCC and correlated with increased gene dosage, it was not significantly higher in samples with gain/amplification or different between normal and cancerous cells (p = 0.12, fold change = 1.3), and therefore, also discounted.
BRF2 Contributes to SqCC Tumorigenesis by Regulating Cell Growth and Proliferation through the Increase of Polymerase III Activity
BRF2 encodes a subunit of a transcription initiation complex responsible for RNA polymerase III (Pol III)-mediated transcription [38],[39]. Pol III transcribes a limited set of genes that encode nontranslated RNAs including 5S rRNA, tRNA, 7SL RNA, and U6 RNA, which are essential for protein synthesis and RNA processing [40]. Because these processes are fundamental determinates of the capacity of a cell to grow, increased activity of Pol III is often observed during cancer development [41]. Indeed, transformed cells express elevated levels of Pol III transcripts, and inhibition of these transcripts limits cell growth and proliferation [42]. It has been proposed that deregulation of Pol III in transformed cells can occur through three different mechanisms: release from cellular repressors, direct activation by oncogenes, and overexpression of transcription factors [40]. In normal cells where growth is tightly controlled, tumor suppressors including RB, p53, and PTEN repress Pol III transcription [43],[44]. Inactivation of these genes or activation of oncogenes such as MYC and ERK reverse this process [42],[44],[45]. Interestingly, the majority of these genes are mutated in lung cancer, representing a potential mechanism of increasing Pol III activity, and subsequently, cell growth potential during tumorigenesis. Transcription factors, however, are often the limiting components of Pol III-mediated transcription and elevated levels of these components have been observed in numerous cancer types [41]. Recently, the overexpression of another Pol III transcription factor BRF1 has been shown to increase Pol III-mediated transcription, resulting in the transformation of cells in vitro and tumor formation in vivo [46],[47]. A study by Marshall et al. was the first to implicate Pol III deregulation as a causative factor in cancer formation [46]; however, no studies have been reported to date of activating mutations in Pol III subunits or associated transcription factors in tumors. Therefore, we hypothesized that the amplification and overexpression of BRF2 may contribute to lung SqCC tumorigenesis by contributing to increased cell growth and proliferation, representing a novel alternative mechanism of increasing Pol III transcription in cancer.
To test this hypothesis (hypothesis 4), we performed complementary loss and gain of function in vitro experiments using lung cancer cell lines and immortalized HBEC lines, respectively. Twenty NSCLC cell lines (16 AC and 4 SqCC) previously analyzed by array CGH were assayed for BRF2 expression by qRT-PCR (Methods). Mirroring the findings from the clinical tumor specimens, BRF2 expression was strongly correlated with gene dosage with the two cell lines with amplification (HCC95 and H520) displaying the highest transcript levels (Figure S2; Table S5). In addition, both these lines were derived from SqCC samples and no AC cell lines contained amplification, re-enforcing the lineage specificity of BRF2 activation. To determine the effect of BRF2 overexpression on BRF2 protein levels, three cell lines were selected for Western blot analysis: a SqCC with amplification (H520), an AC with neutral copy number (H1395), and an AC with loss (H2347) (Figure 3a). Consistent with a role in tumorigenesis, high protein levels were only found in H520.

To determine if increased BRF2 levels lead to higher Pol III activity, we performed Northern blot analysis to assess the expression of Pol III-mediated transcripts. BRF2 is specifically involved in transcription from type 3 (gene external) Pol III promoters, which are responsible for the expression of snRNA genes, two of the best characterized of which are U6 and 7SK [48]–[50]. This finding is in contrast to type 1 and type 2 Pol III promoters (gene internal), which require BRF1 for transcription and are involved in the expression of 5S rRNA and tRNA genes, respectively [48],[50]. Thus, we hypothesized that BRF2 activation would lead to an increase only in type 3 transcripts and not those regulated by BRF1. As expected, assessment of transcript levels in the lung cancer lines showed drastically higher levels of both U6 and 7SK relative to 5S loading control in H520 cells compared to H1395 and H2347 cells, confirming increased BRF2-dependent Pol III transcription upon BRF2 activation (Figure 3a). Furthermore, U6 levels were decreased upon knockdown of BRF2 expression in H520 cells using an shRNA construct (Figure S3). These results match those of a recent study, which showed that BRF2 protein levels correlate with U6 promoter activity [51]. These data suggest that increased BRF2 levels are sufficient to increase Pol III activity, demonstrating the downstream mechanistic effect of gene amplification.
To assess the functional significance of BRF2 amplification and overexpression on SqCC development, RNAi-mediated knockdown was performed in H520 cells. Expression of two different shRNAs) targeting BRF2 substantially reduced transcript levels (Figure 3b) and significantly decreased cell proliferation compared to a negative vector control (Figure 3c). In addition, knockdown of BRF2 expression significantly reduced the ability of these cells to grow in an anchorage-independent manner as measured by colony formation in soft agar (Figure 3d). Similar results on cell proliferation were observed with BRF2 siRNA pool transfection of H520 cells (Figure 3e). In contrast, siRNA knockdown of an AC cell line without BRF2 amplification and overexpression, H1395, did not diminish cell proliferation and resulted in an increase in proliferation relative to transfection with a nontargeting control siRNA pool (Figure 3e). How knockdown of BRF2 could lead to an increase in cell proliferation in this context remains unclear and warrants further investigation. Lastly, to further confirm the specificity of this effect to cell lines with amplification, we also performed knockdown experiments in two SqCC cell lines (HCC15 and HCC2450) without BRF2 amplification (Figure S4). As expected, no significant decrease in proliferation was seen in HCC15 or HCC2450 upon BRF2 inhibition. These results demonstrate a crucial role for BRF2 in contributing to the sustained cellular proliferation and survival of SqCC tumors with gene activation and highlight its cell type specific oncogenic potential in lung cancer.
To further validate its tumorigenic properties, we performed complimentary experiments by overexpressing BRF2 by stable transduction of immortalized HBEC lines (Figure S5) and measured cell growth compared to vector-expressing controls. HBEC lines are immortalized without the use of viral oncoproteins, have minimal genetic changes, and do not exhibit a transformed phenotype [30],[31]. In addition, since they express epithelial markers and morphology and can differentiate into mature airway cells, they represent an attractive model for testing the importance of specific gene alteration found in the initiation of epithelium-derived lung cancer [30],[31]. Strikingly, the introduction of BRF2 alone resulted in a modest but significant increase in cellular growth and saturation density, further supporting a tumorigenic role for this gene (Figure 3f). Furthermore, as p53 is inactivated in ∼50% of NSCLC tumors, and is known to repress Pol III-mediated transcription, we sought to investigate the impact of BRF2 overexpression in conjunction with p53 silencing on HBEC growth. Interestingly, the combination of these two alterations enhanced cell growth greater than each alteration alone (p = 2.36×10−5), suggesting a synergistic role for these alterations in promoting proliferation (Figure 3g). Taken together, our results demonstrate that BRF2 overexpression plays a key role in regulating cell growth and proliferation, confirming the functional significance of BRF2 gene amplification in SqCC.
BRF2 Activation Is an Early Event in SqCC Development
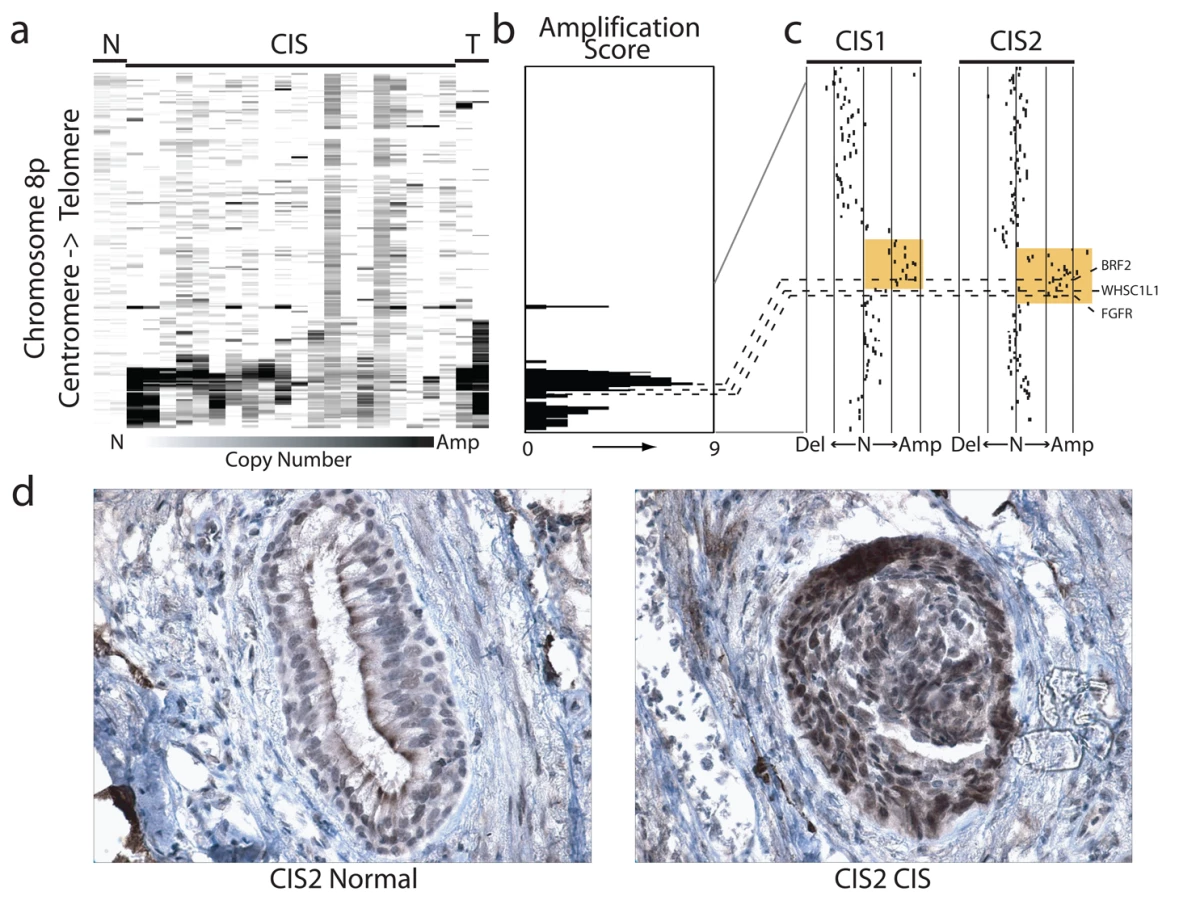
The cell type restricted pattern of activation coupled with its transformation potential strongly implicates BRF2 as a lineage-specific oncogene in lung SqCC. SqCC carcinogenesis is thought to be a multistep process that involves the transformation of normal mucosa though a continuous range of precursor lesions up to CIS before invasive cancer and finally metastasis [52]. However, since most studies focus on clinically evident tumors, little is known about the molecular events preceding the development of lung cancer and the underlying basis of carcinogenesis. Unlike low grade dysplastic lung lesions that rarely progress, the majority of CIS cases will become invasive cancer [52]. Therefore, we hypothesized that critical alterations necessary for disease progression would be evident in preinvasive CIS lesions and persist in invasive tumors. To determine if BRF2 activation occurs early in SqCC development (hypothesis 5), we analyzed gene dosage in a panel of 20 CIS lesions (Table S1, sample set 5) obtained by autofluorescence bronchoscopy (Methods). Remarkably, array CGH revealed BRF2 copy number increases in the majority of CIS cases (Figure 4a) with 35% (seven out of 20) demonstrating high-level amplification (log2 ratio >0.8; Figure 4b). WHSC1L1 and FGFR1 were only amplified five times (five out of 20) and once (one out of 20), respectively, further excluding these genes as primary driver genes of the amplicon (Figure 4b and 4c). To confirm that amplification results in increased expression of BRF2 in preinvasive lesions, we performed immunohistochemistry (IHC) on a CIS sample (CIS2) with amplification (Figure 4c). As expected, BRF2 expression was elevated in CIS epithelia in this sample in comparison to normal epithelia from the same patient (Figure 4d). Strong BRF2 expression was also observed in additional CIS cases with lower levels in earlier stages of neoplastic progression (mild, moderate, and severe dysplasia) and little or no staining in benign lesions (hyperplasia and metaplasia), confirming that gene activation is an early event in SqCC development (Figure 5). Interestingly, the only benign lesion in which BRF2 expression was observed was obtained from a patient that had also developed CIS (Figure 5). The high frequency of activation in preinvasive lesions suggests that BRF2 plays a critical role in the development of SqCC through the increase of cell growth potential. Since patient survival can be significantly improved if the lesions are detected and treated at their preinvasive stage, the identification of genes involved in the development of CIS and invasive SqCC is of vital clinical importance [52],[53]. Our finding that BRF2 is a lineage-specific oncogene amplified early in SqCC development, and not expressed in normal lung tissue, represents a critical step in understanding the development of SqCC, and represents a promising target for therapeutic intervention.

Increased RNA Processing Is Associated with BRF2 Overexpression
To identify other genes and functions that may be associated with BRF2-mediated initiation of tumorigenesis, we performed significance analysis of microarrays (SAM) on a panel of 111 NSCLC tumors (Table S1, sample set 2), followed by gene enrichment analysis using ingenuity pathway assist (IPA) (Methods). This analysis revealed 86 genes, which were significantly increased (78) or decreased (8) (false discovery rate <5%) in tumors with the highest BRF2 expression (Table S6). IPA analysis revealed enrichment for genes with diverse biological functions including RNA post-transcriptional modification, gene expression, cell cycle, and cancer (Table S7). The identification of RNA post-transcriptional modification as the most significantly affected function (p = 1.7×10−06−4.73×10−02, the two significance values refer to a range of specific subfunctions) was significant, as this is one of the main roles of Pol III-related transcripts as stated above. The genes related to this function, which are increased in expression, include FBL, CPSF6, RRP9, SNRPA, SFRS10, CSTF2T, LSM1, and CPSF3, and are involved in the modification, polyadenylation, and processing of both mRNA and rRNA. Since these are fundamental processes necessary for proper protein production and therefore cell growth, upregulation of these components may be associated with the increased proliferative capacity of SqCC cells upon BRF2 activation. However, the exact nature of this association is currently unknown and future studies will be needed to understand the mechanism responsible for BRF2-induced cell growth in SqCC.
Interestingly, as shown above, BRF2 activation leads to increased transcription from type 3 Pol III promoters that are involved in the transcription of snRNA genes including U6 and 7SK [49],[50]. snRNAs are responsible for a range of regulatory functions, including the alteration of gene expression and a potential role for snRNAs in the genomic instability of cancer that has been proposed [54]. In particular, U6 snRNA forms the catalytic core of the spliceosome [55]. The spliceosome performs the splicing of precursor mRNA in eukaryotic cells, removing introns and joining exons. This process is tightly regulated during growth and development and aberrant splicing has been linked to numerous human diseases, including cancer [56]. In fact, many oncogenes demonstrate alternative splicing patterns associated with neoplasia, and splicing regulatory factor expression levels have been shown to increase during cancer progression. Strikingly, many of the genes we identified as being associated with increased BRF2 expression, including SNRPA and SFRS10, are known to interact with snRNAs including U6 in the spliceosome complex. In addition, SNAPC5, which encodes a member of the snRNA-activating complex that is required in conjunction with BRF2 to initiate transcription from snRNA promoters [57], was also found to be increased in samples with high BRF2 expression. Taken together, our data suggest that BRF2-mediated increase of U6 as well as other splicing regulatory factors may contribute to oncogenesis in SqCC with 8p amplification. Future studies of the role BRF2 overexpression plays in spliceosome function will yield insight into this potential function, and its role in the neoplastic transformation of lung epithelium to SqCC.
Association of BRF2 with Clinical-Pathological and Genomic Features
Lastly, to investigate the potential clinical significance of BRF2 activation in patients with lung SqCC and better characterize this subgroup of tumors, we next sought to determine the association between 8p amplification and clinical-pathological and genetic features. For this purpose, we expanded our sample set to include 92 SqCC tumors with well-annotated clinical information that were analyzed by tiling-path array CGH. Overall, increase of BRF2 copy number was found in 43% of the expanded dataset, in concordance with the original frequency of alteration. No associations were found between age, gender, smoking status, or stage and BRF2 amplification in our dataset. Furthermore, no significant associations between the level of BRF2 expression and patient survival were seen in two independent datasets (Figure S6). However, SqCC tumors with and without BRF2 activation showed a unique genome-wide spectrum of DNA amplifications, suggesting that different genetic pathways may be involved in their development (Table S8). Assessment of other clinical and genetic features—for example response to therapy and mutation events—will be necessary in the future to further explore the characteristics of SqCC patients harboring BRF2 amplification.
Discussion
In summary, we show here that the focal amplification of Chromosome 8p12, one of the most frequent amplification events in NSCLC, plays a key role in squamous cell lineage specificity of the disease. Through the integration of genetic and gene expression data for >330 clinical tumor specimens in conjunction with functional cell model studies, we identified BRF2 as the target of this amplification and a cell lineage-specific oncogene, the only such oncogene described for lung SqCC to date. In addition, we highlight the oncogenic potential of BRF2 for the first time and associate its activation with increased Pol III activity, RNA processing, and resultant cell growth potential.
The lineage-dependence model suggests that cancer cells rely on the constitutive activation of lineage-regulating genes involved in normal development for their continued survival and proliferation [7]. BRF2 is unique in that it is not a prototypical lineage-specific oncogene as no role in normal lineage development has been established. These data suggest that lineage-specific oncogenes may span numerous biological functions, and they are not limited only to the established class of transcription factors (lineage survival oncogenes) discovered to date, but also a class of genes selected in tumorigenesis in a cell lineage-specific manner. Recently, a candidate a lineage survival oncogene for lung AC, TITF1 (thyroid transcription factor 1), has also been described by numerous groups [16],[58]–[60]. However, high-level amplification of TITF1 and concordant increase in protein levels occur at approximately the same frequency in both AC and SqCC (∼10%–15%, although protein levels are higher in AC overall) [61],[62]. Thus, BRF2 seems to be even more specific in terms of genetic alteration and its association with an individual subtype. Likewise, SOX2 has been identified as a lineage survival oncogene in lung and esophageal SqCCs [63]. Of interest, both SqCC with and without BRF2 amplification also contains amplification of Chromosome arm 3q targeting SOX2, suggesting that tumors with BRF2 amplification represent a unique subset within the larger SqCC group (Table S8). Nonetheless, our results combined with the recent discoveries of TITF1 and SOX2 suggest that the genes required to initiate tumorigenesis in distinct biological contexts may shape the preferential selection of amplifications and resulting phenotypes specific to different cancers, highlighting the opportunity for treatment design targeting specific cell type.
Supporting Information
Zdroje
1. ParkinDM
BrayF
FerlayJ
PisaniP
2005
Global cancer statistics, 2002.
CA Cancer J Clin
55
74
108
2. JemalA
SiegelR
WardE
HaoY
XuJ
2008
Cancer statistics, 2008.
CA Cancer J Clin
58
71
96
3. MurrayCJ
LopezAD
1997
Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study.
Lancet
349
1498
1504
4. TravisWD
2002
Pathology of lung cancer.
Clin Chest Med
23
65
81, viii
5. KimHS
ParkK
JunHJ
YiSY
LeeJ
2009
Comparison of survival in advanced non-small cell lung cancer patients in the pre - and post-gefitinib eras.
Oncology
76
239
246
6. ScagliottiG
HannaN
FossellaF
SugarmanK
BlatterJ
2009
The differential efficacy of pemetrexed according to NSCLC histology: a review of two Phase III studies.
Oncologist
14
253
263
7. GarrawayLA
SellersWR
2006
Lineage dependency and lineage-survival oncogenes in human cancer.
Nat Rev Cancer
6
593
602
8. GiangrecoA
GrootKR
JanesSM
2007
Lung cancer and lung stem cells: strange bedfellows?
Am J Respir Crit Care Med
175
547
553
9. FisherGH
WellenSL
KlimstraD
LenczowskiJM
TichelaarJW
2001
Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes.
Genes Dev
15
3249
3262
10. GuerraC
MijimolleN
DhawahirA
DubusP
BarradasM
2003
Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context.
Cancer Cell
4
111
120
11. MeuwissenR
LinnSC
van der ValkM
MooiWJ
BernsA
2001
Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene.
Oncogene
20
6551
6558
12. JohnsonL
MercerK
GreenbaumD
BronsonRT
CrowleyD
2001
Somatic activation of the K-ras oncogene causes early onset lung cancer in mice.
Nature
410
1111
1116
13. GarrawayLA
WeirBA
ZhaoX
WidlundH
BeroukhimR
2005
“Lineage addiction” in human cancer: lessons from integrated genomics.
Cold Spring Harb Symp Quant Biol
70
25
34
14. AlbertsonDG
2006
Gene amplification in cancer.
Trends Genet
22
447
455
15. LockwoodWW
ChariR
CoeBP
GirardL
MacaulayC
2008
DNA amplification is a ubiquitous mechanism of oncogene activation in lung and other cancers.
Oncogene
27
4615
4624
16. KendallJ
LiuQ
BaklehA
KrasnitzA
NguyenKC
2007
Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer.
Proc Natl Acad Sci U S A
104
16663
16668
17. TononG
WongKK
MaulikG
BrennanC
FengB
2005
High-resolution genomic profiles of human lung cancer.
Proc Natl Acad Sci U S A
102
9625
9630
18. GarnisC
DaviesJJ
BuysTP
TsaoMS
MacAulayC
2005
Chromosome 5p aberrations are early events in lung cancer: implication of glial cell line-derived neurotrophic factor in disease progression.
Oncogene
24
4806
4812
19. LockwoodWW
CoeBP
WilliamsAC
MacAulayC
LamWL
2007
Whole genome tiling path array CGH analysis of segmental copy number alterations in cervical cancer cell lines.
Int J Cancer
120
436
443
20. BaldwinC
GarnisC
ZhangL
RosinMP
LamWL
2005
Multiple microalterations detected at high frequency in oral cancer.
Cancer Res
65
7561
7567
21. CoeBP
LockwoodWW
GirardL
ChariR
MacaulayC
2006
Differential disruption of cell cycle pathways in small cell and non-small cell lung cancer.
Br J Cancer
94
1927
1935
22. IshkanianAS
MalloffCA
WatsonSK
DeLeeuwRJ
ChiB
2004
A tiling resolution DNA microarray with complete coverage of the human genome.
Nat Genet
36
299
303
23. WatsonSK
deLeeuwRJ
HorsmanDE
SquireJA
LamWL
2007
Cytogenetically balanced translocations are associated with focal copy number alterations.
Hum Genet
120
795
805
24. KhojastehM
LamWL
WardRK
MacAulayC
2005
A stepwise framework for the normalization of array CGH data.
BMC Bioinformatics
6
274
25. ChiB
DeLeeuwRJ
CoeBP
MacAulayC
LamWL
2004
SeeGH–a software tool for visualization of whole genome array comparative genomic hybridization data.
BMC Bioinformatics
5
13
26. ChiB
deLeeuwRJ
CoeBP
NgRT
MacAulayC
2008
MD-SeeGH: a platform for integrative analysis of multi-dimensional genomic data.
BMC Bioinformatics
9
243
27. JongK
MarchioriE
MeijerG
VaartAV
YlstraB
2004
Breakpoint identification and smoothing of array comparative genomic hybridization data.
Bioinformatics
20
3636
3637
28. BildAH
YaoG
ChangJT
WangQ
PottiA
2006
Oncogenic pathway signatures in human cancers as a guide to targeted therapies.
Nature
439
353
357
29. ChariR
LonerganKM
NgRT
MacAulayC
LamWL
2007
Effect of active smoking on the human bronchial epithelium transcriptome.
BMC Genomics
8
297
30. RamirezRD
SheridanS
GirardL
SatoM
KimY
2004
Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins.
Cancer Res
64
9027
9034
31. SatoM
VaughanMB
GirardL
PeytonM
LeeW
2006
Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells.
Cancer Res
66
2116
2128
32. SambrookJ
RussellDW
2001
Molecular cloning: a laboratory manual
New York
Cold Spring Harbor Laboratory Press
33. TusherVG
TibshiraniR
ChuG
2001
Significance analysis of microarrays applied to the ionizing radiation response.
Proc Natl Acad Sci U S A
98
5116
5121
34. LukC
TsaoMS
BayaniJ
ShepherdF
SquireJA
2001
Molecular cytogenetic analysis of non-small cell lung carcinoma by spectral karyotyping and comparative genomic hybridization.
Cancer Genet Cytogenet
125
87
99
35. SySM
WongN
LeeTW
TseG
MokTS
2004
Distinct patterns of genetic alterations in adenocarcinoma and squamous cell carcinoma of the lung.
Eur J Cancer
40
1082
1094
36. CroceCM
2008
Oncogenes and cancer.
N Engl J Med
358
502
511
37. ZhaoX
WeirBA
LaFramboiseT
LinM
BeroukhimR
2005
Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis.
Cancer Res
65
5561
5570
38. SchrammL
PendergrastPS
SunY
HernandezN
2000
Different human TFIIIB activities direct RNA polymerase III transcription from TATA-containing and TATA-less promoters.
Genes Dev
14
2650
2663
39. CabartP
MurphyS
2001
BRFU, a TFIIB-like factor, is directly recruited to the TATA-box of polymerase III small nuclear RNA gene promoters through its interaction with TATA-binding protein.
J Biol Chem
276
43056
43064
40. WhiteRJ
2004
RNA polymerase III transcription and cancer.
Oncogene
23
3208
3216
41. WhiteRJ
2005
RNA polymerases I and III, growth control and cancer.
Nat Rev Mol Cell Biol
6
69
78
42. GoodfellowSJ
WhiteRJ
2007
Regulation of RNA polymerase III transcription during mammalian cell growth.
Cell Cycle
6
2323
2326
43. WoiwodeA
JohnsonSA
ZhongS
ZhangC
RoederRG
2008
PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex.
Mol Cell Biol
28
4204
4214
44. Felton-EdkinsZA
KennethNS
BrownTR
DalyNL
Gomez-RomanN
2003
Direct regulation of RNA polymerase III transcription by RB, p53 and c-Myc.
Cell Cycle
2
181
184
45. Gomez-RomanN
Felton-EdkinsZA
KennethNS
GoodfellowSJ
AthineosD
2006
Activation by c-Myc of transcription by RNA polymerases I, II and III.
Biochem Soc Symp
141
154
46. MarshallL
KennethNS
WhiteRJ
2008
Elevated tRNA(iMet) synthesis can drive cell proliferation and oncogenic transformation.
Cell
133
78
89
47. JohnsonSA
DubeauL
JohnsonDL
2008
Enhanced RNA polymerase III-dependent transcription is required for oncogenic transformation.
J Biol Chem
283
19184
19191
48. SchrammL
HernandezN
2002
Recruitment of RNA polymerase III to its target promoters.
Genes Dev
16
2593
2620
49. SaxenaA
MaB
SchrammL
HernandezN
2005
Structure-function analysis of the human TFIIB-related factor II protein reveals an essential role for the C-terminal domain in RNA polymerase III transcription.
Mol Cell Biol
25
9406
9418
50. DieciG
FiorinoG
CastelnuovoM
TeichmannM
PaganoA
2007
The expanding RNA polymerase III transcriptome.
Trends Genet
23
614
622
51. CabarcasS
JacobJ
VerasI
SchrammL
2008
Differential expression of the TFIIIB subunits Brf1 and Brf2 in cancer cells.
BMC Mol Biol
9
74
52. WistubaII
GazdarAF
2006
Lung cancer preneoplasia.
Annu Rev Pathol
1
331
348
53. SatoM
ShamesDS
GazdarAF
MinnaJD
2007
A translational view of the molecular pathogenesis of lung cancer.
J Thorac Oncol
2
327
343
54. RewDA
2003
Small RNAs: a new class of genome regulators and their significance.
Eur J Surg Oncol
29
764
765
55. ButcherSE
BrowDA
2005
Towards understanding the catalytic core structure of the spliceosome.
Biochem Soc Trans
33
447
449
56. FaustinoNA
CooperTA
2003
Pre-mRNA splicing and human disease.
Genes Dev
17
419
437
57. HenryRW
MittalV
MaB
KobayashiR
HernandezN
1998
SNAP19 mediates the assembly of a functional core promoter complex (SNAPc) shared by RNA polymerases II and III.
Genes Dev
12
2664
2672
58. KweiKA
KimYH
GirardL
KaoJ
Pacyna-GengelbachM
2008
Genomic profiling identifies TITF1 as a lineage-specific oncogene amplified in lung cancer.
Oncogene
27
3635
3640
59. TanakaH
YanagisawaK
ShinjoK
TaguchiA
MaenoK
2007
Lineage-specific dependency of lung adenocarcinomas on the lung development regulator TTF-1.
Cancer Res
67
6007
6011
60. WeirBA
WooMS
GetzG
PernerS
DingL
2007
Characterizing the cancer genome in lung adenocarcinoma.
Nature
450
893
898
61. PernerS
WagnerPL
SoltermannA
LaFargueC
TischlerV
2009
TTF1 expression in non-small cell lung carcinoma: association with TTF1 gene amplification and improved survival.
J Pathol
217
65
72
62. HerbstRS
HeymachJV
LippmanSM
2008
Lung cancer.
N Engl J Med
359
1367
1380
63. BassAJ
2009
SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas.
Nat Genet
41
1238
1242
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2010 Číslo 7
- MUDr. Jiří Vejmelka: Široká diverzita střevní mikrobioty má obrovský potenciál
- Současnost a budoucnost péče o rány
- PPI nezvyšují riziko kardiovaskulárních příhod u pacientů užívajících ASA v antiagregační terapii
- Esomeprazol jako nejúčinnější PPI v léčbě erozivní ezofagitidy a v úlevě od pálení žáhy
- Optimalizace dávkování enoxaparinu u obézních pacientů
Nejčtenější v tomto čísle
- Three Adult Cases of Meningitis in Vietnam
- Left to Their Own Devices: Breakdowns in United States Medical Device Premarket Review
- Social Relationships and Mortality Risk: A Meta-analytic Review
- Evidence-Based Priority Setting for Health Care and Research: Tools to Support Policy in Maternal, Neonatal, and Child Health in Africa
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy