
A rare loss-of-function mutation reduces blood eosinophil counts and protects from asthma
Only a few genes have been found to play a role in asthma. These include the genes IL33 and IL1RL1, and sequence variants in the human genome close to these genes were initially found to affect the number of eosinophils, cells that play a role in inflammation of the airways in asthma. Based on this knowledge, we decided to use high resolution sequencing technology to search for variants in these genes that cause changes in structure and function of the proteins they encode. We found a rare (0.65%) sequence variant in the IL33 gene, that causes less production of the IL33 protein and some of the protein formed lacks the capacity to bind to its receptor on cells and promote inflammation. This rare mutation causes reduced number of eosinophils in blood and protects against asthma. These results suggest that drugs that could interfere with the inflammatory activity of the IL33 protein may be beneficial for treatment of asthma.
Published in the journal:
. PLoS Genet 13(3): e32767. doi:10.1371/journal.pgen.1006659
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1006659
Summary
Only a few genes have been found to play a role in asthma. These include the genes IL33 and IL1RL1, and sequence variants in the human genome close to these genes were initially found to affect the number of eosinophils, cells that play a role in inflammation of the airways in asthma. Based on this knowledge, we decided to use high resolution sequencing technology to search for variants in these genes that cause changes in structure and function of the proteins they encode. We found a rare (0.65%) sequence variant in the IL33 gene, that causes less production of the IL33 protein and some of the protein formed lacks the capacity to bind to its receptor on cells and promote inflammation. This rare mutation causes reduced number of eosinophils in blood and protects against asthma. These results suggest that drugs that could interfere with the inflammatory activity of the IL33 protein may be beneficial for treatment of asthma.
Introduction
Asthma is characterized by airflow obstruction, airway hyper-responsiveness and airway inflammation, that promotes mucus obstruction. The inflammatory cytokine IL-33 is widely expressed[1, 2] and abundantly in the bronchial epithelium. IL-33 resides in the nucleus due to chromatin-binding motifs[3] but is released after exposure to e.g. viruses or allergens. IL-33 binds to its receptor ST2[4] (also called IL1RL1, IL-33R) and activates eosinophils and other immune cells promoting inflammation[5], particularly in the lung[6, 7]. IL-33 also binds to a soluble isofrom IL1RL1-a/sST2, which is thought to act as a decoy receptor and ameliorates airway inflammation[8].
Through GWAS, we previously discovered common sequence variants in IL1RL1 and IL33 that associate strongly with blood eosinophil counts and risk of asthma, consistent with the link between eosinophilic inflammation and asthma [9]. The association between IL1RL1 and IL33 variants and asthma risk is well established [10] and has been replicated in ethnically diverse populations[11, 12], and in severe forms of adult asthma[11] and in particular with early childhood[13] asthma with exacerbations.
The critical role of eosinophilic airway inflammation in asthma[14] together with robust association of eosinophil counts and asthma with common variants at the IL33 and IL1RL1 loci prompted us to search for novel sequence variants at these loci affecting eosinophil counts using high-coverage sequencing [15]. The variants were identified through sequencing of 8,453 Icelanders and imputed into 150,656 chip-typed Icelanders[15]. In light of the role of eosinophils in pathogenesis of asthma and the previously established association of common variants at the IL33 and IL1RL1 loci with eosinophil counts and asthma, individual sequence variants at these loci found to associate significantly with eosinophil counts were further assessed for their effects on asthma.
Results
We focus on the association of sequence variants in two 800kb regions centered on IL33 (chr9 : 5.8–6.6Mb(hg38)) and IL1RL1 (chr2 : 101.9–102.7Mb(hg38)) with eosinophil counts in 103,104 Icelanders[16]. Sequence variants were weighted according to their prior probability of affecting gene function by applying different thresholds for genome-wide (gw) significance that depend on the variant class[17].
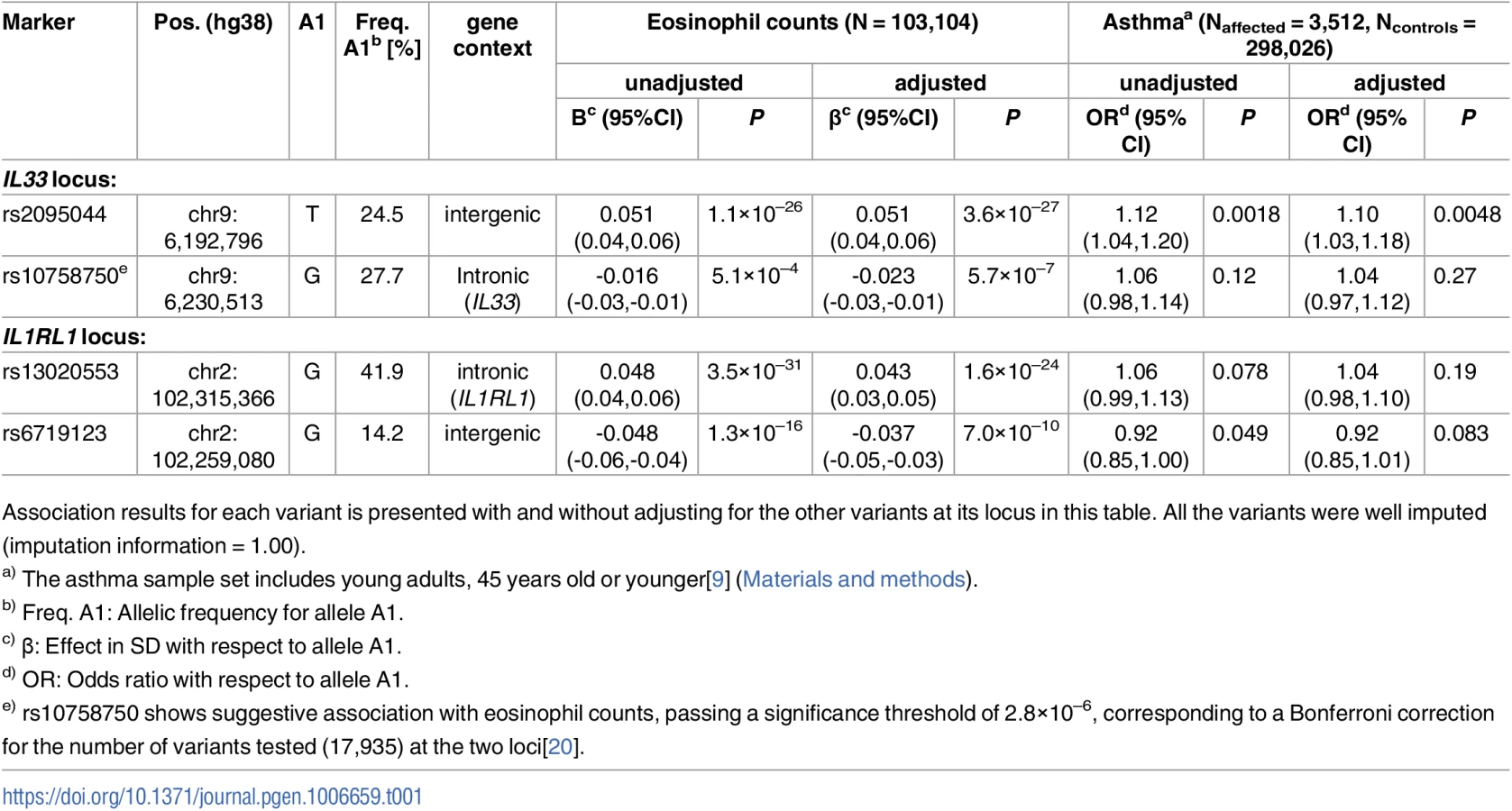
Stepwise conditional analysis at the IL33 locus revealed two significant uncorrelated (pairwise r2<0.02) variants (Fig 1 and S1 Fig, Table 1 and S1 Table). One association is novel: rs146597587-C a rare variant (allelic frequency (AF) = 0.65%) that is predicted to disrupt a canonical splice acceptor site at the beginning of the last exon of IL33[18, 19] (NM_001199640:exon7:c.487-1G>C, βadj = -0.21 SD; Padj = 2.5×10–16) (Table 2). rs146597587-C associates with lower eosinophil counts and is not correlated with previously reported variants at the IL33 locus (S2 Table). The second variant, rs2095044-T (AF = 24.5%), upstream of IL33, is associated with increased eosinophil count (βadj = 0.051 SD; Padj = 3.6×10–27) and is highly correlated with rs2381416 (r2 = 0.94), originally described to associate with eosinophil counts and asthma[9] (Table 1, S3 and S4 Tables). A potential secondary signal was noted, rs10758750-G (AF = 27.7%), an intronic variant in IL33 (βadj = -0.023 SD; Padj = 5.7×10–7) (Table 1) (passing a significance threshold of 2.8×10–6, corresponding to a Bonferroni correction for the number of variants tested (17,935) at the two loci[20]), uncorrelated (pairwise r2<0.02) to the novel variant reported here (Fig 1 and S1 Fig, Table 1 and S1 Table) and to the previously reported variants at the IL33 locus (S2 Table).

![Associations of the <i>IL33</i> splice acceptor variant rs146597587[C] with eosinophil counts and asthma in Iceland and abroad.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/28135666ad5ea8086d606ae343f041cd.png)
Among the two significant variants, the splice acceptor mutation rs146597587-C has the largest effect. The splice acceptor mutation is present in both Europeans (AF = 0.35%) and South-Asians (AF = 0.10%) in the Exome Aggregation Consortium (ExAC) database where it is >10 times more frequent than any other predicted loss-of-function variant in IL33 (S5 Table, URLs). In total we tested six predicted missense, splice region or loss-of-function variants in IL33 (S6 Table) and only the association with the splice acceptor mutation rs146597587-C is significant (P>0.2 for the others). None of the twelve variants highly correlated (r2>0.8) with the splice acceptor mutation were coding, making it most likely to be responsible for the effect (S7 Table). Neither of the two other associating variants, rs2095044 nor rs10758750, were correlated with a coding variant (S4 and S8 Tables).
The IL33 splice acceptor mutation, rs146597587-C was genotyped in 1,370 Dutch samples and its effect on eosinophil counts was replicated (β = -0.48 SD, P = 0.036, AF = 0.69%) (Table 2).
Once the eosinophil count association of the splice acceptor variant was established, we assessed its effect on asthma, based on the prior association of IL33 and with asthma risk and the role of eosinophils in the pathogenesis of asthma. We assessed the effect of rs146597587-C on asthma in Iceland[9], The Netherlands[21–23], Germany [24] and Denmark[25, 26], including Danish children with severe asthma with at least 2 exacerbations leading to hospitalization between 2 and 6 years of age (COPSACexacerbation)[13]. The IL33 splice acceptor mutation protects against asthma (OR = 0.47; 95% CI: 0.32–0.70, P = 1.8×10–4) (Table 2). We did not observe heterogeneity of the effect in the different sample sets (Phet = 0.24, I2 = 26.8). Thus, this rare splice acceptor mutation reduces eosinophil counts and protects against asthma, whereas the minor allele of the common rs2095044-T associates with higher eosinophil counts and increased risk of asthma (Tables 1 and 2).
Stepwise conditional analysis of the association with eosinophil counts (N = 103,104) at the IL1RL1 locus revealed two significant variants, an intronic variant, rs13020553-G (MAF = 41.9%), increasing eosinophil counts (β = 0.043, Padj = 1.6×10–24) and an intergenic variant, rs6719123-G (MAF = 14.2%), decreasing eosinophil counts (β = -0.037, Padj = 7.0×10–10) (Table 1 and S9 Table); no other variant at the locus remained significant after adjusting for these two (Fig 2 and S10 Table). rs13020553 is highly correlated with rs1420101 (r2 = 0.96, D’ = 1.00), originally reported to associate with eosinophil counts and asthma[9] (S3 and S10 Tables). Despite the high correlation, rs1420101-T (that is on the background of rs13020553-G) does not fully explain the eosinophil association of rs13020553, whereas rs13020553 fully explains the eosinophil association for rs1420101 (S10 Table). Together rs13020553 and rs6719123 explain all the reported effects on eosinophil counts and asthma at the locus (S3 Table); however, the reported variants do not fully explain the signal captured by rs13020553 and rs6719123 (S11 Table).

We found and tested 75 coding or splice region variants at the IL1RL1 locus. Among those not in IL1RL1, only one was significant (P>0.001 for all other coding variants not in IL1RL1), rs1420098-C, a splice region variant in IL18R1 (MAF = 42.5%, P = 3.8×10–24, β = 0.042). The eosinophil counts association of rs1420098 was fully accounted for by rs13020553 (r2 = 0.81, Padj = 0.53), whereas the association of rs13020553 could not be accounted for by rs1420098 (Padj = 1.2×10–8, βadj = 0.054). We observed two significant coding signals in IL1RL1 among the sixteen detected and tested (14 missense, one splice region variant and one frameshift variant) (S12 Table). Both coding signals were previously reported to associate with concentration of ST2 in soluble form (sST2)[27]. The first, represented by rs10192157-T (MAF = 39.0%, P = 4.2×10–20; HGVSp: NP_057316.3:p.Thr549Ile), corresponds to five perfectly correlated missense variants (r2 = 1.00, D’ = 1.00 for all pairs) moderately correlated with rs13020553 (r2 = 0.35); the second rs1041973-A (MAF = 17.7%, P = 3.5×10–9, HGVSp: NP_057316.3:p.Ala78Glu) is correlated with rs6719123 (r2 = 0.68) (S13 Table). These two eosinophil counts associations are fully accounted for by rs13020553 and rs6719123 (S14 Table). We conclude that the eosinophil counts signals corresponding to rs13020553 and rs6719123 (correlated variants: S15 and S16 Tables) could not be explained by the observed coding variants or reported variants at the locus. Our results are in agreement with the notion that variants at this locus influence eosinophil counts by affecting IL1RL1; and IL-33 is known to mediate its biological effects through ST2/IL1RL1[4].
The splice acceptor mutation in IL33 changes AG to AC at the 3’ splice junction between the last two exons and is thus predicted to disrupt splicing between them [18, 19]. IL33 is primarily expressed by stromal cells and is expressed at a relatively low level in hematopoetic tissues. Of the two tissues with mRNA sequencing data available to us IL33 is essentially absent from whole blood but is expressed in subcutaneous adipose tissue[1] (median RPKM of 12.9 in 350 samples), according to the GTExV6 database[2]. We therefore analyzed the effect of the splice acceptor mutation on the IL33 transcript quantity and processing using the adipose tissue mRNA sequencing (N = 675) dataset (Materials and methods, and Fig 3). Heterozygote carriers (N = 10) of the splice acceptor mutation showed about 40% lower total IL33 expression than non-carriers (P = 6.8×10–6) (Fig 3). Comparable results were obtained by accessing total IL-33 expression with microarray (S2 Fig). Allele-specific analysis of the RNA sequencing data shows that only 20% of total IL33 transcripts are from the mutated chromosomes (P = 1.3×10–4) indicating that a large fraction of mRNA originating from the mutated chromosomes is likely eliminated through nonsense-mediated decay (NMD)[28]. Analysis of read coverage in the RNA sequencing data, shows retention of the last intron of IL33 in about half of the RNA generated from the mutated chromosome (~11% of total RNA) in heterozygotes compared to 0.6% of that in non-carriers (P = 5.6×10–8). Taken together these data demonstrate that the splice acceptor mutation leads both to elimination of the mutated transcripts and a retention of the last intron, introducing a premature stop codon in about half of the remaining mutated transcripts.

The IL-33 protein generated from the intronic retention transcripts is predicted to lack the last 66 amino acids (out of 270). IL-33 folds as a 12-stranded “β-barrel” structure, prototypical for the IL-1 family. Elimination of the last 66 amino acids removes 5 of the 12 core β-strands and is predicted to disrupt the tertiary structure of the protein. To determine whether IL-33 lacking these residues is functional we expressed recombinant IL-33 lacking the last exon (termination at residue 204, Fig 4). IL-331–204 was detectable in transfected mammalian cellular lysates as a 27kD protein, compared to the 35 kD wild type IL-331–270 (S3 Fig). IL-331–204 was in the nucleus in a manner indistinguishable from IL-331–270 (Fig 4), indicating that the truncation does not disrupt nuclear trafficking.

Computational modeling [29] indicates that truncated IL-33 lacks core structural folds and key surface residues predicted to contribute to receptor binding (Fig 4A). To determine whether the truncated form binds ST2 we generated IL-3395–270 (wild type) and IL-3395–204 (truncated mutant) recombinant proteins. The N-terminus starting residue at amino acid position 95 was chosen based on natural processing of IL-33 before receptor binding[30] (S4 Fig).
Using surface plasmon resonance we found that IL-3395–270 bound rapidly to IL-33R/ST2 (Fig 4), consistent with high affinity binding to IL-33R that we previously reported[31], whereas IL-3395–204 showed no interaction signal, indicating a complete lack of receptor binding. Furthermore, IL-3395–270 induced a concentration - and IL-33R-dependent CCL1 release in human mast cell line expressing IL-33R (Fig 4C and S5 Fig), whereas IL-3395–204 did not at any concentration tested, consistent with a complete loss of cytokine activity. The mutant IL-33 was also inactive compared to wild type inducing IFN-γ release from human CD4+ T cells (Fig 5).

In il33 knock-out mice, 30% reduced fertility by has been reported[32]. Nine imputed rs146597587-C homozygotes were found in the Icelandic data and they neither show reduced life expectancy nor reduced fertility (S16 Table), indicating that a predicted very low level of IL-33 is compatible with long life and healthy reproduction. Moreover, rs146597587-C homozygosity (recessive model) does not confer risk of any other disease that was tested in the Icelandic data[15, 33], indicating that IL-33 is largely dispensable. Two homozygotes of the splice acceptor mutation are reported in ExAC (S5 Table, URLs). An association of il33 polymorphism with eosinophil numbers in rats has been reported[34] and we found that deletion of il33 leads to significant reduction in blood eosinophils in unchallenged mice (P = 0.014 males, P = 0.00010 females) (Fig 6), mimicking our human observations.

Discussion
Resequencing of 100 genes implicated in asthma only revealed a few coding variants associating with asthma[35]. Since eosinophils are known to play a key role in inflammation of the airway in asthma[12] we used high-coverage sequencing [13] to search for novel sequence variants affecting eosinophil counts at the well established asthma loci, the IL33 and IL1RL1 loci, and tested their effects on asthma. Our results indicate that variants at the IL1RL1 locus affect eosinophil counts and the risk of asthma most likely by affecting IL1RL1 itself. IL-33 is known to mediate its biological effects through ST2/IL1RL1[4] and sensitized mice with il1rl1 (ST2-/-) knocked out show less eosinophil numbers in bronchoalveolar lavage fluid upon allergen exposure than wild-type mice, reduced levels of Th2 cytokines and chemoattractants in the lungs, and reduced goblet cell hyperplasia around the peripheral airways in murine models of allergy and asthma[36, 37].
We report a rare variant, rs146597587, in IL33 representing a loss-of-function mutation in this known asthma gene. This splice acceptor mutation associates with lower eosinophil counts and protection against asthma, with the largest protective effect from severe asthma with frequent exacerbations in young Danish children (OR = 0.24, Table 2). This is in agreement with increasing risk conferred by the common IL33 variant rs2381416 with increasing severity of asthma in these children, with OR from 1.27 to 1.69 for severity groups 1 to 4 (S17 Table). The splice acceptor mutation caused reduced expression of IL33 transcripts, likely due to NMD, and production of truncated IL-33 that lacks the cytokine function due to lack of binding to IL-33R/ST2 resulting in abrogation of IL-33R-dependent release of CCL1 from mast cells and IFN-γ release from human CD4+ T cells. We therefore infer that asthma risk is mediated through IL-33. Accordingly, common variants in IL1RL1 that associate with increased asthma risk associate with reduced expression of soluble ST2[38, 39], with the predicted effect being increased IL-33 activity, due to reduced level of this decoy receptor. Thus, human genetics support the rationale for therapeutically inhibiting the IL-33-ST2 pathway in an attempt at containing asthma.
Materials and methods
Ethics statement
The Icelandic study was approved by the National Bioethics Committee (VSN_14–099) and the Data Protection Authority (no. PV_2014060841/ÞS) in Iceland. All participating subjects who donated blood provided informed consent. Personal identities of the participants and biological samples were encrypted by a third-party system approved and monitored by the Icelandic Data Protection Authority.
The Denmark-1 study was approved by the local ethics committee of Copenhagen, Denmark (Approval no. KF01-400/98 and KF01-074/01). All participants signed informed consent.
The COPSAC study research protocol was approved by The Danish National Ethical Committee on Health Research (KF 01-289/96, H-B-2008-093, H-16039498 and H-B-2998-103) and is in accordance with the ethical scientific principles of the Helsinki Declaration II. All parents in the cohorts signed informed consent.
The German study was approved by the Ethical Commission of the University of Freiburg (Approval no. 96/05). All participants signed informed consent.
The Dutch European ancestry asthma study was approved by the Medical Ethics Committee of the University Hospital Groningen (Approvals no. MEC 90/09/178, MEC 96/04/077, MEC 97/10/184). All participants signed informed consent. The Dutch Vlagtwedde/Vlaardingen study protocol was approved by the local university medical hospital ethics committee, University of Groningen, University Medical Center Groningen, The Netherlands and all participants gave their written informed consent. In 1984, the Committee on Human Subjects in Research of the University of Groningen reviewed the study and affirmed the safety of the protocol and study design.
The mouse experiments were performed at Amgen Inc. All animal use procedures were in accordance with Amgen animal use and care guidelines and approved by IACUC.
Study sample sets for eosinophil count and asthma
Iceland
We obtained eosinophil counts from three of the largest laboratories in Iceland (measurements performed between the years 1993 and 2015). The median eosinophil counts were 200/μl (lower and upper quartiles: 100/μl and 300/μl). The circulating eosinophil counts were standardized to a standard normal distribution using quantile-quantile standardization and then adjusted for sex, year of birth and age at measurement, as previously described[9, 40]. A total of 103,104 individuals with eosinophil counts were included in the study, where 82,642 were chip-typed and directly imputed; the remaining 20,462 were first and second degree relatives of chip-typed individuals and had their genotypes inferred based on genealogy[16].
Icelandic asthma patients over 18 years of age who attended an asthma clinic or emergency room at the National University Hospital of Iceland or the Icelandic Medical Center (Laeknasetrid) during the years 1977 to 2014 were recruited[9]. Asthma diagnosis was based on a combination of physician’s diagnosis, a positive reply to the question: „Has a doctor confirmed your asthma diagnosis?“, questionnaires pertaining to asthma symptoms and ICD diagnosis when receiving emergency care[9, 40]. Atopy status (defined by at least one positive response to allergens) determined by skin prick testing was available for part of the cohort. A total of 3,512 (1,842 chip-typed) asthma cases 45 years old or younger were used in the study (18.5% with known age of onset 18 years of age or younger) [9, 40] and 298,026 (134,762 chip-typed) controls. Icelandic controls were participants from various deCODE genetics programs without known asthma.
The study was approved by the National Bioethics Committee (VSN_14–099) and the Data Protection Authority (no. PV_2014060841/ÞS) in Iceland. All participating subjects who donated blood provided informed consent. Personal identities of the participants and biological samples were encrypted by a third-party system approved and monitored by the Icelandic Data Protection Authority.
Denmark 1
Asthma subjects and controls from three studies at the Copenhagen Research Unit, aged 14 to 44 years and resided in Copenhagen, Denmark, were used in the analysis[25, 26]. The asthma patients had doctor diagnosed asthma by a respiratory specialist and had undergone a physical examination and spirometry. Danish control subjects were recruited through the Danish Blood Donor Corps in the Copenhagen area.
Denmark 2 (COPSAC)
Cases were from the COPSACexacerbation cohort. Children with repeated acute hospitalizations (cases) were identified in the Danish National Patient Register covering all diagnoses of discharges from Danish hospitals[41]. Information on birth-related events was obtained from the national birth register. Inclusion criteria were at least two acute hospitalizations for asthma (ICD-8-code 493, ICD-10 codes J45-46) from 2 to 6 years of age, as previously described[13]. Duration of hospitalization had to be more than 1 day, and two hospitalizations had to be separated by at least 6 months. Exclusion criteria were side diagnosis during hospitalization, registered chronic diagnosis considered to affect risk of hospitalization for asthma, low birth weight (<2.5 kg) or gestational age of under 36 weeks at birth. Control subjects were children without asthma exacerbations in the COPSAC2000 and COPSAC2010 birth cohorts. Study participants gave informed consent for use of their biological samples for genetic studies. The research protocol was approved by The Danish National Ethical Committee on Health Research and is in accordance with the ethical scientific principles of the Helsinki Declaration II.
Germany
The German case-control population consisted of children (aged 5–18 years) with suspected asthma recruited from the southwestern part of Germany between July 2000 and January 2005[24]. The probands were characterized at the Centre of Pediatrics and Adolescent Medicine, Freiburg, Germany. The diagnosis of asthma was based on a history of respiratory symptoms, the use of anti-asthmatic medication and the presence of bronchial hyperresponsiveness (defined as a fall of at least 15% in baseline FEV1 after either inhalation of ≤8 mg/ml histamine or ≤6 minutes of exercise provocation). Participants were asked in advance to discontinue any asthma or allergy medication before the clinical testing. Atopy status was determined by skin prick tests to 17 common allergens. Measurement of total serum IgE was carried out by using an enzyme allergosorbent test (Phadezym; Pharmacia, Uppsala, Sweden). The control sample consisted of subjects (aged 19–40 years) randomly chosen from the same area in the southwestern part of Germany and children recruited from clinics at the Centre of Pediatrics and Adolescent Medicine, Freiburg, Germany. No medical history was taken, and no medical testing was performed on the adult controls whereas the pediatric controls had no previous history of asthma, recurrent wheezing, atopic dermatitis or atopy. Approval was granted by the Ethical Commission of the University of Freiburg. A statement of informed consent was signed by all participants or signed by their parents in the case of children.
The Netherlands
The Dutch cases have been previously described[21–23]. Briefly asthma patients of European ancestry were initially studied between 1962 and 1975. At the time of recruitment and initial testing, all patients had asthma symptoms, were hyperresponsive to histamine (PC20 histamine <32 mg/ml), and were younger than 45 years of age. Between 1990 and 1999, patients were invited for follow-up tests: including pulmonary function, bronchial responsiveness to histamine, and total and specific serum IgE and skin tests to 16 common aero allergens [22, 23]. Participants were asked in advance to discontinue asthma or allergy medication, therapy with oral corticosteroids was however continued.
A second independent Dutch asthmatic population was ascertained between 1998 and 2001, consisting of probands with asthma, ascertained through local hospitals and media appeals. Controls were derived from Vlagtwedde/Vlaardingen cohort study, a population-based cohort of adult subjects of European ancestry in the Netherlands, recruited from 1965 with a follow-up of over 25 years. Surveys were performed every 3 years, in which information was collected on respiratory symptoms, smoking status, FEV1, and allergy skin tests [42].
Whole-genome sequencing and imputation
Genotyping of all Icelandic samples was carried out at deCODE genetics in Reykjavik, Iceland, using methods recently described[15]. In brief, whole-genome sequencing was performed for 8,453 Icelanders who were recruited as part of various genetic programs at deCODE genetics, to an average depth of at least 10× (median 32×) using Illumina technology. The sequencing was performed using the following three different library preparation methods and sequencing instruments from Illumina: (i) standard TruSeq DNA library preparation method; Illumina GAIIx and/or HiSeq 2000 sequencers; (ii) TruSeq DNA PCR-free library preparation method; Illumina HiSeq 2500 sequencers; and (iii) TruSeq Nano DNA library preparation method; Illumina HiSeq X sequencers (S1 Method).
SNPs and indels identified in the whole-genome sequencing data were identified using the Genome Analysis Toolkit HaplotypeCaller (GATK version 3.3.0)[43] and imputed into 150,656 Icelanders who had been genotyped with various Illumina SNP chips and their genotypes phased using long-range phasing chip-genotyped individuals using long-range phasing[44]. In addition, using the Icelandic genealogical database, genotype probabilities were calculated for first - and second-degree relatives of chip-genotyped individuals[15]. The effects of sequence variants on protein-coding RefSeq genes[18] were annotated using the Variant Effect Predictor (VEP) version 80[19].
Genotyping of single variants
Single SNP genotyping in the replication sample sets was carried out by deCODE Genetics in Reykjavik, Iceland, applying the Centaurus (Nanogen) platform[45] except that genotyping single SNPs in the Danish cohort of children with severe asthma was carried out at the AROS Applied Biotechnology AS center (http://arosab.com/services/microarrays/genotyping/), in Aarhus, Denmark, applying the Illumina Infinium HumanOmniExpressExome Bead chip platform and genome studio software[46].
Association testing in Iceland
Generalized linear regression models were used to test for associations between sequence variants and quantitative traits, assuming an additive genetic model. Let y be the vector of quantitative measurements, and let g be the vector of expected allele counts for the sequence variant being tested. We assume the quantitative measurements follow a normal distribution with a mean that depends linearly on the expected allele at the variant and a variance covariance matrix proportional to the kinship matrix:


Association summary statistics are provided in supplementary tables for the IL33 locus (S18 Table) and the IL1RL1 locus (S19 Table).
LD score regression
To account for inflation in test statistics due to cryptic relatedness and stratification, we applied the method of LD score regression[47]. With a set of 1.1M variant we regressed the χ2 statistics from GWAS scans against LD score and used the intercept as a correction factor. The LD scores were downloaded from an LD score database (see URLs). The correction factors were 1.2844 for the eosinophil counts, and 1.1120 for asthma.
Gene expression microarrays
Samples of RNA from human peripheral blood and adipose tissue were hybridized to Agilent Technologies Human 25K microarrays as described previously[48] and the effect of SNPs on the IL33 expression in adipocytes evaluated. We quantified expression changes between two samples as the mean logarithm (log10) expression ratio (MLR) compared to a reference pool RNA sample. In comparing expression levels between groups of individuals with different genotypes, we denoted the expression level for each genotype as 10 (average MLR), where the MLR is averaged over individuals with the particular genotype. We determined s.e.m. and significance by regressing the MLR values against the number of risk alleles carried. We took into account the effects of age, gender and differential cell type count in blood as explanatory variables in the regression.
RNA sequencing
The effect of on the IL33 expression in adipocytes was evaluated by RNA sequencing as previously described[20].
Preparation of Poly-A cDNA sequencing libraries
The quality and quantity of isolated total RNA samples was assessed using the Total RNA 6000 Nano chip for the Agilent 2100 Bioanalyzer. cDNA libraries derived from Poly-A mRNA were generated using Illumina‘s TruSeq RNA Sample Prep Kit. Briefly, Poly-A mRNA was isolated from total RNA samples (1–4 μg input) using hybridizaton to Poly-T beads. The Poly-A mRNA was fragmented at 94°C, and first-strand cDNA was prepared using random hexamers and the SuperScript II reverse transcriptase (Invitrogen). Following second-strand cDNA synthesis, end repair, addition of a single A base, adaptor ligation, AMPure bead purification, and PCR amplification, the resulting cDNA was measured on a Bioanalyzer using the DNA 1000 Lab Chip.
Sequencing
Samples were clustered on to flowcells using Illumina‘s cBot and the TruSeq PE cluster kits v2, respectively. Paired-end sequencing was performed with either GAIIx instruments using the TruSeq SBS kits v5 from Illumina or HiSeq 2000 instruments using TruSeq v3 flowcells/SBS kits; read lengths were 2x76, 2x101 or 2x125 cycles.
Read alignment
RNA sequencing reads were aligned to Homo sapiens Build 38 with TopHat[49] version 2.0.12 with a supplied set of known transcripts in GTF format (RefSeq hg38; Homo sapiens, NCBI, build 38). TopHat was configured such that it attempts first to align reads to the provided transcriptome then, for reads that do not map fully to the transcriptome, it attempts to map them onto the genome. Read mapping statistics used for read count normalization were calculated using the CollectRnaSeqMetrics tool in Picard version 1.79 (http://broadinstitute.github.io/picard/command-line-overview.html#CollectRnaSeqMetrics). After the read alignment step by TopHat, gene expression estimation in FPKM values (Fragments Per Kilobase of transcript per Million mapped reads) was done using Cufflinks[49, 50] version 2.2.1 for the same set of transcripts used in the read alignment by TopHat.
Mammalian expression
IL-33 variant-expressing vectors as shown in Fig 4D were transfected into HEK293–EBNA1 cells (obtained from National Research Council of Canada) using the protocols described elsewhere[51]. At 24 hours post transfection, cells were either seeded onto polylysine-coated glass coverslips and cultured for 24 hours for imaging, or fed with Difco yeastolate cell culture supplement (BD Biosciences). Cell culture media were collected and whole cell lysates were prepared 2, 4, and 7 days post transfection.
Cell imaging
At 48 hrs post transfection, cells were fixed with 4% paraformaldehyde in 0.1 M sodium phosphate, pH 7.2, for 30 minutes at room temperature. After a wash in PBS containing 0.1 M glycine, the fixed cells were incubated with permeabilization buffer (PBS containing 0.4% saponin, 1% BSA, 5% fish gelatin) for 15 minutes, followed by incubation with primary antibody in permeabilization buffer for 60 min. Primary antibodies included anti-IL33 mature domain (R&D Systems), anti-FLAG (clone M2, Amgen), anti-GAPDH (clone 6C5, Amgen), anti-gianin (Covance) and anti-GM130 (BD Transduction Lab). After 3 washes in permeabilization buffer, the cells were incubated with Alexa Fluor 488 - or 594-conjugated secondary antibody in permeabilization buffer for 60 minutes. Slides were analyzed using a Nikon Eclipse 80i microscope using a 100× or 60× CFI Plan Apo oil objective lens. Images were acquired using a Cool SNAP HQ2 digital camera (Photometrics) and Nikon Elements imaging software.
Western blotting
Harvested cell culture media and processed cell pellets were heated for 5 min at 90°C in lithium dodecyl sulfate sample buffer (Life Technologies) containing 5% (v/v) beta-mercaptoethanol. NuPAGE 4–12% Bis–Tris gradient gel and the accompanying running buffer system (both from Life Technologies) were used to perform SDS-PAGE. Resolved proteins were electrotransferred to a nitrocellulose membrane, blocked with fluorescent Western blocking buffer (Rockland), and probed with primary antibodies (described above). After 3–4 washes in PBS containing 0.05% (v/v) Tween-20, the nitrocellulose membranes were incubated with AlexaFluor 680 - conjugated secondary antibodies (Life Technologies), followed by 2–3 additional washing steps in PBS containing 0.05% (v/v) Tween-20. The fluorescent Western images were acquired by using an Odyssey infrared imaging system from LI-COR Biosciences.
E. coli expression and purification
IL-33 was cloned into the pET24a expression plasmid in frame with an amino terminus poly-His tag and thrombin cleavage site such that thrombin cleavage would release IL-33 proteins corresponding to residues 95–270 (wild type) or 95–204 (mutant).
For IL-3395–270, following E. coli culture, cell pellets were lysed and the supernatants were collected and purified on a nickel HisTrap column (GE Healthcare Life Sciences). The material was eluted from the column using an imidazole gradient. Appropriate fractions were pooled and further purified by preparative size exclusion chromatography (SEC) using a Superdex 75 column (GE Healthcare Life Sciences) with PBS, pH 7.2, 1 mM DTT as the running buffer. Recovered material was cleaved using the Thrombin CleanCleave Kit (Sigma) with a 1 hour incubation at 4°C. Preparative SEC was used to remove the free His tag and any aggregated material, followed by an additional SEC step to further reduce endotoxin contamination. IL-3395–270 was formulated in PBS, pH 7.2, 1 mM DTT, 1 mM EDTA and filtered through a 0.2 μm filter. Endotoxin was undetectable.
IL-3395–204 was not recoverable from the cell pellet supernatant due to low expression in the soluble fraction. Instead, inclusion bodies were solubilized 1/10 (w/v) with 8 M guanidine, 50 mM Tris, 8mM DTT, pH 9.0 for 1 hour. This was followed by dilution 1/25 (v/v) with PBS buffer, pH 7.6 and the material was concentrated and loaded onto a Superdex 75 column using PBS, pH 7.2 as the running buffer. The huIL-33 peak was then buffer-exchanged into 1X thrombin cleavage buffer (50 mM Tris-HCl, pH 8.0, 100 mM CaCl2) and concentrated to 1 mg/mL. 0.1 mL of thrombin agarose resin (Sigma) was added to 1 mg of huIL-33 and rotated at 4°C for 1 hour. Cleaved huIL-33 was then passed over a HisTrap column to capture free cleaved His tag. The flow-through material was further purified using preparative SEC, as for IL-3395–270, prior to formulation in PBS, pH 7.2, 1 mM EDTA. Fractions were analyzed by SDS-PAGE and Coomassie staining (total protein) or Western blot (with anti-IL-33) before and after thrombin cleavage to confirm cleavage and IL-33 identity (S4 Fig).
Computational modeling
The structure model of the ST2/IL-33 complex was provided by Lingel et al.[29]. The receptor and ligand are shown with a molecular surface, computed with default parameters in the Molecular Operating Environment (MOE, Chemical Computing Group Inc., Montreal, QC, Canada, H3A 2R7, 2011. (n.d.)), with ST2 shown in gold and IL-33 shown in blue, with amino acids 205–270, which are derived from exon 7, shown in magenta (Fig 4A).
Surface plasmon resonance
All experiments were performed using a Biacore T200 optical biosensor (Biacore AB). HBS-P buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% Surfactant P20) was purchased from Teknova. The CM5 sensor chip, sodium dodecyl sulphate (SDS) (0.5% w/v), NaOH (50 mM), coupling reagents (N-ethyl-N’-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC)/ N-hydroxy-succinimide(NHS)), ethanolamine (1.0 M Ethanolamine-HCl, pH 8.5), Acetate 5.0 (10 mM sodium acetate) and Glycine 1.5 (10 mM glycine, pH 1.5) were purchased from GE Healthcare Life Sciences. The HCl solution (100 mM) was purchased from Bio Rad Labs. The penta-His antibody (BSA-free) was purchased from 5Prime. All other reagents were prepared by Amgen.
Biosensor analysis was conducted at 25°C in HBS-P buffer. The CM5 sensor chip was conditioned with twice with serial injections (30 seconds) of SDS (0.1%), NaOH (10 mM), HCl (10 mM) and again with SDS (0.1%) at a flow rate of 30 uL/min (first two injections) or 60 ul/min (last three injections). The penta-His antibody was diluted (200 ug/mL) in water and then buffer exchanged via desalting spin column (Zeba, 0.5 mL, 40 K) into Acetate 5.0 buffer. This buffer-exchanged antibody was immobilized to flow cells 1 (3148 RU) and 2 (7554 RU) of the sensor chip via standard amine coupling (EDC/NHS) and ethanolamine blocking[52]. Human ST2-Flag-His (8 ug/mL in HBS-P, Amgen) was injected (300s at 10 uL/min) over flow cell 2. This captured between 600 and 900 RU of antibody. After the capture step, the human E. coli-derived IL-33 variants (200 nM) were injected (180s, 50 ul/min) over flow cells 1–2 to observe the association (180 s) of human IL-33 to human ST2. Each flow cell was then flushed with running buffer to observe the dissociation of human IL-33 (300 s) from the chip surface. Each sample of IL-33 was tested individually as a single replicate. A blank buffer injection was tested before the first sample injection and the surface was regenerated (15 ul at 50 uL/min) with 10 mM glycine pH 1.5 and a reloading of ST2 was captured to the chip surface before each sample injection.
The data was prepared and analyzed with Scrubber 2.0 software (BioLogic Software Pty Ltd, Campbell, Australia) as follows. The raw data from each experiment was x and y-axis normalized just prior to the injection of IL-33 and then cropped to include the ST2 capture step and the association/dissociation of IL-33.
Mast cell bioassay
LAD2 human mast cells were seeded into 48-well tissue culture plates at 250,000 cells/375 μL media (Stem-Pro-34 serum-free media (Life Technologies, Grand Island, NY, USA) supplemented with 2 mM L-glutamine, 100 IU/ml penicillin, 50 μg/ml streptomycin (complete SFM) and 100ng/ml SCF (Peprotech). The following day cells were stimulated with medium only or increasing concentrations (10 pg/mL – 100 ng/mL) of wild-type (95–270) or mutant (95–204) IL-33 for 24 hours at 37°C. Culture supernatants were then collected followed by removal of cell debris by centrifugation and stored at -80°C until ready for use. The concentration CCL1 in the collected supernatants was determined using an R&D Human CCL1 DuoSet ELISA kit under manufacturer’s specifications.
To demonstrate the ST2-dependence of the assay, stimulations were performed as described with the addition of a final concentration of 20 ug/mL human IgG2 isotype control (Amgen) or human anti-huST2 blocking antibody (Amgen) 30 minutes prior to addition of 100 ng/mL IL-33.
Human CD4 T cell bioassay
Highly purified human CD4+ T cells were seeded at 200,000 cells/well in 96-well round bottom plates. E. coli-produced human IL-33, either beginning at amino acid 95, as described above, or using IL-33 (112–270) (R&D Systems) was either titrated as a dose-response or added to a final concentration of 10ng/mL along with human IL-12 and human IL-2, each at a final concentration of 10ng/mL. For ST2 blocking, anti-huST2 IgG2 or IgG2 control was added to a final concentration of 25 ug/mL 30 minutes prior to addition of cytokines. Cultures were incubated for 72hrs at 37°C in a 5% CO2 incubator. Cell-free supernatants were collected and IFN-γ was quantitated by ELISA (R&D Systems).
Mouse eosinophil enumeration
Blood was collected from 10–12 week old wild-type and il33-deficient Bl/6 under isoflurane anesthesia and analyzed on a Siemens Advia 120 hematology analyzer with mouse-specific software. Blood smears were air-dried and stained with Wright-Giemsa. Automated differentials generated on the Advia 120 were confirmed by slide evaluation for any sample with eosinophil counts greater than 4% or with evidence of platelet clumps on the Advia scatterplots or on the blood smear.
URLs
Predicted loss-of-function variants in IL33 and their genotype counts in the Exome Aggregation Consortium (ExAC) database were retrieved from http://exac.broadinstitute.org/gene/ENSG00000137033, accessed November 24, 2015.
LD Score database (accessed 23 June 2015), ftp://atguftp.mgh.harvard.edu/brendan/1k_eur_r2_hm3snps_se_weights.RDS
Supporting Information
Zdroje
1. Wood IS, Wang B, Trayhurn P. IL-33, a recently identified interleukin-1 gene family member, is expressed in human adipocytes. Biochemical and biophysical research communications. 2009;384(1):105–9. Epub 2009/04/28. doi: 10.1016/j.bbrc.2009.04.081 19393621
2. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–60. Epub 2015/05/09. doi: 10.1126/science.1262110 25954001
3. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(1):282–7. Epub 2006/12/23. doi: 10.1073/pnas.0606854104 17185418
4. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–90. Epub 2005/11/16. doi: 10.1016/j.immuni.2005.09.015 16286016
5. Makrinioti H, Toussaint M, Jackson DJ, Walton RP, Johnston SL. Role of interleukin 33 in respiratory allergy and asthma. The Lancet Respiratory medicine. 2014;2(3):226–37. Epub 2014/03/14. doi: 10.1016/S2213-2600(13)70261-3 24621684
6. Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. The Journal of allergy and clinical immunology. 2008;121(6):1484–90. Epub 2008/06/10. doi: 10.1016/j.jaci.2008.04.005 18539196
7. Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol. 2010;185(6):3472–80. Epub 2010/08/10. doi: 10.4049/jimmunol.1000730 20693421
8. Lee HY, Rhee CK, Kang JY, Byun JH, Choi JY, Kim SJ, et al. Blockade of IL-33/ST2 ameliorates airway inflammation in a murine model of allergic asthma. Experimental lung research. 2014;40(2):66–76. Epub 2014/01/23. doi: 10.3109/01902148.2013.870261 24446582
9. Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir GM, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nature genetics. 2009;41(3):342–7. Epub 2009/02/10. doi: 10.1038/ng.323 19198610
10. Grotenboer NS, Ketelaar ME, Koppelman GH, Nawijn MC. Decoding asthma: translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. The Journal of allergy and clinical immunology. 2013;131(3):856–65. Epub 2013/02/06. doi: 10.1016/j.jaci.2012.11.028 23380221
11. Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nature genetics. 2011;43(9):887–92. Epub 2011/08/02. doi: 10.1038/ng.888 21804549
12. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. The New England journal of medicine. 2010;363(13):1211–21. Epub 2010/09/24. doi: 10.1056/NEJMoa0906312 20860503
13. Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nature genetics. 2014;46(1):51–5. Epub 2013/11/19. doi: 10.1038/ng.2830 24241537
14. Brusselle GG, Maes T, Bracke KR. Eosinophils in the spotlight: Eosinophilic airway inflammation in nonallergic asthma. Nature medicine. 2013;19(8):977–9. Epub 2013/08/08. doi: 10.1038/nm.3300 23921745
15. Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-scale whole-genome sequencing of the Icelandic population. Nature genetics. 2015;47(5):435–44. Epub 2015/03/26. doi: 10.1038/ng.3247 25807286
16. Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A, et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature. 2013;497(7450):517–20. Epub 2013/05/07. doi: 10.1038/nature12124 23644456
17. Sveinbjornsson G, Albrechtsen A, Zink F, Gudjonsson SA, Oddson A, Masson G, et al. Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nature genetics. 2016;48(3):314–7. Epub 2016/02/09. doi: 10.1038/ng.3507 26854916
18. Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, et al. The Sequence Ontology: a tool for the unification of genome annotations. Genome biology. 2005;6(5):R44. Epub 2005/05/17. doi: 10.1186/gb-2005-6-5-r44 15892872
19. McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70. Epub 2010/06/22. doi: 10.1093/bioinformatics/btq330 20562413
20. Gretarsdottir S, Helgason H, Helgadottir A, Sigurdsson A, Thorleifsson G, Magnusdottir A, et al. A Splice Region Variant in LDLR Lowers Non-high Density Lipoprotein Cholesterol and Protects against Coronary Artery Disease. PLoS genetics. 2015;11(9):e1005379. Epub 2015/09/04. doi: 10.1371/journal.pgen.1005379 26327206
21. Jongepier H, Koppelman GH, Nolte IM, Bruinenberg M, Bleecker ER, Meyers DA, et al. Polymorphisms in SPINK5 are not associated with asthma in a Dutch population. The Journal of allergy and clinical immunology. 2005;115(3):486–92. Epub 2005/03/09. doi: 10.1016/j.jaci.2004.12.013 15753894
22. Koppelman GH, Stine OC, Xu J, Howard TD, Zheng SL, Kauffman HF, et al. Genome-wide search for atopy susceptibility genes in Dutch families with asthma. The Journal of allergy and clinical immunology. 2002;109(3):498–506. Epub 2002/03/19. 11897998
23. Postma DS, Meyers DA, Jongepier H, Howard TD, Koppelman GH, Bleecker ER. Genomewide screen for pulmonary function in 200 families ascertained for asthma. American journal of respiratory and critical care medicine. 2005;172(4):446–52. Epub 2005/05/20. doi: 10.1164/rccm.200407-864OC 15901612
24. Heinzmann A, Jerkic SP, Ganter K, Kurz T, Blattmann S, Schuchmann L, et al. Association study of the IL13 variant Arg110Gln in atopic diseases and juvenile idiopathic arthritis. The Journal of allergy and clinical immunology. 2003;112(4):735–9. Epub 2003/10/18. doi: 10.1016/S0091 14564352
25. Hansen JW, Thomsen SF, Nolte H, Backer V. Rhinitis: a complication to asthma. Allergy. 2010;65(7):883–8. Epub 2010/02/04. doi: 10.1111/j.1398-9995.2009.02290.x 20121767
26. Hansen JW, Thomsen SF, Porsbjerg C, Rasmussen LM, Harmsen L, Johansen JS, et al. YKL-40 and genetic status of CHI3L1 in a large group of asthmatics. European clinical respiratory journal. 2015;2 : 25117. Epub 2015/12/18. doi: 10.3402/ecrj.v2.25117 26672955
27. Ho JE, Chen WY, Chen MH, Larson MG, McCabe EL, Cheng S, et al. Common genetic variation at the IL1RL1 locus regulates IL-33/ST2 signaling. The Journal of clinical investigation. 2013;123(10):4208–18. Epub 2013/09/04. doi: 10.1172/JCI67119 23999434
28. Miller JN, Pearce DA. Nonsense-mediated decay in genetic disease: friend or foe? Mutation research Reviews in mutation research. 2014;762 : 52–64. Epub 2014/12/09. doi: 10.1016/j.mrrev.2014.05.001 25485595
29. Lingel A, Weiss TM, Niebuhr M, Pan B, Appleton BA, Wiesmann C, et al. Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors—insight into heterotrimeric IL-1 signaling complexes. Structure. 2009;17(10):1398–410. Epub 2009/10/20. doi: 10.1016/j.str.2009.08.009 19836339
30. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(5):1673–8. Epub 2012/02/07. doi: 10.1073/pnas.1115884109 22307629
31. Palmer G, Lipsky BP, Smithgall MD, Meininger D, Siu S, Talabot-Ayer D, et al. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine. 2008;42(3):358–64. Epub 2008/05/03. doi: 10.1016/j.cyto.2008.03.008 18450470
32. Wu J, Carlock C, Zhou C, Nakae S, Hicks J, Adams HP, et al. IL-33 is required for disposal of unnecessary cells during ovarian atresia through regulation of autophagy and macrophage migration. J Immunol. 2015;194(5):2140–7. Epub 2015/01/27. doi: 10.4049/jimmunol.1402503 25617473
33. Sulem P, Helgason H, Oddson A, Stefansson H, Gudjonsson SA, Zink F, et al. Identification of a large set of rare complete human knockouts. Nature genetics. 2015;47(5):448–52. Epub 2015/03/26. doi: 10.1038/ng.3243 25807282
34. Luo H, Higuchi K, Matsumoto K, Mori M. An interleukin-33 gene polymorphism is a modifier for eosinophilia in rats. Genes and immunity. 2013;14(3):192–7. Epub 2013/03/01. doi: 10.1038/gene.2013.7 23446743
35. Torgerson DG, Capurso D, Mathias RA, Graves PE, Hernandez RD, Beaty TH, et al. Resequencing candidate genes implicates rare variants in asthma susceptibility. American journal of human genetics. 2012;90(2):273–81. Epub 2012/02/14. doi: 10.1016/j.ajhg.2012.01.008 22325360
36. Morita H, Arae K, Ohno T, Kajiwara N, Oboki K, Matsuda A, et al. ST2 requires Th2-, but not Th17-, type airway inflammation in epicutaneously antigen - sensitized mice. Allergology international: official journal of the Japanese Society of Allergology. 2012;61(2):265–73. Epub 2012/03/01.
37. Zoltowska AM, Lei Y, Fuchs B, Rask C, Adner M, Nilsson GP. The interleukin-33 receptor ST2 is important for the development of peripheral airway hyperresponsiveness and inflammation in a house dust mite mouse model of asthma. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2016;46(3):479–90. Epub 2015/11/27.
38. Akhabir L, Berube JC, Bosse Y, Laviolette M, Hao K, Nickle DC, et al. Lung expression quantitative trait loci data set identifies important functional polymorphisms in the asthma-associated IL1RL1 region. The Journal of allergy and clinical immunology. 2014;134(3):729–31. Epub 2014/04/22. doi: 10.1016/j.jaci.2014.02.039 24746754
39. Savenije OE, Kerkhof M, Reijmerink NE, Brunekreef B, de Jongste JC, Smit HA, et al. Interleukin-1 receptor-like 1 polymorphisms are associated with serum IL1RL1-a, eosinophils, and asthma in childhood. The Journal of allergy and clinical immunology. 2011;127(3):750–6 e1–5. Epub 2011/02/02. doi: 10.1016/j.jaci.2010.12.014 21281963
40. Halapi E, Gudbjartsson DF, Jonsdottir GM, Bjornsdottir US, Thorleifsson G, Helgadottir H, et al. A sequence variant on 17q21 is associated with age at onset and severity of asthma. European journal of human genetics: EJHG. 2010;18(8):902–8. Epub 2010/04/08. doi: 10.1038/ejhg.2010.38 20372189
41. Lynge E, Sandegaard JL, Rebolj M. The Danish National Patient Register. Scandinavian journal of public health. 2011;39(7 Suppl):30–3. Epub 2011/08/04. doi: 10.1177/1403494811401482 21775347
42. van Diemen CC, Postma DS, Vonk JM, Bruinenberg M, Schouten JP, Boezen HM. A disintegrin and metalloprotease 33 polymorphisms and lung function decline in the general population. American journal of respiratory and critical care medicine. 2005;172(3):329–33. Epub 2005/05/10. doi: 10.1164/rccm.200411-1486OC 15879414
43. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20(9):1297–303. Epub 2010/07/21. doi: 10.1101/gr.107524.110 20644199
44. Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, et al. Parental origin of sequence variants associated with complex diseases. Nature. 2009;462(7275):868–74. Epub 2009/12/18. doi: 10.1038/nature08625 20016592
45. Kutyavin IV, Milesi D, Belousov Y, Podyminogin M, Vorobiev A, Gorn V, et al. A novel endonuclease IV post-PCR genotyping system. Nucleic acids research. 2006;34(19):e128. Epub 2006/10/03. doi: 10.1093/nar/gkl679 17012270
46. Loisel DA, Du G, Ahluwalia TS, Tisler CJ, Evans MD, Myers RA, et al. Genetic associations with viral respiratory illnesses and asthma control in children. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2016;46(1):112–24. Epub 2015/09/25.
47. Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature genetics. 2015;47(3):291–5. Epub 2015/02/03. doi: 10.1038/ng.3211 25642630
48. Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452(7186):423–8. Epub 2008/03/18. doi: 10.1038/nature06758 18344981
49. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7(3):562–78. Epub 2012/03/03. doi: 10.1038/nprot.2012.016 22383036
50. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28(5):511–5. Epub 2010/05/04. doi: 10.1038/nbt.1621 20436464
51. Stoops J, Byrd S, Hasegawa H. Russell body inducing threshold depends on the variable domain sequences of individual human IgG clones and the cellular protein homeostasis. Biochimica et biophysica acta. 2012;1823(10):1643–57. Epub 2012/06/26. doi: 10.1016/j.bbamcr.2012.06.015 22728328
52. Johnsson B, Lofas S, Lindquist G. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Analytical biochemistry. 1991;198(2):268–77. Epub 1991/11/01. 1724720
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2017 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- CUZD1 is a critical mediator of the JAK/STAT5 signaling pathway that controls mammary gland development during pregnancy
- A rare loss-of-function mutation reduces blood eosinophil counts and protects from asthma
- A variant in the gene in a dog with ichthyosis
- Fishing for adaptive epistasis using mitonuclear interactions
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy