
Intracellular Zn(II) Intoxication Leads to Dysregulation of the PerR Regulon Resulting in Heme Toxicity in
Zinc (Zn(II)) is often considered to be a “first among equals” in metal ion homeostasis. Zn(II) is critically important to the proper function of many cellular processes, yet is toxic at high levels. The molecular basis for Zn(II) intoxication is poorly understood. Using a forward genetic approach in B. subtilis, we demonstrate that elevated levels of external Zn(II) inhibit the electron transport chain, whereas intracellular Zn(II) intoxication is due to dysregulation of heme biosynthesis. Since the host immune system utilizes both Zn(II) sequestration and toxicity as a means of responding to pathogens, these findings contribute to our understanding of host-microbe interactions.
Published in the journal:
. PLoS Genet 12(12): e32767. doi:10.1371/journal.pgen.1006515
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1006515
Summary
Zinc (Zn(II)) is often considered to be a “first among equals” in metal ion homeostasis. Zn(II) is critically important to the proper function of many cellular processes, yet is toxic at high levels. The molecular basis for Zn(II) intoxication is poorly understood. Using a forward genetic approach in B. subtilis, we demonstrate that elevated levels of external Zn(II) inhibit the electron transport chain, whereas intracellular Zn(II) intoxication is due to dysregulation of heme biosynthesis. Since the host immune system utilizes both Zn(II) sequestration and toxicity as a means of responding to pathogens, these findings contribute to our understanding of host-microbe interactions.
Introduction
Approximately 30% of proteins require a metal cofactor. Unlike iron (Fe(II)), which can generate cell damaging hydroxyl radicals in the presence of hydrogen peroxide (Fenton reaction), Zn(II) is not redox reactive. As a result, Zn(II) is favored as a structural cofactor that facilitates folding of a large number of proteins, and is also widely used as a Lewis acid catalyst. Total cellular Zn(II) (the Zn(II) quota) must be high enough to perform these essential roles. Yet, since Zn(II) binds much more avidly to common protein ligands than Fe(II) or Mn(II) (an observation codified in the Irving-Williams series; [1]) excess Zn(II) may result in mismetallation of proteins requiring these other metals. Thus, cellular Zn(II) is highly regulated at multiple levels: in Bacillus subtilis the total intracellular concentration at equilibrium is ~0.8 mM, and much of this is sequestered in metalloproteins. A subset of intracellular Zn(II) comprises a labile pool which buffers the thermodynamically free Zn(II) concentration in the picomolar range (< 1 Zn(II) per cell) [2,3], thereby ensuring that only physiologically relevant Zn(II) metalloproteins are normally metallated by Zn(II).
The narrow range of free Zn(II) in Bacillus subtilis is set by the transcription repressors Zur, the sensor of Zn(II) limitation, and CzrA, the sensor of Zn(II) excess [4–6]. B. subtilis contains one high affinity uptake system (znuABC) and two efflux systems (cadA and czcD). Under conditions of Zn(II) sufficiency, Zn(II)-binds to Zur [7], which represses transcription of the Zn(II) uptake systems. Upon Zn(II) deficiency, transcription is depressed and Zn(II) is imported into the cell [4]. When Zn(II) is in excess, CzrA binds Zn(II) and is inactivated [8], leading to depression of CadA and CzcD and Zn(II) efflux [5]. These metalloregulators sense the labile Zn(II) pool consisting of Zn(II) bound to small molecules, proteins and other macromolecules in a kinetically accessible form. In B. subtilis and related low G+C Firmicutes, the abundant LMW thiol, bacillithiol (BSH), serves as a major buffer of the labile Zn(II) pool [3]. These buffering systems maintain labile Zn(II) concentrations high enough for metallation of Zn(II) containing proteins, but low enough to reduce mismetallation.
The specific targets of zinc intoxication are not well defined. In this study, we take advantage of the well characterized Zn(II) homeostasis mechanisms in the model Gram-positive bacterium, B. subtilis, and use a forward genetic approach to investigate the underlying causes of Zn(II) intoxication. Our results suggest that in wild type cells, which are competent for export of Zn(II) from the cytosol, Zn(II) intoxication results from inactivation of the electron transport chain due to inhibition of the major aerobic cytochrome aa3 oxidase. Zn(II) resistant suppressors arise that either reduce access of Zn(II) to the cell surface or increase expression of the alternative anaerobic cytochrome bd oxidase due to inactivation of Rex, a NAD+/NADH sensing transcription factor. Conversely, in a Zn(II) efflux deficient mutant (cadA czcD), Zn(II) intoxication results from mismetallation of cytosolic proteins. Here, we identify heme accumulation as a major consequence of intracellular Zn(II) intoxication, which in turn results from mismetallation and consequent dysregulation of the PerR regulon.
Results
Isolation of Zn(II) resistant suppressors in wild-type B. subtilis
To identify potential targets of Zn(II) toxicity and genes involved in Zn(II) resistance, we isolated Zn(II) resistant mutants. A mariner transposon library was generated in wild-type cells and plated on a Petri plate containing LB medium and a continuous Zn(II) gradient (0–5 mM). Colonies able to grow in the highest Zn(II) concentrations were isolated and the location of the transposon insertion was identified. We isolated multiple independent transposon insertions in rex, ykuI, and the fla-che operon (Table 1). We backcrossed the transposon insertions into the parental strain by chromosomal DNA transformation. These reconstructed strains, as well as targeted gene deletions, phenocopied the originally isolated Zn(II) resistant transposon mutants, suggesting that the observed Zn(II) resistance is linked to the transposon insertion rather than a second site mutation.

YkuI is a c-di-GMP binding protein [9] known to affect production of extracellular matrix (ECM) in B. cereus [10]. The fla-che operon contains genes encoding components of the flagella and chemotaxis machinery, as well as the alternative sigma factor, σD [11]. ECM production is inversely controlled with respect to flagellar motility in B. subtilis [12,13]. We therefore hypothesized that the ykuI and fla-che disruptions prevent Zn(II) intoxication by increasing production of ECM which can prevent access of Zn(II) to the cell, rather than by altering a target of mismetallation. In contrast, Rex is a regulator of anaerobic metabolism and is not known to affect ECM production.
To test whether the ykuI and the fla-che transposon mutants serve to restrict access of Zn(II) to the cell, we monitored intracellular Zn(II) levels after Zn(II) shock in each of the isolated suppressors (S1 Fig). We reasoned that mutations that restrict access of Zn(II) to the cell, and thereby reduce uptake, would not accumulate Zn(II). Conversely, those that allow the cell to circumvent metabolic pathways intoxicated by Zn(II) would still accumulate Zn(II) upon Zn(II) shock. In contrast with wild-type, strains with transposon insertions in ykuI and the fla-che operon did not accumulate intracellular Zn(II) upon shock, whereas those with insertions in rex did (S1 Fig). These results support the notion that ykuI and fla-che operon insertions restrict access of Zn(II) to the cell, presumably by increasing ECM production. Since our goal in this study is to define mechanisms of Zn(II) intoxication, we focus here on the role of rex in Zn(II) resistance.
Derepression of cydAB is critical for Zn(II) resistance in wild-type B. subtilis
Rex is a DNA-binding transcription repressor that senses the intracellular NAD/NADH+ ratio and regulates genes involved in growth under anaerobic conditions in many Gram positive bacteria such as S. coelicolor and B. subtilis, including the cytochrome bd terminal oxidase (cydABCD), lactate dehydrogenase (lctP-ldh), and a putative nitrate transporter (ywcJ) [14–16]. Since a transposon insertion in rex leads to increased Zn(II) resistance, we hypothesized that derepression of one or more members of the Rex regulon contributes to Zn(II) resistance.
We constructed mutants in which each Rex-regulated gene was individually deleted in a wild-type or Δrex background. Deletion of cydABCD resulted in a Zn(II) sensitive phenotype in a wild-type background (Fig 1A), while there was no Zn(II) phenotype associated with deletion of any other member of the Rex regulon. Additionally, deletion of cydABCD in a Δrex background completely reversed the Zn(II) resistance phenotype of the Δrex mutant, consistent with the idea that derepression of cydABCD confers Zn(II) resistance (Fig 1B). Furthermore, expression of the Rex regulon is derepressed under conditions of Zn(II) intoxication as measured by qRTPCR of cydA and ldh expression (Fig 2).


B. subtilis encodes three terminal oxidases, cytochrome caa3, aa3, and bd [17]. Cytochrome caa3 and aa3 are heme-copper oxidases, whereas the relatively less efficient cytochrome bd oxidase does not utilize copper. The major cytochrome oxidase used during exponential growth is cytochrome aa3 [18]. Interestingly, expression of either cytochrome aa3 or cytochrome bd is required for viability [18]. Thus, during Zn(II) intoxication, the expression of the relatively Zn(II) insensitive cytochrome bd terminal oxidase may be required since the major aerobic system, cytochrome aa3, is inhibited by Zn(II) (Fig 1C). This is consistent with prior findings in Escherichia coli and Streptomyces coelicolor that suggest a similar extracellular target of Zn(II) intoxication [14,19]. Since the ability of Zn(II) to inhibit cytochrome oxidases is well established, we next decided to repeat our selection of Zn(II) resistant mutants using an efflux deficient strain in which Zn(II) intoxication presumably occurs by mismetallation of cytosolic targets.
Isolation of Zn(II) resistant suppressors in a Zn(II) efflux deficient mutant
We selected spontaneous Zn(II) resistant mutants in a Zn(II) efflux mutant background that lacks the genes encoding the CadA and CzcD Zn(II) efflux pumps. Since the cadA czcD mutant displays Zn(II) toxicity at concentrations well below the MIC for wild-type, we reasoned that suppressors isolated from this background would reveal intracellular targets of Zn(II) intoxication. Using whole-genome resequencing, seven independently isolated strains were found to contain nonsense or frameshift mutations in aroB and aroC (Table 1). No additional mutations were identified in the suppressed strains, suggesting that the Zn(II) resistant phenotype is linked to the inactivation of aroB or aroC. Consistent with this prediction, strains in which aroB or aroC were inactivated by insertion of an antibiotic resistant cassette phenocopied the evolved Zn(II) resistant mutants (S2 Fig).
Interestingly, aroB and aroC were not recovered as suppressors of Zn(II) intoxication in the wild-type background (Table 1). This indicates the presence of distinct extracellular and intracellular targets of Zn(II) intoxication. Indeed, each suppressor mutation conferred Zn(II) resistance only in the genetic background in which it was isolated (Fig 3). For example, deletion of rex (isolated in wild-type) did not confer Zn(II) resistance to the Zn(II) efflux mutant, and mutation of aroB (isolated in the Zn(II) efflux mutant background) did not confer resistance to wild-type (Fig 3).

The aroB and aroC genes are involved in the biosynthesis of chorismate, a precursor for aromatic amino acid biosynthesis. Since our selection was performed on rich media, it is unlikely that the cells are limited for amino acids. However, chorismate is also a precursor for menaquinone biosynthesis [20]. We therefore hypothesized that Zn(II) resistance may have resulted from a decrease in cellular pools of menaquinone. In B. subtilis, null mutations affecting late steps in menaquinone biosynthesis fail to form colonies on LB medium [21], whereas the aroB and aroC null mutants grow well. This suggests that LB medium provides a precursor (perhaps chorismate) that can be used to maintain some level of menaquinone synthesis, and likely accounts for the failure to recover insertions affecting later steps in this pathway.
Transposon insertions in aroB and aroC, as well as other menaquinone biosynthesis genes, were previously identified in S. aureus in a selection for heme resistance [22]. The authors suggested a model in which heme toxicity results when superoxide is generated by the redox cycling of membrane-associated heme and reduced quinone molecules [22]. We therefore hypothesized that Zn(II) intoxication in B. subtilis may also be related to menaquinone and perhaps to heme.
Zn(II) intoxication requires both menaquinone and heme
To determine if the relevant effect of the aroB and aroC mutations is a reduction of menaquinone levels, we measured Zn(II) sensitivity in media supplemented with menaquinone or its precursor 1,4-dihydroxy-2-napthoate (DHNA). We observed that external menaquinone or DHNA supplementation reverses the Zn(II) resistance phenotype of the cadA czcD aroB mutant. However, menaquinone or DHNA supplementation does not affect a cadA czcD hemA mutant, which is defective in heme biosynthesis (Fig 4). Overall, these data support the hypothesis that an aroB mutation leads to Zn(II) resistance by reducing cellular menaquinone levels.

If the role of menaquinone is to act as an electron donor to membrane-localized heme, as proposed for S. aureus [22], we reasoned that a loss of heme synthesis would also lead to Zn(II) resistance. Indeed, a cadA czcD hemA mutant is as Zn(II) resistant as a cadA czcD aroB mutant (Fig 4). This strain forms small colonies and is slow growing, thus it not surprising that mutations affecting heme biosynthesis were not isolated in our initial Zn(II) resistance selection. Importantly, the effects of the aroB and hemA mutations are not additive (compare the czcD cadA aroB hemA quadruple mutant with the triple mutants), and this epistasis implies that they act in the same genetic pathway.
A Zn(II) efflux mutant accumulates heme under conditions of Zn(II) excess
To determine if heme accumulates under conditions of Zn(II) intoxication, we used a fluorescence based assay to monitor heme levels. The level of heme increases more than two-fold in the cadA czcD mutant background, but not in wild-type (Fig 5A). Although B. subtilis can utilize heme as an Fe(II) source, it is not known to encode heme uptake or efflux systems. However, B. subtilis does encode two heme monooxygenases, HmoA and HmoB, that bind and degrade heme in vitro, although a physiological role for these proteins has not yet been demonstrated [23]. HmoB belongs to the well-characterized IsdG family and is not regulated by Fe(II), whereas the Fur-regulated HmoA protein represents a poorly characterized subgroup of monooxygenases found in several pathogenic bacteria [23,24]. Since Zn(II) intoxication leads to heme accumulation, we reasoned that HmoA and HmoB may contribute to Zn(II) tolerance. We tested the Zn(II) sensitivity of cadA czcD mutant strains where hmoA and hmoB were deleted individually or in combination. While deletion of either gene does not have a significant effect, simultaneous deletion of both heme monoxygenases reveals an increased Zn(II) sensitivity in a cadA czcD mutant background (Fig 5B). Interestingly, expression of either hmoA or hmoB from an inducible promoter is able to complement Zn(II) sensitivity of the cadA czcD hmoA hmoB mutant. These results suggest that HmoA and HmoB are both active and that they are functionally redundant in vivo. Collectively, these data support the hypothesis that intracellular Zn(II) intoxication is due to the toxic redox cycling of excess intracellular heme, as suggested previously in S. aureus [22]. Next, we sought to find a link between elevated intracellular Zn(II) levels and the observed increase in intracellular heme.

The PerR regulon is dysregulated under conditions of Zn(II) excess
The heme biosynthesis genes are encoded by the hemAXCDBL operon. This operon is repressed by PerR, a metal-cofactored, DNA-binding repressor that serves as a sensor of peroxide stress [25]. In addition, PerR also regulates the expression of catalase (katA)[26], the major vegetative catalase in B. subtilis and an abundant heme binding protein. Coordinate regulation of catalase and heme biosynthesis by PerR ensures sufficient heme availability for catalase function under conditions of oxidative stress.
PerR contains both a structural Zn(II) binding site and a regulatory metal binding site. PerR represses transcription when its regulatory site is associated with either Mn(II) or Fe(II), but only the Fe(II) bound form responds to H2O2 [27–29]. Since PerR requires a bound regulatory metal ion in order to bind DNA, it also senses conditions of Fe(II) and Mn(II) depletion. In vitro data suggest that Zn(II) can also populate the PerR metal sensing site [30], and it may thereby affect the expression of PerR regulated genes. This raises the possibility that under conditions of intracellular Zn(II) intoxication, PerR may become mismetallated with Zn(II), leading to dysregulation of its target genes and to intracellular heme accumulation.
To investigate this hypothesis, we monitored expression of the hemA and katA genes by qRT-PCR. Upon Zn(II) intoxication, hemA mRNA levels increased whereas katA mRNA levels decreased in the cadA czcD mutant, whereas they were relatively unaffected in a wild-type background (Fig 2). Additionally, catalase activity is also decreased under these same conditions (Fig 5C). This discoordinate regulation contrasts sharply with the documented effects of Fe(II) and Mn(II), which both lead to coordinate regulation of these two operons to allow for sufficient heme production to support catalase activity [31]. These data suggest that the PerR regulon, particularly genes involved in heme biosynthesis and usage, is dysregulated upon Zn(II) intoxication.
To test if this effect is due to direct mismetallation of PerR with Zn(II), we used a fluorescence anisotropy assay to monitor DNA-binding activity. Although PerR binds DNA when bound with either Mn(II) or Fe(II), we routinely use Mn(II) to allow measurement of DNA-binding activity under aerobic conditions [32,33]. As expected, Mn(II)-cofactored PerR binds tightly to 6-carboxyfluorescein-labeled DNA fragments containing binding sites for PerR as judged by an increase in the fluorescence anisotropy signal [34]. Here, we used DNA containing the known PerR operators for the hemA operon (encoding heme biosynthesis function), katA (encoding the major vegetative catalase), and mrgA (encoding a mini-ferritin that sequesters Fe(II) under oxidative stress conditions). The mrgA operator is the first identified [35] and the best characterized Per box [36]. Upon titration with Zn(II), a decrease in anisotropy was observed indicative of PerR dissociation from the hemA and mrgA operator sites (Fig 6A). Interestingly, addition of Zn(II) did not lead to full dissociation of PerR from the katA operator site. Collectively, these data suggest that elevated intracellular Zn(II) can lead to the mismetallation of the Fe(II)/Mn(II) sensing transcription factor PerR resulting in a loss of coordination in synthesis of heme and catalase, a major heme-containing protein (Fig 6B).
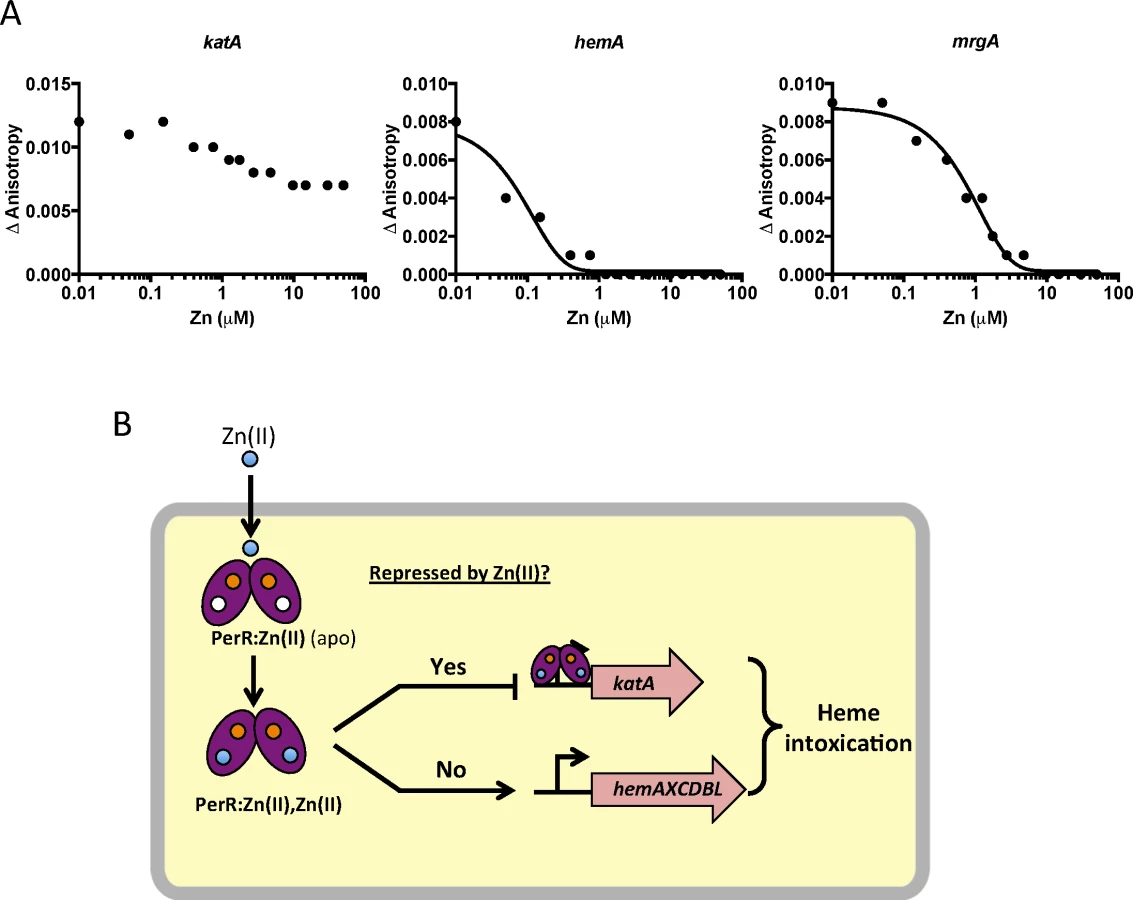
Discussion
Metal ion homeostasis relies on the precise control of metal uptake and the complementary action of metal efflux pumps, which prevent intracellular intoxication. Exposure to excess Zn(II), either environmentally or imposed by the innate immune system during infection, can result in growth inhibition [37]. Because of the high efficiency of efflux, Zn(II) often inhibits bacterial growth by binding to extracytoplasmic targets. Two major extracellular targets have been identified previously. In S. pneumoniae, Zn(II) exerts its toxic effects by preventing the uptake of the essential metal Mn(II) by binding to the Mn(II) solute binding protein, PsaA [38], which delivers Mn(II) to the specific PsaBCD ATP binding cassette (ABC) transporter [39]. Inhibition of PsaA by Zn(II) leaves S. pneumoniae more sensitive to oxidative stress and more susceptible to killing by the host immune response. Previous studies, and now this work, suggest that a major extracellular target of Zn(II) toxicity in bacteria and mammalian mitochondria is the major aerobic cytochrome oxidase [19,40]. Cytochrome oxidases are key enzymes in aerobic respiration, responsible for establishment of the proton gradient required for ATP synthesis. Measurement of Zn(II) inhibition of the Rhodobacter sphaeroides and mitochondrial cytochrome c oxidase indicates an extracellular Zn(II) binding site with Ki of 2–5 μM [1,41–43].
We and others have shown that the expression of the alternate anaerobic cytochrome oxidase (cydBD) is upregulated under conditions where the major cytochrome oxidase is inhibited by anaerobic conditions [2,14], excess Zn(II) [4,5,14], or excess sulfide [4,44]. Additionally, expression of the cydBD cytochrome oxidase is important for survival within a host for S. aureus [5,45], M. tuberculosis [46] and E. coli [47]. Proteomic studies of S. aureus cells internalized by macrophages reveal an increased level of the anaerobic cydBD cytochrome oxidase [48], suggestive of an important role in survival in the host. It is notable that in the case of Zn(II), mutation of rex and derepression of the cydBD cytochrome oxidase significantly increased Zn(II) tolerance as judged by a zone-of-inhibition assay (Fig 1). Since excess Zn(II) leads to induction of the Rex regulon (Fig 2), we infer that induction of the cydBD operon in the presence of Zn(II) does not confer a similar level of Zn(II) tolerance. This suggests that assembly of the CydBD system may be impaired in the presence of excess Zn(II), and what would be an appropriate adaptive response to inhibition of the major aerobic oxidase(s) is, in this case, inadequate.
Bacteria form biofilms and increase the production of extracellular matrix (ECM) in response to a variety of environmental stresses [49]. In both Xylella fastidiosa and E. coli toxic levels of Zn(II) trigger increased ECM production [50,51]. Here, we isolated transposon insertions in ykuI and the fla-che operon that likely had a similar effect. YkuI is a c-di-GMP binding protein previously implicated in regulation of ECM [9], and mutations affecting flagellar motility have been shown to increase poly-γ-glutamate, a component of the ECM, in B. subtilis [12,13]. It remains to be seen if biofilm formation and increased ECM synthesis is be a normal physiological response to Zn(II) stress in B. subtilis.
The importance of intracellular Zn(II) homeostasis suggests the presence of an intracellular target for Zn(II) toxicity. In Group A Streptococcus, key glycolytic enzymes are mismetallated by Zn(II) and production of capsule polysaccharides is inhibited [52]. In E. coli, intracellular Zn(II) toxicity under conditions of oxidative stress results from mismetallation of the iron sulfur clusters of dehydratases, which are critical to key metabolic processes [53,54].
We sought to identify additional proteins that could be mismetallated by Zn(II) and the underlying mechanisms of Zn(II) intoxication in B. subtilis. By isolation of suppressors of zinc toxicity in a Zn(II) efflux mutant, we identified PerR as a target of mismetallation by Zn(II). PerR has been implicated in Zn(II) homeostasis in S. pneumoniae since it regulates expression of the P1B4-type ATPase encoded by pmtA [55]. In a perR null mutant, overexpression of PmtA leads to induction of a Zn(II) starvation response, implying that this transporter may export Zn(II). However, PmtA is not known to be induced by Zn(II) stress, and its physiological role is most likely related to peroxide resistance (as evidenced by its regulation by PerR). Indeed, recent studies of PmtA orthologs in B. subtilis [56], L. monocytogenes [57], and M. tuberculosis [58] (PfeT, FrvA, and CtpD, respectively) suggest that the primary substrate of these P1B4-type ATPases is Fe(II), and this is also a likely role for PmtA consistent with the key role this protein plays in peroxide resistance [55].
It is presently unclear why Zn(II) inhibits binding of PerR to some operons, but not others. Prior work suggests that many Fur family proteins may utilize the cooperative binding to effect repression [32,59]. For example, Corynebacterium diptheriae DtxR can bind DNA cooperatively as a tetramer [60], and a similar model has been proposed for E. coli Zur [61]. We note that Zn(II) leads to complete dissociation of PerR from the hemA operon regulatory site, but not from katA (Fig 6). The regulatory region of hemA contains multiple PerR binding sites [28], whereas the katA regulatory region includes one very strong consensus site [26]. Therefore, we can speculate that perhaps Zn(II)-metallated PerR is defective in cooperative binding, but can still bind strong consensus sites as a dimer. Regardless of the molecular details, Zn(II) clearly leads to dysregulation of the PerR regulon and an increase in intracellular heme levels. Our work supports the model of heme toxicity proposed by the Skaar lab which suggests that heme toxicity is caused by the generation of superoxide as membrane associated heme and reduced quinones form a redox cycle [22].
Previous studies support the idea that mismetallation of metalloregulatory proteins can have dire consequences. The Fe(II) sensing transcription factor Fur is known to be mismetallated by Mn(II) under Mn(II) intoxication conditions or when Fur levels increase [62]. Recently, Cd(II) intoxication in S. pneumoniae was linked to dysregulation of Zn(II) homeostasis, resulting from inhibition of Zn(II) uptake gene expression and activation of Zn(II) efflux gene expression [63]. However, direct interaction of Cd(II) with the Zn(II) sensing transcription factor AdcR and SczA has not yet been shown. Additionally, as a shown in Group A Streptococcus, under conditions of Mn(II) intoxication, the Fe(II)/Mn(II) sensing transcription factor PerR is in its Mn(II)-cofactored form [64]. As a result, the PerR regulon is unable to be induced by H202, resulting in increased sensitivity to oxidative stress. Similarly, excess Mn(II) blocks catalase de-repression and prevents the increase in H2O2 tolerance that is typical of the entry of B. subtilis cells into stationary phase [26].
Understanding the mechanisms of Zn(II) intoxication may contribute to the development of novel antibacterial treatments. Only recently has the role of host-mediated Zn(II) toxicity as an antimicrobial mechanism been widely appreciated: regulated Zn(II) trafficking to the phagosome appears to play an active role in antimicrobial responses in several systems. Phagosomal Zn(II) levels rise dramatically upon Mycobacterium tuberculosis [65] or Streptococcus pyogenes [66] infection, and Zn(II) containing vesicles have been observed in macrophages upon Salmonella enterica Typhimurium infection [67]. Furthermore, many pathogenic bacteria require Zn(II) efflux pumps to avoid killing by macrophages [65,66,68]. Such evidence suggests that high levels of Zn(II) may exert a direct bactericidal effect within macrophages.
Methods
Bacterial strains, plasmids, and growth conditions
Strains used in this study are listed in S1 Table. Bacteria were grown in the media described in the following sections. When necessary, antibiotics were used at the following concentrations: chloramphenicol (10 μg ml-1), kanamycin (15 μg ml-1), spectinomycin (100 μg ml-1), and tetracycline (5 μg ml-1). Gene deletions were constructed using long flanking homology PCR as previously described [69]. Chromosomal DNA transformation was performed as described.
Disk diffusion assays
Strains were grown in LB at 37°C with vigorous shaking to an OD600~0.4. A 100 μl aliquot of these cultures was added to 4 ml of LB soft agar (0.7% agar) and poured on to prewarmed LB agar plates. The plates were then allowed to solidify for 10 minutes at room temperature in a laminar flow hood. Filter disks (6 mm) were placed on top of the agar and 10 μl of Zn(II) (50 mM) was added to the disks and allowed to absorb for 10 minutes. The plates were then incubated at 37°C for 16–18 hours. The diameter of the zone of inhibition was measured. The data shown represent the values (diameter of the zone of inhibition minus diameter of the filter disk) and standard deviation of three biological replicates.
Quantification of total Zn(II) quota by ICP-MS
For quantification of the total Zn(II) quota, cells were grown in 5 ml LB medium in the presence or absence of 200 μM ZnCl2 to mid-log phase. For fractionation experiments, 25 ml of cells were grown in presence or absence of 200 μM Zn(II). All samples were prepared as described previously. Briefly, samples were washed once with buffer 1 (1X PBS buffer, 0.1 M EDTA) then twice with buffer 2 (1X chelex-treated PBS buffer). Cell pellets were resuspended in 400 μl buffer 3 (1X chelex-treated PBS buffer, 75 mM NaN3, 1% Triton X-100) and incubated at 37°C for 90 min for cell lysis. Lysed samples were centrifuged and subject to Bradford assay to quantify the total protein content. Then, samples were mixed with 600 μl buffer 4 (5% HNO3, 0.1% (v/v) Triton X-100) and heated in a 95°C sand bath for 30 min. Samples were centrifuged and supernatants were diluted in 1% HNO3. Levels of intracellular Zn were analyzed by Perkin-Elmer ELAN DRC II ICP-MS. Gallium was used as an internal standard. The total concentration of metal ions is expressed as μg ion per gram of protein. The data shown represent the average and standard deviation of three biological replicates.
qRT-PCR
Cells were grown at 37°C in LB medium with rigorous shaking till OD600 ~0.4. 1 ml aliquots were treated with ZnCl for 10 min. Total RNA from both treated and untreated samples were extracted RNeasy Mini Kit following the manufacturer's instructions (Qiagen Sciences, Germantown, MD). RNA samples were then treated with Turbo-DNA free DNase (Ambion) and precipitated with ethanol overnight. RNA samples were re-dissolved in RNase-free water and quantified by NanoDrop spectrophotometer. 2 μg total RNA from each sample was used for cDNA synthesis with TaqMan reverse transcription reagents (Applied Biosystems). qPCR was then carried out using iQ SYBR green supermix in an Applied Biosystems 7300 Real Time PCR System. 23S rRNA was used as an internal control and fold-changes between treated and untreated samples were plotted.
Catalase activity
Cells were grown on LB agar plates overnight. A colony was picked from the fresh plate and placed in a drop of dilute hydrogen peroxide. Strains resulting in the formation of bubbles were determined to possess catalase activity. For quantitative determinations of catalase activity, cells were assayed for the rate of H2O2 decomposition spectrophotometrically (240 nm) as described previously [70].
Overexpression and purification of PerR
pET16b plasmids carrying PerR between the NcoI and BamHI restriction sites were used for overexpression in Escherichia coli. A single colony was inoculated into 10 ml of LB and grown overnight at 37°C. Cells were then diluted into 1 L of LB medium containing 0.5% glucose. 1 mM IPTG was added to the culture when OD600 reached ∼0.6 and cells were harvested after 2 h of induction by centrifugation. After resuspension and sonication in buffer A [20 mM Tris, pH 8.0, 100 mM NaCl, 5% (v/v) glycerol] with 10 mM EDTA (ethylenediaminetetraacetic acid), the supernatant were loaded onto a heparin column pre-equilibrated with the same buffer. Proteins were eluted with a linear gradient of NaCl from 0.1 to 1 M and fractions containing PerR were combined and concentrated to load onto a Superdex 200 size exclusion column. The recovered PerR eluted was further purified using a Mono-Q column with a linear gradient of 0.1–1 M NaCl in buffer A containing 10 mM EDTA. Fractions containing PerR were concentrated and dialyzed extensively against buffer A to remove EDTA. The purified PerR was then aliquoted and frozen at −80°C. The purified PerR was determined to be ∼85% active, respectively, based on the amount of protein required to stoichiometrically bind 100 nM DNA.
Fluorescence anisotropy
A 6-carboxyfluorescein (6-FAM)—labeled DNA was used and fluorescence anisotropy (FA) was measured with λex = 492 nm and λem = 520 nm. For Zn(II) binding, increasing amounts of ZnCl2 were mixed with 120μl buffer A containing 100 nM labeled DNA, 100 nM active PerR dimer, and 10 μM MnCl2. FA measurements of each sample were performed immediately after transferring to a quartz cuvette. The data were fit using a nonlinear regression model with GraphPad Prism.
Fluorescence-based PPIX and heme assays
Strains were grown overnight in LB medium and then subcultured with 1 : 100 ratio in fresh LB medium to an OD600 of 0.4. OD600 was recorded and aliquots of 5 ml of cell culture were harvested. Cell pellets were resuspended in 1 ml 50 mM Tris-HCl buffer (pH 7.4) containing 100 μg ml−1 of lysozyme and were incubated at 37°C for 30 min to lyse the cells. Cell debris was removed by centrifugation. Heme was extracted from 500 μl of the clear lysates using 500 μl acidic acetone (20% (v/v) 1.6 M HCl). Precipitate was removed by centrifugation and the supernatant was analyzed by fluorescence spectroscopy. The fluorescence emission of heme was scanned from 400 nm to 500 nm with excitation at 380 nm as described. Fluorescence intensity at 450 nm (peak) was normalized and plotted.
Supporting Information
Zdroje
1. Irving H, Williams R. 637. The stability of transition-metal complexes. J Chem Soc. (Resumed). 1953 : 3192–3210.
2. Outten CE, O'Halloran TV. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science. 2001;292 : 2488–2492. doi: 10.1126/science.1060331 11397910
3. Ma Z, Chandrangsu P, Helmann TC, Romsang A, Gaballa A, Helmann JD. Bacillithiol is a major buffer of the labile zinc pool in Bacillus subtilis. Mol Microbiol. 2014;94 : 756–770. doi: 10.1111/mmi.12794 25213752
4. Gaballa A, Helmann JD. Identification of a zinc-specific metalloregulatory protein, Zur, controlling zinc transport operons in Bacillus subtilis. J Bacteriol. 1998;180 : 5815–5821. 9811636
5. Moore CM, Gaballa A, Hui M, et al. Genetic and physiological responses of Bacillus subtilis to metal ion stress. Mol Microbiol. 2005;57 : 27–40. doi: 10.1111/j.1365-2958.2005.04642.x 15948947
6. Harvie DR, Andreini C, Cavallaro G, Meng W, Connolly BA, Yoshida K-I, et al. Predicting metals sensed by ArsR-SmtB repressors: allosteric interference by a non-effector metal. Mol Microbiol. 2006;59 : 1341–1356. doi: 10.1111/j.1365-2958.2006.05029.x 16430705
7. Ma Z, Gabriel SE, Helmann JD. Sequential binding and sensing of Zn(II) by Bacillus subtilis Zur. Nucleic Acids Res. 2011;39 : 9130–9138. doi: 10.1093/nar/gkr625 21821657
8. Pennella MA, Shokes JE, Cosper NJ, Scott RA, Giedroc DP. Structural elements of metal selectivity in metal sensor proteins. Proc Natl Acad Sci USA. 2003;100 : 3713–3718. doi: 10.1073/pnas.0636943100 12651949
9. Minasov G, Padavattan S, Shuvalova L, et al. Crystal structures of YkuI and its complex with second messenger cyclic Di-GMP suggest catalytic mechanism of phosphodiester bond cleavage by EAL domains. J Biol Chem. 2009;284 : 13174–13184. doi: 10.1074/jbc.M808221200 19244251
10. Fagerlund A, Smith V, Røhr ÅK, Lindbäck T, Parmer MP, Andersson KK, et al. Cyclic diguanylate regulation of Bacillus cereus group biofilm formation. Mol Microbiol. 2016;101 : 471–494. doi: 10.1111/mmi.13405 27116468
11. Márquez-Magaña LM, Chamberlin MJ. Characterization of the sigD transcription unit of Bacillus subtilis. J Bacteriol. 1994;176 : 2427–2434. 8157612
12. Cairns LS, Marlow VL, Bissett E, Ostrowski A, Stanley-Wall NR. A mechanical signal transmitted by the flagellum controls signalling in Bacillus subtilis. Mol Microbiol. 2013;90 : 6–21. doi: 10.1111/mmi.12342 23888912
13. Chan JM, Guttenplan SB, Kearns DB. Defects in the flagellar motor increase synthesis of poly-γ-glutamate in Bacillus subtilis. J Bacteriol. 2014;196 : 740–753. doi: 10.1128/JB.01217-13 24296669
14. Brekasis D, Paget MSB. A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J. 2003;22 : 4856–4865. doi: 10.1093/emboj/cdg453 12970197
15. Larsson JT, Rogstam A, Wachenfeldt von C. Coordinated patterns of cytochrome bd and lactate dehydrogenase expression in Bacillus subtilis. Microbiology (Reading, Engl). 2005;151 : 3323–3335.
16. Gyan S, Shiohira Y, Sato I, Takeuchi M, Sato T. Regulatory loop between redox sensing of the NADH/NAD(+) ratio by Rex (YdiH) and oxidation of NADH by NADH dehydrogenase Ndh in Bacillus subtilis. J Bacteriol. 2006;188 : 7062–7071. doi: 10.1128/JB.00601-06 17015645
17. Wachenfeldt von C, Hederstedt L. Molecular biology of Bacillus subtilis cytochromes. FEMS Microbiology Letters. 1992;100 : 91–100. 1335950
18. Winstedt L, Wachenfeldt von C. Terminal oxidases of Bacillus subtilis strain 168: one quinol oxidase, cytochrome aa(3) or cytochrome bd, is required for aerobic growth. J Bacteriol. 2000;182 : 6557–6564. 11073895
19. Beard SJ, Hughes MN, Poole RK. Inhibition of the cytochrome bd-terminated NADH oxidase system in Escherichia coli K-12 by divalent metal cations. FEMS Microbiology Letters. 1995;131 : 205–210. 7557331
20. Hoch JA, Nester EW. Gene-enzyme relationships of aromatic acid biosynthesis in Bacillus subtilis. J Bacteriol. 1973;116 : 59–66. 4200844
21. Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci USA. 2003;100 : 4678–4683. doi: 10.1073/pnas.0730515100 12682299
22. Wakeman CA, Hammer ND, Stauff DL, et al. Menaquinone biosynthesis potentiates haem toxicity in Staphylococcus aureus. Mol Microbiol. 2012;86 : 1376–1392. doi: 10.1111/mmi.12063 23043465
23. Gaballa A, Helmann JD. Bacillus subtilis Fur represses one of two paralogous haem-degrading monooxygenases. Microbiology (Reading, Engl). 2011;157 : 3221–3231.
24. Anzaldi LL, Skaar EP. Overcoming the heme paradox: heme toxicity and tolerance in bacterial pathogens. Infect Immun. 2010;78 : 4977–4989. doi: 10.1128/IAI.00613-10 20679437
25. Bsat N, Chen L, Helmann JD. Mutation of the Bacillus subtilis alkyl hydroperoxide reductase (ahpCF) operon reveals compensatory interactions among hydrogen peroxide stress genes. J Bacteriol. 1996;178 : 6579–6586. 8932315
26. Chen L, Keramati L, Helmann JD. Coordinate regulation of Bacillus subtilis peroxide stress genes by hydrogen peroxide and metal ions. Proc Natl Acad Sci USA. 1995;92 : 8190–8194. 7667267
27. Faulkner MJ, Ma Z, Fuangthong M, et al. Derepression of the Bacillus subtilis PerR Peroxide Stress Response Leads to Iron Deficiency. J Bacteriol. 2012;194 : 1226–1235. doi: 10.1128/JB.06566-11 22194458
28. Herbig AF, Helmann JD. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol Microbiol. 2001;41 : 849–859. 11532148
29. Fuangthong M, Herbig AF, Bsat N, et al. Regulation of the Bacillus subtilis fur and perR genes by PerR: not all members of the PerR regulon are peroxide inducible. J Bacteriol. 2002;184 : 3276–3286. doi: 10.1128/JB.184.12.3276-3286.2002 12029044
30. Lee J-W, Helmann JD. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature. 2006;440 : 363–367. doi: 10.1038/nature04537 16541078
31. Helmann JD. Specificity of metal sensing: iron and manganese homeostasis in Bacillus subtilis. J Biol Chem. 2014;289 : 28112–28120. doi: 10.1074/jbc.R114.587071 25160631
32. Lee J-W, Helmann JD. Functional specialization within the Fur family of metalloregulators. Biometals. 2007;20 : 485–499. doi: 10.1007/s10534-006-9070-7 17216355
33. Ma Z, Lee J-W, Helmann JD. Identification of altered function alleles that affect Bacillus subtilis PerR metal ion selectivity. Nucleic Acids Res. 2011;39 : 5036–5044. doi: 10.1093/nar/gkr095 21398634
34. Lee J-W, Helmann JD. Biochemical characterization of the structural Zn2+ site in the Bacillus subtilis peroxide sensor PerR. J Biol Chem. 2006;281 : 23567–23578. doi: 10.1074/jbc.M603968200 16766519
35. Chen L, Helmann JD. Bacillus subtilis MrgA is a Dps(PexB) homologue: evidence for metalloregulation of an oxidative-stress gene. Mol Microbiol. 1995;18 : 295–300. 8709848
36. Fuangthong M, Helmann JD. Recognition of DNA by three ferric uptake regulator (Fur) homologs in Bacillus subtilis. J Bacteriol. 2003;185 : 6348–6357. doi: 10.1128/JB.185.21.6348-6357.2003 14563870
37. Djoko KY, Ong C-LY, Walker MJ, et al. Copper and zinc toxicity and its role in innate immune defense against bacterial pathogens. Journal of Biological Chemistry. 2015.
38. McDevitt CA, Ogunniyi AD, Valkov E, et al. A molecular mechanism for bacterial susceptibility to zinc. PLoS Pathog. 2011;7: e1002357. doi: 10.1371/journal.ppat.1002357 22072971
39. Dintilhac A, Alloing G, Granadel C, Claverys JP. Competence and virulence of Streptococcus pneumoniae: Adc and PsaA mutants exhibit a requirement for Zn and Mn resulting from inactivation of putative ABC metal permeases. Mol Microbiol. 1997;25 : 727–739. 9379902
40. Skulachev VP, Chistyakov VV, Jasaitis AA, Smirnova EG. Inhibition of the respiratory chain by zinc ions. Biochem Biophys Res Commun. 1967;26 : 1–6. 4291553
41. Lee D-W, Khoury El Y, Francia F, et al. Zinc inhibition of bacterial cytochrome bc(1) reveals the role of cytochrome b E295 in proton release at the Q(o) site. Biochemistry. 2011;50 : 4263–4272. doi: 10.1021/bi200230e 21500804
42. Vygodina TV, Zakirzianova W, Konstantinov AA. Inhibition of membrane-bound cytochrome c oxidase by zinc ions: high-affinity Zn2+-binding site at the P-side of the membrane. FEBS Lett. 2008;582 : 4158–4162. doi: 10.1016/j.febslet.2008.11.018 19041645
43. Kuznetsova SS, Azarkina NV, Vygodina TV, Siletsky SA, Konstantinov AA. Zinc ions as cytochrome C oxidase inhibitors: two sites of action. Biochemistry Mosc. 2005;70 : 128–136. 15807649
44. Korshunov S, Imlay KRC, Imlay JA. The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol Microbiol. 2016.
45. Hammer ND, Reniere ML, Cassat JE, Zhang Y, Hirsch AO, Indriati Hood M, et al. Two heme-dependent terminal oxidases power Staphylococcus aureus organ-specific colonization of the vertebrate host. MBio. 2013;4.
46. Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, et al. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc Natl Acad Sci USA. 2005;102 : 15629–15634. doi: 10.1073/pnas.0507850102 16227431
47. Jones SA, Chowdhury FZ, Fabich AJ, Anderson A, Schreiner DM, House AL, et al. Respiration of Escherichia coli in the mouse intestine. Infect Immun. 2007;75 : 4891–4899. doi: 10.1128/IAI.00484-07 17698572
48. Surmann K, Michalik S, Hildebrandt P, Gierok P, Depke M, Brinkmann L, et al. Comparative proteome analysis reveals conserved and specific adaptation patterns of Staphylococcus aureus after internalization by different types of human non-professional phagocytic host cells. Front Microbiol. 2014;5 : 392. doi: 10.3389/fmicb.2014.00392 25136337
49. Vlamakis H, Chai Y, Beauregard P, Losick R, Kolter R. Sticking together: building a biofilm the Bacillus subtilis way. Nat Rev Microbiol. 2013;11 : 157–168. doi: 10.1038/nrmicro2960 23353768
50. Navarrete F, La Fuente De L. Response of Xylella fastidiosa to zinc: decreased culturability, increased exopolysaccharide production, and formation of resilient biofilms under flow conditions. Appl Environ Microbiol. 2014;80 : 1097–1107. doi: 10.1128/AEM.02998-13 24271184
51. Wang D, Fierke CA. The BaeSR regulon is involved in defense against zinc toxicity in E. coli. Metallomics. 2013;5 : 372–383. doi: 10.1039/c3mt20217h 23446818
52. Ong C-LY, Walker MJ, McEwan AG. Zinc disrupts central carbon metabolism and capsule biosynthesis in Streptococcus pyogenes. Sci Rep. 2015;5 : 10799. doi: 10.1038/srep10799 26028191
53. Anjem A, Imlay JA. Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. Journal of Biological Chemistry. 2012;287 : 15544–15556. doi: 10.1074/jbc.M111.330365 22411989
54. Xu FF, Imlay JA. Silver(I), mercury(II), cadmium(II), and zinc(II) target exposed enzymic iron-sulfur clusters when they toxify Escherichia coli. Appl Environ Microbiol. 2012;78 : 3614–3621. doi: 10.1128/AEM.07368-11 22344668
55. Brenot A, Weston BF, Caparon MG. A PerR-regulated metal transporter (PmtA) is an interface between oxidative stress and metal homeostasis in Streptococcus pyogenes. Mol Microbiol. 2007;63 : 1185–1196. doi: 10.1111/j.1365-2958.2006.05577.x 17238923
56. Guan G, Pinochet-Barros A, Gaballa A, Patel SJ, Argüello JM, Helmann JD. PfeT, a P1B4 -type ATPase, effluxes ferrous iron and protects Bacillus subtilis against iron intoxication. Mol Microbiol. 2015;98 : 787–803. doi: 10.1111/mmi.13158 26261021
57. Pi H, Patel SJ, Argüello JM, Helmann JD. The Listeria monocytogenes Fur-regulated virulence protein FrvA is an Fe(II) efflux P1B4 -type ATPase. Mol Microbiol. 2016;100 : 1066–1079. doi: 10.1111/mmi.13368 26946370
58. Raimunda D, Long JE, Padilla-Benavides T, Sassetti CM, Argüello JM. Differential roles for the Co(2+) /Ni(2+) transporting ATPases, CtpD and CtpJ, in Mycobacterium tuberculosis virulence. Mol Microbiol. 2014;91 : 185–197. doi: 10.1111/mmi.12454 24255990
59. Baichoo N, Wang T, Ye R, Helmann JD. Global analysis of the Bacillus subtilis Fur regulon and the iron starvation stimulon. Mol Microbiol. 2002;45 : 1613–1629. 12354229
60. Tao X, Zeng HY, Murphy JR. Transition metal ion activation of DNA binding by the diphtheria tox repressor requires the formation of stable homodimers. Proc Natl Acad Sci USA. 1995;92 : 6803–6807. 7624323
61. Gilston BA, Wang S, Marcus MD, Canalizo-Hernández MA, Swindell EP, Xue Y, et al. Structural and mechanistic basis of zinc regulation across the E. coli Zur regulon. PLoS Biol. 2014;12: e1001987. doi: 10.1371/journal.pbio.1001987 25369000
62. Ma Z, Faulkner MJ, Helmann JD. Origins of specificity and cross-talk in metal ion sensing by Bacillus subtilis Fur. Mol Microbiol. 2012;86 : 1144–1155. doi: 10.1111/mmi.12049 23057863
63. Begg SL, Eijkelkamp BA, Luo Z, et al. Dysregulation of transition metal ion homeostasis is the molecular basis for cadmium toxicity in Streptococcus pneumoniae. Nat Commun. 2015;6 : 6418. doi: 10.1038/ncomms7418 25731976
64. Turner AG, Ong C-LY, Gillen CM, et al. Manganese homeostasis in group A Streptococcus is critical for resistance to oxidative stress and virulence. MBio. 2015;6.
65. Botella H, Peyron P, Levillain F, et al. Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe. 2011;10 : 248–259. doi: 10.1016/j.chom.2011.08.006 21925112
66. Ong C-LY, Gillen CM, Barnett TC, et al. An antimicrobial role for zinc in innate immune defense against group A streptococcus. J Infect Dis. 2014;209 : 1500–1508. doi: 10.1093/infdis/jiu053 24449444
67. Kapetanovic R, Bokil NJ, Achard MES, Ong C-LY, Peters KM, Stocks CJ, et al. Salmonella employs multiple mechanisms to subvert the TLR-inducible zinc-mediated antimicrobial response of human macrophages. FASEB J. 2016.
68. Abrantes MC, Kok J, Silva Lopes M de F. Enterococcus faecalis zinc-responsive proteins mediate bacterial defence against zinc overload, lysozyme and oxidative stress. Microbiology (Reading, Engl). 2014;160 : 2755–2762.
69. Mascher T, Margulis NG, Wang T, Ye RW, Helmann JD. Cell wall stress responses in Bacillus subtilis: the regulatory network of the bacitracin stimulon. Mol Microbiol. 2003;50 : 1591–1604. 14651641
70. Faulkner MJ, Ma Z, Fuangthong M, Helmann JD. Derepression of the Bacillus subtilis PerR peroxide stress response leads to iron deficiency. J Bacteriol. 2012;194 : 1226–1235. doi: 10.1128/JB.06566-11 22194458
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2016 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Intracellular Zn(II) Intoxication Leads to Dysregulation of the PerR Regulon Resulting in Heme Toxicity in
- Craniofacial Ciliopathies and the Interpretation of Hedgehog Signal Transduction
- Mutations in the Heme Exporter Cause Sensory Neurodegeneration with Loss of Pain Perception
- Shared Information between Residues Is Sufficient to Detect Pairwise Epistasis in a Protein
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy