
Pendredův syndrom v České republice
Pendred Syndrome in the Czech Republic
Pendred syndrome (PS, OMIM #274600) is an autosomal recessive genetic disease, which becomes manifest in a combination of sensoric-neural deafness and dyshormonogenetic goitre. In the PS we frequently encounter abnormal formation of inner ear structures – enlarged vestibular aqueduct (EVA) and/or Mondini dysplasia (MD). PS is caused by mutations in the SLC26A4 gene (called also PDS gene, OMIM *605646), which encodes the anion transporter pendrin, expressed especially in thyroid gland and the inner ear. Pendrin in the thyroid gland is localized on the apical pole of thyreocytes. Its main function is the transport of iodine into colloids in follicle lumen, where it is subsequently bound to organic molecules. The affection of thyroid gland in the Pendred syndrome becomes manifest mostly in the second decade of life under the picture of euthyroid and hypothyroid goitre, rarely there is dyshormogenesis at birth and the disease is then diagnosed in the newborn screening for congenital hypothyreosis. The mutation in the SLC26A4 gene causes also a non-syndrome hearing loss associated with EVA (formerly called DFNB4, OMIM #600791). Only recently the digenetic heredity has been established – mutation in the SLC26A4 gene in combination with a mutation in the FOXI1 gene (OMIM *601093) or KCNJ10 gene (OMIM *602208).
PS represents the most frequent syndrome deafness and the SLC26A4 gene is apparently the second most frequent gene responsible for AR non-syndrome hearing loss (after GJB2 gene for Connexin 26, which is responsible for 40% of AR non-syndrome hearing loss). A recently undertake study in the Czech Republic confirmed mutation in the SLC26A4 gene in 8.5% of predominantly child patients with inborn hearing loss; however only less than 27% of these children displayed a completely expressed PS (after puberty in all of them). Thanks to molecular genetic examination it is therefore possible to predict with high probability the development of thyroid gland disorder and to prevent the development of goitre by an early intervention. The examination of the SLC26A4 gene in presently available in the Czech Republic.
Key words:
Pendred syndrome, EVA syndrome, Mondini dysplasia, dyshormonogenesis, goitre, SLC26A4gene, PDS.
Autoři:
R. Katra 1; R. Pourová 4,5; P. Dytrych 1
![]() ; H. Jelínková 3
; H. Jelínková 3
![]() ; Z. Kabelka 1; M. Dvořáková 6; J. Astl 2
; Z. Kabelka 1; M. Dvořáková 6; J. Astl 2
Působiště autorů:
Klinika zobrazovacích metod 2. LF UK a FN Motol, Praha
; DNA laboratoř Kliniky dětské neurologie, UK 2. LF a FN Motol, Praha
; Ústav biologie a lékařské genetiky, UK 2. LF a FN Motol
; Klinika otorinolaryngologie a chirurgie hlavy a krku
Pardubická krajská nemocnice, a. s.
; Klinika ORL a chirurgie hlavy a krku 1. LF UK a FN Motol, Praha
Katedra otorinolaryngologie IPVZ, Praha
; Klinika ušní, nosní a krční 2. LF UK a FN Motol, Praha
Subkatedra dětské otorinolaryngologie IPVZ, Praha
; přednosta doc. MUDr. Z. Kabelka, Ph. D.
1; přednosta prof. MUDr. J. Betka, DrSc.
2; přednosta MUDr. J. Mejzlík, Ph. D.
3; přednosta prof. MUDr. M. Macek, DrSc.
4; přednosta prof. MUDr. V. Komárek, CSc.
5; přednosta doc. MUDr. M. Roček, CSc.
6
Vyšlo v časopise:
Otorinolaryngol Foniatr, 60, 2011, No. 2, pp. 103-111.
Kategorie:
Přehledový článek
Souhrn
Pendredův syndrom (PS, OMIM #274600) je autosomálně recesivně dědičné onemocnění, projevující se kombinací senzorineurální hluchoty a dyshormonogenetické strumy. U PS pravidelně nalézáme abnormální utváření struktur vnitřního ucha – rozšířený vestibulární akvadukt (EVA, Enlarged Vestibular Aqueduct) a/nebo Mondiniho dysplazii (MD). PS je způsoben mutacemi v genu SLC26A4 (nazývaný také PDS gen, OMIM *605646), který kóduje aniontový transportér pendrin, exprimovaný hlavně ve štítné žláze a vnitřním uchu. Pendrin ve štítné žláze je lokalizován na apikálním pólu tyreocytů. Jeho hlavní funkcí je přenos jodu z tyreocytů do koloidů v lumen folikulů, kde je následně jodid organifikován. Postižení štítné žlázy u Pendredova syndromu se manifestuje nejčastěji ve druhé dekádě života pod obrazem eutyroidní nebo hypotyroidní strumy, vzácně se projeví dyshormonogeneze již při narození a onemocnění je pak diagnostikováno novorozeneckým screeningem pro kongenitální hypotyreózu. Mutace v genu SLC26A4 způsobují kromě PS také nesyndromovou ztrátu sluchu spojenou s EVA (dříve nazývaná DFNB4, OMIM #600791). Nedávno byla u těchto chorob prokázána i digenická dědičnost – mutace v genu SLC26A4 v kombinaci s mutací v genu FOXI1 (OMIM *601093) nebo genu KCNJ10 (OMIM *602208). PS je nejčastější syndromovou hluchotou a gen SLC26A4 je zřejmě druhým nejčastějším genem zodpovědným za AR nesyndromovou ztrátu sluchu (po genu GJB2 pro Connexin 26, který je zodpovědný až za 40% AR nesyndromové ztráty sluchu). V ČR nedávno proběhla studie, která potvrdila mutace v genu SLC26A4 u 8,5 % převážně dětských pacientů s vrozenou ztrátou sluchu, ale jen méně než 27 % z nich mělo kompletně vyjádřený PS (všichni postpubertálně). Díky molekulárně genetickému vyšetření je tedy možné u pacientů s vysokou pravděpodobností předpovědět rozvoj poruchy štítné žlázy a včasným zásahem předejít rozvoji strumy. Vyšetření genu SLC26A4 je nově dostupné i v ČR.
Klíčová slova:
Pendredův syndrom, EVA syndrom, Mondiniho dysplazie, dyshormonogeneze, struma, gen SLC26A4, PDS.
ÚVOD
V roce 1896 popsal poprvé Vaughan Pendred (37) hluchotu spojenou s nálezem strumy. Přesně o 100 let později identifikovali Everett a spolupracovníci gen SLC26A4 (PDS) (16) a odhalili tak společnou molekulárně genetickou podstatu PS a nesyndromové ztráty sluchu vázané k lokusu 7q31 popsané Baldwinem 1995) (10), tzv. DFNB4. V návaznosti na toto zjištění popisuje Reardon rozšíření vestibulárního aqueduktu (původně Large Vestibular Aqueduct syndrom (LVAS) popsaný Valvassorim 1978 (56) jako typický znak Pendredova syndromu, ale zároveň poukazuje, že porucha štítné žlázy je nekonstantním znakem, označuje tedy jakýkoliv výskyt EVA sdružený s mutacemi v genu SLC26A4 za Pendredův syndrom – bez ohledu na přítomnost poruchy štítné žlázy (Reardon, Q J M 2000). LVAS byl původně jednotkou zahrnující rozšíření vestibulárního aqueduktu (EVA) a/nebo Mondiniho dysplazii (MD, aplazie posledního závitu kochley), ztrátu sluchu a zpravidla vestibulární poruchu. V současné době jsou za jednotky způsobené mutacemi v genu SLC26A4 považovány:
1. Pendredův syndrom - porucha sluchu s EVA/MD a porucha štítné žlázy, OMIM #274600. 2. Enlarged Vestibular Aqueduct; EVA - porucha sluchu s EVA/MD bez poruchy štítné žlázy, OMIM #600791 (dříve DFNB4 a LVAS). Rozlišení tohoto spektra klinických obrazů komplikuje fakt, že porucha štítné žlázy se u PS vyskytuje až postpubertálně (tradičně v druhé dekádě života), EVA/DFNB4 může tedy v průběhu života u jednoho pacienta přecházet v PS. Incidence Pendredova syndromu, uváděná Taybim a Lachmannem (55), je 1 až 8 na 100 000 obyvatel, ostatní autoři uvádějí incidenci velice podobnou. Pendredův syndrom je nejčastější příčinou syndromové hluchoty a je zodpovědný až za 10 % případů vrozené nedoslýchavosti (17). Oproti tomu nejčastější příčinou nesyndromové ztráty sluchu (NSZS) jsou mutace v GJB2, způsobující v ČR až 40 % případů prelingvální NSZS (46 ). O poruchách vývoje štítné žlázy v ČR referují již Astl a spol. (7), včetně jednoho z prvních popisů rodiny s Pendredovým syndromem v roce 1996 (8), diagnostikováné na základě klinických parametrů. U rodiny byly později molekulárně geneticky prokázány dvě mutace v genu SLC26A4 (dosud nepublikováno).
DEFINICE
Pendredův syndrom je tedy definován neendemickou strumou spojenou s nedoslýchavostí a geneticky autozomálně recesivním mechanismem přenosu. Porucha sluchu je prelingvální, těžká či progredující a je doprovázena strukturálními abnormalitami vnitřního ucha (EVA, MD). Vestibulární funkce jsou patologické u části nemocných a projevují se epizodami vertiga, které mohou být spojeny s náhlým zhoršením sluchu či fluktujícím průběhem ztráty sluchu. Eutyreoidní či hypofunkční struma s poruchou organifikace jodu může být prokázána perchlorátovým testem. U menší části pacientů se projeví dyshormonogeneze již při narození a onemocnění je pak diagnostikováno celoplošným novorozeneckým screeningem pro kongenitální hypotyreózu. Struma se obvykle nemanifestuje ihned po narození, ale v pubertě či u více jak poloviny nemocných v časné dospělosti.
Kongenitální hypotyreóza
Kongenitální hypotyreóza (KH) je nejčastější vrozenou endokrinní poruchou s frekvencí 1 : 3000 až 1 : 4000 novorozenců (35 ). U dívek se vyskytuje 2–4krát častěji než u chlapců. V dnešní době je součástí plošného novorozeneckého screeningu vyšetření suché krevní kapky odebrané z vpichu do patičky novorozence se stanovením TSH metodou fluoroimunoanalýzy. Existují dvě základní formy KH – endemická, jako následek jodového deficitu matky a plodu během těhotenství, a sporadická KH, jejíž příčina souvisí nečastěji s anatomickými poruchami při vývoji štítné žlázy (anatomický defekt tvorby a/nebo sestupu štítné žlázy), nebo s poruchami syntézy tyreoidálních hormonů (24, 35). Ve 20-25 % je příčinou KH autozomálně recesivní dyshormonogeneze, tj. porucha biosyntézy tyreoidálních hormonů způsobená poruchou transportu jodu, jeho organifikace, poruchou syntézy tyreoglobulinu nebo poruchou dehalogenace (35). Nedoslýchavost je u dětí s vrozenou hypotyreózou nejčastější sdruženou funkční vadou, která může být způsobena mutací v genu pro pendrin, ale častěji přímým vlivem nedostatku hormonů štítné žlázy na pre - i postnatální vývoj sluchového aparátu (17). V ČR je cca 1,5 % případů pacientů s pozitivním neonatálním screeningem způsobeno PS (11).
Molekulárně genetická charakteristika Pendredova syndromu a EVA/DFNB4
Gen, jehož porucha vede k rozvinutí Pendredova syndromu, je označován historicky PDS (Pendred syndrome gene), ale také podle funkční příslušnosti SLC26A4 (member 4 of solute carrier family 26) (16). Je lokalizován na 7. chromozomu, lokus 7q31, a má 21 exonů. PDS/SLC26A4 gen patří ke skupině genů, jenž zahrnuje 26 genů (solute carrier family 26) se statisticky významnou homologií 13 proteinů, které jsou sulfátovými nosiči. Tyto geny zahrnují velké systematické rozpětí, včetně obratlovců, rostlin a kvasinek, zahrnují však dva geny, které jsou s PDS genem příbuzné, a to lidský (down-regulated adenoma geny) a DTD gen (dystrofická dysplazie). Gen DRA a PDS mají postavení konec ke konci a jsou odděleny pouhými 48 kb, a proto Everett a spol. (16) uvažují o jejich evoluční závislosti. Ne zcela opominutelný je fakt blízké lokalizace PDS/ /SLC26A4 genu a genu cystické fibrózy CFTR na 7. chromozomu.
Normální genový produkt PDS genu je protein tvořený 780 aminokyselinami (86 kDa), pojmenovaný jako pendrin, jeho mRNA je asi 5 kb dlouhá, tvořená 2343 páry bází. Pendrin je hydrofobní protein s 12 transmembránovými doménami (16), který slouží jako chloridojodidový transportér (symportér). Pendrin byl detekován ve štítné žláze, vnitřním uchu, ledvinách (16), placentě (12) a prsní žláze (42) metodou RT-PCR i v endometriu, plicích, prostatě a varleti (30).
Abnormální genový produkt: Dosud bylo detekováno více než 100 různých variant v sekvenci PDS/SLC26A4 genu, které byly nalezeny u manifestního Pendredova syndromu nebo EVA/DFNB4. Z těchto abnormit je přibližně 15 mutací nacházeno opakovaně (29). Mutační spektrum se významně liší u různých populací. Mezi Asiaty převládají mutace IVS7-2 A>G a H723R, zatímco v evropské populaci dominují L236P, T416P, a 1001+1G>A, které dohromady tvoří cca 50 % všech mutací (3 ) a jsou těsně následovány mutacemi V138F, E384G, R409H, T410M, L445W a Y530H. Mutační spektrum v ČR odpovídá spektru evropské populace s nejčastější mutací V138F, bylo však nalezeno již 6 nových mutací (11, 39) a jsou nacházeny stále nové. Tento fakt zpochybňuje u pacientů s podezřením na PS nebo EVA význam tzv. „hluchotových čipů“ (43), které se zaměřují pouze na omezené množství již známých mutací, a naopak podporuje význam sekvenování celého genu, které je sice časově i materiálně náročné, lze ho však považovat za jedinou aktuálně dostupnou metodu dostatečně spolehlivou k nalezení nebo vyloučení mutací v genu SLC26A4. Russo a spol. (44) sledovali souvislost výskytu sodíko-jodidových transportérů a chlorido-jodidového transportéru, resp. pendrinu. Prokazují vyšší expresi těchto transportérů v hyperfunkčních uzlech oproti „studeným uzlům“. Usuzují proto, že i v normální žláze budou tyto transportéry exprimovány.
Poznámky k fyziologii a patofyziologii štítné žlázy
Buňky štítné žlázy mají schopnost aktivně vychytávat a akumulovat jod, čímž se výrazně liší od buněk ostatních tkání. Tento mechanismus je realizován tzv. jodidovou pumpou. Akumulace jodu je proces aktivní, energeticky náročný a je nazýván trapping. Jodid je ve funkční štítné žláze 20-40x koncentrovanější než v krevní plazmě vázán v podobě tyreoglobulinu. Jod je základním prvkem potřebným k syntéze aktivních hormonů – tyreoidální hormony obsahují 59–65 % jodu. Denní potřeba jodu je 150–200 μg/den (6). Příjem pod 50 μg/den je uváděn jako kritický pro vznik a růst strumy. Organifikace probíhá konverzí jodu na jodid, jod je následně inkorporován do tyrozolového kruhu. Tak je syntetizován monojodtyrozin (MIT), dijodtyrozin (DIT), trijodtyrozin a tyroxin (T3,T4). Při nadbytku jodu dochází zpočátku ke zvýšené organifikaci s následnou inhibicí jodidové pumpy. Nadměrná organifikace může vést k inhibici hormonogeneze – tzv. Wolffův-Chaikoffův efekt – v důsledku inhibice tyreoidální peroxidázy. Tento efekt je však jen přechodný. Za patologické situace může nadbytek jodu vyvolat hypertyreózu („jodbasedow“) – tento jev může vzniknout nejen u nemocných s Graves-Basedowovou nemocí, ale také u polynodózní strumy, vzácněji i u jiných onemocnění štítné žlázy. Následně probíhá sekrece T3 a T4 do krve, kde je konvertován méně účinný T4 na účinný T3. Jod obsažený v séru je vázán na glykoprotein tyreoglobulin (Tgb). Fyziologická hladina tyreoglobulinu je nižší než 50 ng/ml, u atyreózních nemocných (po totální tyroidektomii) méně jak 9 ng/ml. Obsah jodu je v jeho molekule 0,1-1% molekulové váhy. Tyreoglobulin s nízkým obsahem jodu se hydrolyzuje rychleji než tyreoglobulin s vysokým obsahem jodu. Proteolýzu tyreoglobulinu inhibuje vysoká hladina jodu a lithium. Nedostatek jodu v potravě má vliv na funkci, ale i morfologii štítné žlázy. V oblastech s nedostatkem jodu byl pozorován vznik tzv. endemického kretenismu v důsledku hypotyreózy (kongenitální). Kretenismus je spojen s typickým výrazem v obličeji, nálezem strumy, mentálním postižením, poruchou celkového somatického vývoje, poruchou sluchu a projevy hypotyreózy (24). V dnešní době stále existují oblasti, kde je nedostatečný přívod jodu v potravě, a to i ve vyspělých zemích Evropy, Asie i Ameriky. Nedostatečný přívod jodu a poruchy funkce štítné žlázy jsou známy i v ČR. Nejzávažnější je nedostatečný přívod jodu v době těhotenství, kde důsledky pro vyvíjející se plod jsou nejzávažnější. V důsledku nedostatku jodu dochází ke zvětšení štítné žlázy. Struma z nedostatečného přívodu jodu vzniká i v dospělosti. Nedostatek jodu se projevuje v různém věku rozdílně. V době intrauterinního vývoje se projeví abortivním efektem, vznikem vrozených vývojových vad, zvýšenou mortalitou plodu, vzniká kretenismus a psychomotorické poruchy. U novorozenců vzniká struma, hypotyreóza, zpožděný psychomotorický a somatický vývoj. V dětském věku a u dospívajících se projevuje zpoždění mentálního vývoje, nedostatečný vývoj intelektu a inteligence, dětská juvenilní hypotyreóza, struma, porucha somatického vývoje; nedoslýchavost. U dospělých vzniká struma – nodózní struma – mechanický syndrom, hypotyreóza, jodová tyreotoxikóza, rizikový faktor maligního bujení štítné žlázy. Hypotyreóza má různé projevy dle příčiny. Nejvýznamněji se podílí na hypotyreóze snížený přívod jodu a/nebo zánět štítné žlázy (chronický, subakutní, vzácně akutní). Zvláštní skupinou je hypotyreóza po operaci štítné žlázy. Autoimunitní onemocnění jsou ve vztahu k hypofunkci štítné žlázy především spojovány s polyglandulárním syndromem, inzulin dependentním diabetes mellitus, Schmidtovým syndromem, Addisonovou chorobou, ovariální dysfunkcí a Hashimotovou tyreoiditis. Tato onemocnění jsou spojena s poruchou organifikace jodu, kde je zjištěna enzymatická porucha syntézy hormonů štítné žlázy jako jsou: porucha vychytávání jodu, porucha organifikace jodu (Pendredův, Hollanderův syndrom), porucha syntézy dijodtyrosinu a porucha syntézy T3 aT4, syntéza abnormních T3 a T4, porucha proteolýzy, defektní tyreoglobulin, rezistence na TSH a periferní rezistence na tyreoidální hormony (tab. 1).

Klinický obraz Pendredova syndromu (EVA/DFNB4) v otorinolaryngologii
Pendredův syndrom je charakterizován vrozenou, pomalu progredující těžkou senzorineurální nedoslýchavostí, anomáliemi v temporální kosti a vývojem eutyroidní či hypofunkční strumy v pozdním dětství či časné dospělosti. Ve velmi malém procentu je záchyt onemocnění spojen s kongenitální hypotyreózou, u většiny pacientů však nastupuje porucha štítné žlázy až postpubertálně, protože dyshormonogenze (v důsledku poruchy organizikace jodu) je v raném dětství kompenzována jinými mechanismy. Lze tedy konstatovat, že Pendredův syndrom je vyjádřen postižením senzorineurálně-endokrinním.
Porucha sluchu: Ve většině případů oboustranná percepční (event. kombinovaná) nedoslýchavost, charakterizována jako těžká vrozená nedoslýchavost. Progresivní deteriorace sluchu se vyskytuje častěji až po vývoji řeči (37 ), ale tíže postižení je individuálně variabilní a může kolísat. I bilaterální postižení sluchu může být asymetrické. U několika jedinců byla zjištěna progresivní nedoslýchavost, která se vyvinula po úrazu hlavy. V souvislosti s úrazem je předpokladem, že u těchto osob byla přítomna malformace vnitřního ucha (rozšíření akvaduktu) či se na tíži sluchového postižení podílela i složka hypomikro-cirkulační. U některých pacientů jsou postiženy hlavně vyšší frekvence, k bazokochleárnímu postižení se přidává postupně i apikochleární, resp. pankochleární. Problematická může být diagnostika u dětí s vrozenou hypotyreózou, kde je nedoslýchavost jedním z projevů tohoto stavu (19). Průkaz mutace genu SLC26A4 je pak jedinou možností jak odlišit u těchto dětí jednotlivé geneticky podmíněné poruchy (11). Kongenitální hypotyreóza zahrnuje více mutací, včetně průkazů mutací transkripčních faktorů v době embryogeneze TTF 1, 2 a PAX 8. (2, 31).
Vestibulární příznaky: Byla zjištěna vestibulární dysfunkce u 12-66 % nemocných s Pendredovým syndromem, jejichž projevy byly charakterizovány jednostranným postižením polokruhových kanálků. Klinicky se projevuje epizodami vertiga (39, 50-52). Zjednodušeně je možné přirovnat poruchu rovnováhy u PS k obrazu Menièrovy nemoci. V některých případech, kdy se objeví epizoda náhlého zhoršení sluchu a rotačního vertiga, může po určité době dojít k přetrvávání poruch rovnováhy (50). Obvykle je přítomna oboustranná hyporeflexie.
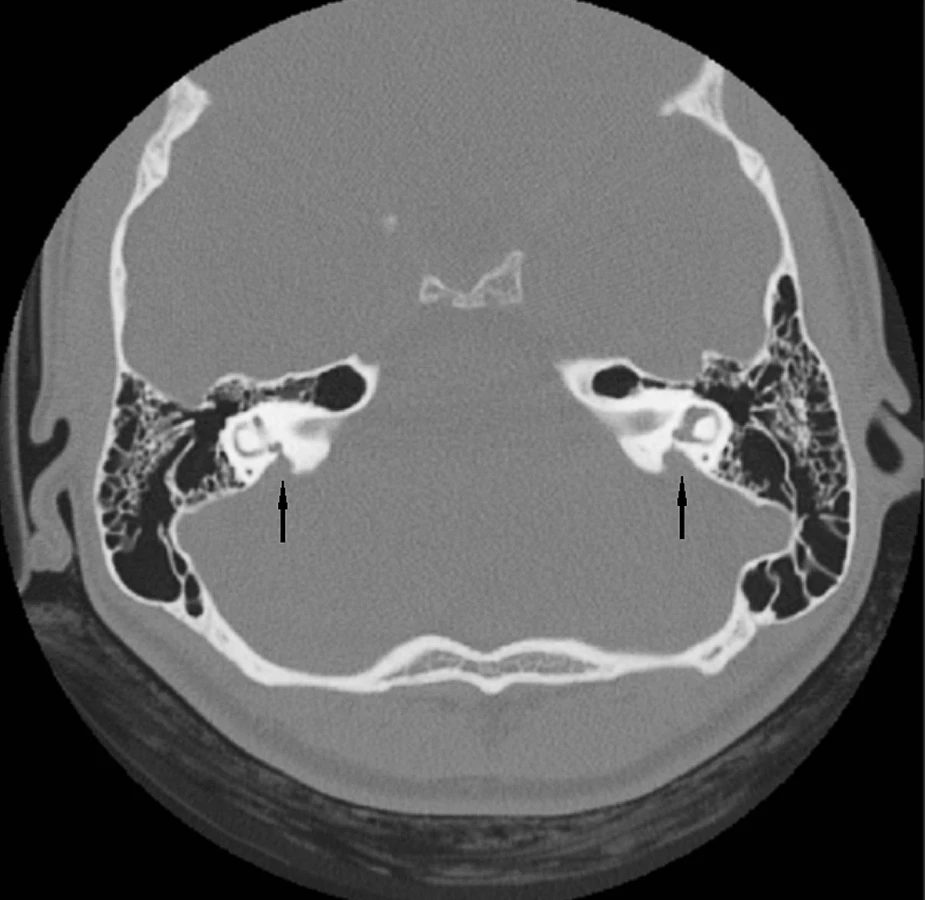
Abnormality temporální kosti: Patologický radiologický obraz temporální kosti (HRCT) je zjištěn v 85 případů s prokázaným Pendredovým syndromem. Nejčastěji je zjišťována EVA a Mondiniho malformace (hlemýžď má jen jeden a půl závitu a vzniká zde patologická komunikace mezi scala vestibuli a scala tympani se současným rozšířením saccus a ductus endolymfaticus (18, 38) (obr. 1). Ve většině případů je zjišťována oboustranná dilatace vestibulárního akvaduktu (obr. 2). Vestibulární akvadukt byl považován za rozšířený, pokud jeho průměr přesáhl 1,5 mm uprostřed jeho délky. Dominující zobrazovací metodou je HRCT pyramid se zaměřením na struktury vnitřního ucha a šíři akvaduktu (20). V individuálních případech je indikována MRI (47). Dle Fugazzola a spol. (18) jsou malformace membranózního labyrintu zobrazitelné při MRI považovány za znak s vůbec nejvyšší diagnostickou hodnotou. Také v ČR se EVA na HRCT či MRI ukázaly jako velmi efektivní diagnostický znak – u pacientů s bilaterální EVA byla alespoň jedna mutace v genu SLC26A4 zachycena u 69,2 % pacientů (u 46 % pacientů byly nalezeny obě mutace). Všichni pacienti s oběma nalezenými mutacemi a 75 % pacientů s jednou mutací, u kterých bylo zhodnoceno HRCT, měli bilaterální EVA, zatímco u pacientů s unilaterální EVA a MD nebyla nalezena žádná mutace (39). Preferujeme zobrazení vysokorozlišovacím CT vyšetřením se zaměřením na oblast vnitřního ucha (obr. 3).


Struma: Přibližně 75 % nemocných má v době diagnózy zjištěnu klinicky přítomnou strumu většinou ve formě eufunkční. Porucha funkce štítné žlázy je pozorována někdy již v dětství, avšak pacienti jsou v mnoha případech v pásmu eutyreózy (13). Asi u 40 % postižených se struma vyvíjí v pozdním dětství či v počátku dospělosti, dominuje období puberty, při existenci významné intrafamiliární variability exprese těchto obtíží (37, 55). Většina nemocných je léčena substituční terapií, pouze 10 % postižených má abnormální funkci štítné žlázy spojenou s vzestupem sérových hladin TSH (40). Zpočátku se jedná o difuzní postižení štítnice, později se objevuje nodulární přestavba, při které může i narůstat riziko maligní transformace (49). Zde je třeba zdůraznit, že porucha štítné žlázy se u přibližně 20 % pacientů nemusí vůbec klinicky projevit (40). Perchlorátový test prokazuje zhoršenou organifikaci jodu, jelikož důsledkem poruchy jodidového transportního mechanismu je parciální defekt organifikace. Úroveň organifikace jodu vykazuje velmi vysokou interindividuální variabilitu (např. podílem jiných apikálních jodidových transportérů či dalších mechanismů částečně kompenzujících defekt pendrinu). U pacientů s Pendredovým syndromem je patologický či hraniční perchlorátový test ve 25 až 50 % (17, 23, 45). Citlivost Perchlorátového testu je tedy omezena, dnes je považován za málo senzitivní a s ohledem na vyšší radiační zátěž jej lze z hlediska přesnosti nahradit genetickým testem mutace genů pro pendrin (23). Perchlorátový test nelze doporučit především u žen ve fertilním věku a u dětí. Nezbytné je odlišit endemický kretenismus, který je také spojen s poruchou sluchu, ale také získanou hypotyreózu v koincidenci s jinou poruchou sluchu. Banghová a spol. z roku 2008 (11) konstatují nález dvou případů s potvrzenými mutacemi v genu SLC26A4 ve skupině kongenitální hypotyreózy ve dvacetiletém intervalu sledování. Jde tedy v této skupině spíše o výjimečný nález. Poruchy sluchu společně s onemocněním štítné žlázy jsou popsány a jsou spojeny se 3 základními typy tyreopatií : endemickým kretenismem, Pendredovým syndromem a hypotyreózou vznikající v dospělém věku (15, 32). Byla popsána také hypertyreóza spojená s nedoslýchavostí (54). Pozorován byl i výskyt karcinomu štítné žlázy a Pendredova syndromu (1) a heterotopie štítné žlázy na kořeni jazyka u Pedredova syndromu (57) (tab. 2).

Prevalence Pendredova syndromu není dostatečně známa. Literární údaje udávají, že tato vývojová vada postihuje 7,5 % - 10 % všech postižených s kongenitální hluchotou (2, 22, 28, 33, 36, 53). Pro takto vysokou četnost však nesvědčí údaje dalších autorů: Gorlina (21), Newtona (34), Arnose a spol. (4, 5). Část pacientů, a to i v ČR, je zařazena do skupiny tzv. nesyndromové senzorineurální nedoslýchavosti (NSHL non-syndrome hearing loss). Reardon a kol. (41) zastávají názor, že struma není konstantním znakem PS a postižení štítné žlázy může být subklinické. Proto je velmi pravděpodobné, že přítomnost PS je častější, než je diagnostikován, či je na něj vysloveno podezření. Mnohem výmluvnějším příznakem je proto rozšíření vestibulárního akvaduktu (25, 58). Tato kostní abnormalita je nejčastějším radiologickým nálezem u dětí s progredující senzorineurální poruchou sluchu (14) a je nalézána až u 15 % všech pacientů s poruchou sluchu. Proto je diskutováno zavedení vyhledávání SLC26A4, resp. PDS genu, jako příčiny poruchy sluchu u pacientů s nesyndromovou ztrátou sluchu, která je zjištěna zpravidla u jednoho z 600 novorozenců (48). Kabakkaya a spol. (27) uvádějí, že až 5 % pacientů s kongenitální hluchotou lze diagnostikovat jako Pendredův syndrom. U neslyšících je tedy nutné myslet na vyšetření štítné žlázy při sebemenším náznaku její dysfunkce. Mondiniho typ dysplazie a současný výskyt rozšířeného akvaduktu je zjištěn u 85 % postižených, přesto jej nelze považovat za specifický (26, 58). Významnějším nálezem je výskyt rozšířeného vestibulárního akvaduktu, který je dnes považován za zásadní malformaci v oblasti vnitřního ucha a za diagnostický znak tohoto syndromu. Je nutno odlišovat Pendredův syndrom či kretenismus spojený s hluchotou, od hypotyreózy s nedoslýchavostí vzniklé v pozdějším věku. Pro PS svědčí nedoslýchavost, resp. progredující těžká porucha sluchu až hluchota od dětství, porucha funkce štítné žlázy, demonstrující se většinou jako parciální porucha organifikace jodu od dětství, či nález strumy s laboratorní eufunkcí / hypofunkcí, kterou nelze ovlivnit podáváním substituční terapie. Častý je výskyt kochley Mondiniho typu a nález rozšířeného akvaduktu na HRCT je téměř pravidlem. Je zřejmé, že autosomální mechanismus dědičnosti povede k postižení obou pohlaví stejným dílem. Z uvedených faktů je zřejmé, že žádný znak, uvedený v charakteristice Pendredova syndromu, nemá 100% penetranci a konstantní výskyt. Proto se diagnostika Pendredova syndromu opírá o společný výskyt několika z uvedených znaků – struma, percepční nedoslýchavost, porucha organifikace jodu, rozšíření vestibulárního akvaduktu a/nebo Mondiniho dysplazie spojené s nálezem jedné mutace v genu SLC26A4/PDS. Za jednoznačný průkaz považujeme nález dvou mutací v genu SLC26A4/PDS. Pendredův syndrom byl popsán také u pacientů v České republice (8, 9), ale do současné doby se jednalo spíše pouze o kazuistická sdělení. Vzhledem k možnosti včasné diagnostiky lze předpokládat možnost individualizování programů rehabilitace sluchu a prognózování vývoje nedoslýchavosti. Za nezanedbatelnou součást pak považujeme možnost dokonalejšího genetického poradenství u postižených osob. Spojení změn exprese tohoto genu u osob s některými onkologickými onemocněními štítné žlázy je předpokladem k využití genu SLC26A4 v diagnostice, léčbě či prognózování osudu i u těchto onemocnění. Současný stav problematiky lze podpořit pracemi Pourové a spol., kde lze považovat za zásadní, že PS, resp. mutace genu SLC26A4, byly zjištěny u více než 8 % neslyšících (39). V ČR se tak podařilo po téměř 15 letech od popsání prvního klinického případu rodiny s Pendredovým syndromem prokázat, že Pendredův syndrom není v ČR pouze raritním úkazem, ale naopak příčinou nedoslýchavosti u značné části postižených. U těchto nemocných se především projevuje ztrátou sluchu, v jejíž léčbě není možné očekávat příznivý vývoj a tito postižení spějí do skupiny neslyšících, kterým může kochleární implantace zprostředkovat obnovení funkce sluchu. Dalším podstatným aspektem je diagnostika Pendredova syndromu. Protože byla v ČR zavedena molekulárně genetická analýza SLC26A4, je možné jednoznačně popsat nemocné s mutací tohoto genu a tak je jasně vyčlenit ze skupiny nesyndromových nedoslýchavostí. Tyto skutečnosti poukazují na nutnost provádění správného genetického poradenství, které pak ovlivňuje nejen prognózu a další léčbu nositelů této mutace, ale i jejich rodiny. Pro otolaryngology a foniatry je tak vyřešena otázka správné diagnostiky této nedoslýchavosti a včasné zařazení postižených do programů péče o nedoslýchavé, resp. do programů kochleárních implantací, a to zvláště u nemocných v dětském věku. Právě o diagnostiku mutací SLC26A4 se rozšířil okruh možností pro nemocné, u kterých nebyla příčina nedoslýchavosti, resp. hluchoty, objasněna na genetickém podkladě po analýze mutací v genu pro Connexin 26 (tab. 3).

ZÁVĚR
Výskyt nemocných s mutací SLC26A4 ve skupině českých neslyšících lze považovat za prokázaný. Současné práce nám umožňují vyslovit hypotézu, že incidence těchto mutací v populaci ČR bude podobná jako v ostatních zemích a ve skupině neslyšících bude do 10 %. Výskyt Pendredova syndromu, resp. EVA/DFNB4 v populaci ČR, včetně možností molekulárně genetické analýzy, je možno považovat za novou kapitolu pro otolaryngology a foniatry.
Práce vznikla za částečné podpory IGA NS 9913-4 a VZ MZOFNM2005.
MUDr. Rami Katra
Klinika ušní, nosní a krční 2. LF UK a FNM
V Úvalu 84
150 06 Praha 5
e-mail: rami.katra@centrum.cz
Zdroje
1. Abs, R., Verhelst, J., Schoofs, E. et al.: Hyperfunctioning metastatic follicular thyroid carcinoma in Pendred’s syndrome. Cancer, 67, 1991, s. 2191-2193.
2. Al Taji, E., Biebermann, H., Limanova, Z. et al.: Screening for mutations in transcription factors in a Czech cohort of 170 patients with congenital and early-onset hypothyroidism: identification of a novel PAX8 mutation in dominantly inherited early-onset non-autoimmune hypothyroidism. Eur. J. Endocrinol., 156, 2007, s. 521-529.
3. Albert, S., Blons, H., Jonard, L. et al.: SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur. J. Hum. Genet., 14, 2006, s. 773-779.
4. Arnos, K. S., Cunningham, M., Israel, J. et al.: Innovative approach to genetic counseling services for the deaf population. Am. J. Med. Genet., 44, 1992, s. 345-351.
6. Astl, J.: Chirurgická léčba štítné žlázy. Maxdorf Jessenius, 2007, s. 42-47.
7. Astl, J., Betka, J., Slavíček, A.: Heterotopics thyroid tissue. Head and Neck Diseases, 2, 1993, s. 55-57.
8. Astl, J., Štolbová, D., Skřivan, J.: Struma spojená s nedoslýchavostí. Otorinolaryngologie a foniatrie, 45, 1996, s. 31-34.
9. Astl, J., Veselý, D., Jablonický, P.: Pendredův syndrom - poznámky k problematice vrozené autosomálně recesivní percepční nedoslýchavosti spojené se strumou. Otorinolaryngologie a foniatrie, 53, 2004, s. 55-59.
10. Baldwin, C. T., Weiss, S., Farrer, L. A. et al.: Linkage of congenital, recessive deafness (DFNB4) to chromosome 7q31 and evidence for genetic heterogeneity in the Middle Eastern Druze population. Hum. Mol. Genet., 4, 1995, s. 1637-1642.
11. Banghova, K., Al Taji, E., Cinek, O. et al.: Pendred syndrome among patients with congenital hypothyroidism detected by neonatal screening: identification of two novel PDS/SLC26A4 mutations. Eur. J. Pediatr., 167, 2008, s. 777-783.
12. Bidart, J. M., Lacroix, L., Evain-Brion, D. et al.: Expression of Na+/I - symporter and Pendred syndrome genes in trophoblast cells. J. Clin. Endocrinol. Metab., 85, 2000, s. 4367-4372.
13. Caksen, H., Kurtoglu, S., Yuksel, S. et al.: Do not overlook Pendred’s syndrome in children with sensorineural hearing loss. Ear Nose Throat. J., 80, 2001, s. 760.
14. Colvin, I. B., Beale, T., Harrop-Griffiths, K.: Long-term follow-up of hearing loss in children and young adults with enlarged vestibular aqueducts: relationship to radiologic findings and Pendred syndrome diagnosis. Laryngoscope, 116, 2006, s. 2027-2036.
15. Elamin, A.: Goitre and deaf-mutism. Ups. J. Med. Sci., 96, 1991, s. 213-218.
16. Everett, L. A., Glaser, B., Beck, J. C., et al.: Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Genet., 17, 1997, s. 411-422.
17. Fraser, G. R.: Association of congenital deafness with goitre (Pendred’s syndrome) a study of 207 families. Ann. Hum. Genet., 28, 1965, s. 201-249.
18. Fugazzola, L., Mannavola, D., Cerutti, N. et al.: Molecular analysis of the Pendred’s syndrome gene and magnetic resonance imaging studies of the inner ear are essential for the diagnosis of true Pendred’s syndrome. J. Clin. Endocrinol. Metab., 85, 2000, s. 2469-2475.
19. Gaudino, R., Garel, C., Czernichow, P. et al.: Proportion of various types of thyroid disorders among newborns with congenital hypothyroidism and normally located gland: a regional cohort study. Clin. Endocrinol. (Oxf.), 62, 2005, s. 444-448.
20. Goldfeld, M., Glaser, B., Nassir, E. et al.: CT of the ear in Pendred syndrome. Radiology, 235, 2005, s. 537-540.
21. Gorlin, R. J.: Genetic hearing loss associated with endocrine and metabolic disorders. In: Gorlin R. J., Torielle, H. V., Cohen, M. M. Jr. (eds) Hereditary hearing loss and its syndromes. Oxford Monographs on Medical Genetics, 28, Oxford University Press, New York, 1995.
22. Grimaldi, R., Capuano, P., Miranda, N. et al.: Pendrin: physiology, molecular biology and clinical importance. G. Ital. Nefrol., 24, 2007, s. 288-294.
23. Gross, M., Hahn, K., Biesalski, H. K.: The diagnosis of the Pendred syndrome in children by the perchlorate discharge test with 123I (author’s transl). Hno, 29, 1981, s. 95-97.
24. Hníková, O.: Kongenitální hypotyreóza. Pediatrie pro praxi, 3, 2005, s. 123-126.
25. Chen, A. H., Mueller, R. F., Prasad, S. D. et al.: Presymptomatic diagnosis of nonsyndromic hearing loss by genotyping. Arch. Otolaryngol. Head Neck Surg., 124, 1998, s. 20-24.
26. Johnsen, T., Jorgensen, M. B., Johnsen, S.: Mondini cochlea in Pendred’s syndrome. A histological study. Acta Otolaryngol., 102, 1986, s. 239-247.
27. Kabakkaya, Y., Bakan, E., Yigitoglu, M. R. et al.: Pendred’s syndrome. Ann. Otol. Rhinol. Laryngál., 102, 1993, s. 285-288.
28. Kopp, P., Pesce, L., Solis, S. J.: Pendred syndrome and iodide transport in the thyroid. Trends Endocrinol. Metab., 19, 2008, s. 260-268.
29. Laboratory, M. O. R., Clinics, U. O. I. H. A., Department of Otolaryngology - Head and Neck Surgery, P. B. H., Http://Www.Healthcare.Uiowa.Edu/Labs/Pendredandbor/Slcmutations.Htm.
30. Lacroix, L., Mian, C., Caillou, B. et al.: Na(+)/I(-) symporter and Pendred syndrome gene and protein expressions in human extra-thyroidal tissues. Eur. J. Endocrinol., 144, 2001, s. 297-302.
31. Lacroix, L., Michiels, S., Mian, C. et al.: HEX, PAX-8 and TTF-1 gene expression in human thyroid tissues: a comparative analysis with other genes involved in iodide metabolism. Clin. Endocrinol. (Oxf.), 64, 2006, s. 398-404.
32. Li, X. C., Everett, L. A., Lalwani, A. K. et al.: A mutation in PDS causes non-syndromic recessive deafness. Nat. Genet., 18, 1998, s. 215-217.
33. Maciaszczyk, K., Lewinski, A.: Phenotypes of SLC26A4 gene mutations: Pendred syndrome and hypoacusis with enlarged vestibular aqueduct. Neuro Endocrinol. Lett., 29, 2008, s. 29-36.
34. Newton, V. E.: Aetiology of bilateral sensori-neural hearing loss in young children. J. Laryngál. Otol. Suppl., 10, 1985, s. 1-57.
35. Park, S. M., Chatterjee, V. K.: Genetics of congenital hypothyroidism. J. Med. Genet., 42, 2005, s. 379-389.
36. Pearce, J. M.: Pendred’s syndrome. Eur. Neurol., 58, 2007, s. 189-190.
37. Penderd, V.: Deaf-mutism and goitre. Lancet., 2, 1896, s. 532.
38. Phelps, P. D., Coffey, R. A., Trembath, R. C. et al.: Radiological malformations of the ear in Pendred syndrome. Clin. Radiol., 53, 1998, s. 268-273.
39. Pourova, R., Janousek, P., Jurovcik, M. et al.: Spectrum and frequency of SLC26A4 mutations among Czech patients with early hearing loss with and without Enlarged Vestibular Aqueduct (EVA). Ann. Hum. Genet., 74, 2010, s. 299-307.
40. Reardon, W., Coffey, R., Chowdhury, T. et al.: Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J. Med. Genet., 36, 1999, s. 595-598.
41. Reardon, W., Coffey, R., Phelps, P. D. et al.: Pendred syndrome—100 years of underascertainment? Qjm, 90, 1997, s. 443-447.
42. Rillema, J. A., Hill, M. A.: Pendrin transporter carries out iodide uptake into MCF-7 human mammary cancer cells. Exp. Biol. Med. (Maywood), 228, 2003, s. 1078-1082.
43. Rodriguez-Paris, J., Pique, L., Colen, T. et al.: Genotyping with a 198 mutation arrayed primer extension array for hereditary hearing loss: assessment of its diagnostic value for medical practice. PLoS One, 5, 2010, s. e11804.
44. Russo, D., Bulotta, S., Bruno, R. et al.: Sodium/iodide symporter (NIS) and pendrin are expressed differently in hot and cold nodules of thyroid toxic multinodular goiter. Eur. J. Endocrinol., 145, 2001, s. 591-597.
45. Scinicariello, F., Murray, H. E., Smith, L. et al.: Genetic factors that might lead to different responses in individuals exposed to perchlorate. Environ Health Perspect, 113, 2005, s. 1479-1484.
46. Seeman, P., Malikova, M., Raskova, D. et al.: Spectrum and frequencies of mutations in the GJB2 (Cx26) gene among 156 Czech patients with pre-lingual deafness. Clin. Genet., 66, 2004, s. 152-157.
47. Sharghi, S., Haghpanah, V., Heshmat, R. et al.: Comparison of MRI findings with traditional criteria in diagnosis of Pendred syndrome. Int. J. Audio., 46, 2007, s. 69-74.
48. Schrijver, I.: Hereditary non-syndromic sensorineural hearing loss: transforming silence to sound. J. Mol. Dian., 6, 2004, s. 275-284.
49. Skubis-Zegadlo, J., Nikodemska, A., Przytula, E. et al.: Expression of pendrin in benign and malignant human thyroid tissues. Br. J. Cancer, 93, 2005, s. 144-151.
50. Stinckens, C., Huygen, P. L., Joosten, F. B. et al.: Fluctuant, progressive hearing loss associated with Meniere like vertigo in three patients with the Pendred syndrome. Int. J. Pediatr. Otorhinolaryngol, 61, 2001, s. 207-215.
51. Stinckens, C., Huygen, P. L., Van Camp, G. et al.: Pendred syndrome redefined. Report of a new family with fluctuating and progressive hearing loss. Adv. Otorhinolaryngol, 61, 2002, s. 131-141.
52. Sugiura, M., Sato, E., Nakashima, T. et al.: Long-term follow-up in patients with Pendred syndrome: vestibular, auditory and other phenotypes. Eur. Arch. Otorhinolaryngol, 262, 2005, s. 737-743.
53. Suzuki, K., Yoshida, A., Fukata, S.: Pendred syndrome. Nippon Rinsho, Suppl. 1, 2006, s. 371-373.
54. Štolbová, D., Možný, J., Límanová, Z.: Syndrom familiární hyperthyreosy a kochleární nedoslýchavosti s manifestací v mladém věku. Čs. Otolaryng., 32, 1983, s. 34-39.
55. Taybi, H., Lachmann, A.: Radiology of syndromes, metabolic disoders and sceletal dysplasias. Year Book Medical Publishers., 1990, s. 993.
56. Valvassori, G. E., Clemis, J. D.: Abnormal vestibular aqueduct in cochleovestibular disorders. Adv. Otorhinolaryngol, 24, 1978, s. 100-105.
57. Wetke, R.: Goiter of the tongue base and bilateral perceptive impairment of hearing. Ugeskr Laeger, 151, 1989, s. 2734-2735.
58. Yang, J. J., Tsai, C. C., Hsu, H. M. et al.: Hearing loss associated with enlarged vestibular aqueduct and Mondini dysplasia is caused by splice-site mutation in the PDS gene. Hear Res., 199, 2005, s. 22-30.
Štítky
Audiologie a foniatrie Dětská otorinolaryngologie OtorinolaryngologieČlánek vyšel v časopise
Otorinolaryngologie a foniatrie

2011 Číslo 2
- Moderní přístupy zvyšující efektivitu antibiotické léčby v nemocniční praxi
- Primární a sekundární imunodeficience, přehled a klasifikace
- Diagnostika primárních imunodeficiencí
- Poruchy komplementového systému
- Protilátkové imunodeficience
Nejčtenější v tomto čísle
- Změna komunikace je u pacientů s trvalou tracheostomií největším hendikepem
- Extraezofageální refluxní choroba - mezioborový konsenzus
- Pendredův syndrom v České republice
-
Nádorová trombóza v. jugularis interna
Kazuistika a přehled jejích epidemiologických, patogenetických a klinických aspektů
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy