
Capture-based next-generation sequencing reveals multiple actionable mutations in cancer patients failed in traditional testing
Background:
Targeted therapies including monoclonal antibodies and small molecule inhibitors have dramatically changed the treatment of cancer over past 10 years. Their therapeutic advantages are more tumor specific and with less side effects. For precisely tailoring available targeted therapies to each individual or a subset of cancer patients, next-generation sequencing (NGS) has been utilized as a promising diagnosis tool with its advantages of accuracy, sensitivity, and high throughput.
Methods:
We developed and validated a NGS-based cancer genomic diagnosis targeting 115 prognosis and therapeutics relevant genes on multiple specimen including blood, tumor tissue, and body fluid from 10 patients with different cancer types. The sequencing data was then analyzed by the clinical-applicable analytical pipelines developed in house.
Results:
We have assessed analytical sensitivity, specificity, and accuracy of the NGS-based molecular diagnosis. Also, our developed analytical pipelines were capable of detecting base substitutions, indels, and gene copy number variations (CNVs). For instance, several actionable mutations of EGFR, PIK3CA,TP53, and KRAS have been detected for indicating drug susceptibility and resistance in the cases of lung cancer.
Conclusion:
Our study has shown that NGS-based molecular diagnosis is more sensitive and comprehensive to detect genomic alterations in cancer, and supports a direct clinical use for guiding targeted therapy.
Keywords:
Next-generation sequencing, molecular diagnosis, cancer panel, targeted therapy.
Authors:
Jing Xie 1,2; †; Xiongxiong Lu 1,3; †; Xue Wu 4; Xiaoyi Lin 5; Chao Zhang 4; Xiaofang Huang 4; Zhili Chang 4; Xinjing Wang 1,3,6; Chenlei Wen 1,3,6; Xiaomei Tang 1,3,6; Minmin Shi 1,3,6; Qian Zhan 1,3; Hao Chen 1,3,6; Xiaxing Deng 1,3,6; Chenghong Peng 1,3,6; Hongwei Li 3; Yuan Fang 1,3,6,*; Yang Shao 4,7,*; Baiyong Shen 1,3,6,*
Authors place of work:
Research Institute of Pancreatic Disease, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
1; Department of Pathology, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
2; Pancreatic Disease Centre, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
3; Department of Research and Development, Geneseeq Technology Inc., Toronto, Ontario, Canada
4; Department of Laboratory Medicine, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
5; Shanghai Institute of Digestive Surgery, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
6; Department of Medical Biophysics, University of Toronto, Toronto, Ontario, Canada
7
Published in the journal:
Molecular Genetics & Genomic Medicine 2016; 4(3)
Category:
Original article
doi:
https://doi.org/10.1002/mgg3.201
© 2016 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals, Inc.
This is an open access article under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Summary
Background:
Targeted therapies including monoclonal antibodies and small molecule inhibitors have dramatically changed the treatment of cancer over past 10 years. Their therapeutic advantages are more tumor specific and with less side effects. For precisely tailoring available targeted therapies to each individual or a subset of cancer patients, next-generation sequencing (NGS) has been utilized as a promising diagnosis tool with its advantages of accuracy, sensitivity, and high throughput.
Methods:
We developed and validated a NGS-based cancer genomic diagnosis targeting 115 prognosis and therapeutics relevant genes on multiple specimen including blood, tumor tissue, and body fluid from 10 patients with different cancer types. The sequencing data was then analyzed by the clinical-applicable analytical pipelines developed in house.
Results:
We have assessed analytical sensitivity, specificity, and accuracy of the NGS-based molecular diagnosis. Also, our developed analytical pipelines were capable of detecting base substitutions, indels, and gene copy number variations (CNVs). For instance, several actionable mutations of EGFR, PIK3CA,TP53, and KRAS have been detected for indicating drug susceptibility and resistance in the cases of lung cancer.
Conclusion:
Our study has shown that NGS-based molecular diagnosis is more sensitive and comprehensive to detect genomic alterations in cancer, and supports a direct clinical use for guiding targeted therapy.
Keywords:
Next-generation sequencing, molecular diagnosis, cancer panel, targeted therapy.
Introduction
The greatly improved understanding of molecular etiology of cancer (Chin and Gray 2008; Stratton et al. 2009; International Cancer Genome C et al., 2010; Mardis 2012) has changed systemic cancer treatment by using molecularly targeted drugs prescribed to an individual patient. Targeted therapies that are intended to be safer and more efficacious block the growth and spread of cancer by interfering with the molecules that are involved in growth, progression, and metastasis. Many targeted cancer therapies have been approved by the Food and Drug Administration (FDA) to treat specific types of cancer, such as Trastuzumab (Herceptin) in ERBB2 (also known as HER2; OMIM*164870)-amplified breast cancer, Imatinib (Gleevec) in BCR-ABL (OMIM*151410; *189980) fusion-positive chronic myelogenous leukemia (CML), Erlotinib (Tarceva) in EGFR (OMIM*131500)mutated non-small-cell lung cancer (NSCLC), and Vemurafenib (Zelboraf) in BRAF-V600E (OMIM*164757) mutant melanoma (Stegmeier et al. 2010). More personalized cancer therapy will be achieved as there are now thousands of compounds in preclinical testing and clinical trials targeting hundreds of genomic alterations in cancer-related genes involving innumerous cellular pathways (Barretina et al. 2012; Garnett et al. 2012). Moreover, certain somatic mutations can also impact the sensitivity or resistance to specific cancer therapies (Diaz et al. 2012; Camidge et al. 2014). In order to precisely match each individual or a subset of cancer patients with available targeted therapies, comprehensive molecular diagnosis tests need to be developed to characterize the genomic alterations occurring within individual tumors. Several technologies, including PCR, Sanger sequencing, mass spectrometric genotyping, fluorescence in situ hybridization (FISH), and immunohistochemistry (IHC) (Thomas et al.2007; MacConaill et al. 2009; Dias-Santagata et al. 2010; Ross 2011; McCourt et al. 2013), are currently in clinical use for the molecular assessment. However, due to technical limitations, none of these methodologies can be scaled to address the increasing number and variety of therapeutically relevant genomic alterations that occur across hundreds of cancer-related genes (Cancer Genome Atlas N, 2012; Cancer Genome Atlas Research N, 2012; Nik-Zainal et al. 2012a,b; Stephens et al. 2012).
Next-generation sequencing (NGS), also known as massively parallel sequencing, is therefore becoming an attractive clinical diagnostic tool since it is able to accurately detect most genomic alterations in a single assay (Roychowdhury et al. 2011; Liang et al. 2012; Craig et al.2013; Frampton et al. 2013). However, the clinical practice of this technology as a routine diagnostic test is still challenging. Firstly, the majority of cancer specimens are formalin-fixed, paraffin-embedded (FFPE), a process can damage DNA in different extends depending on the pathology processing protocol and the age of the sample (Hadd et al. 2013). Therefore, robust DNA extraction and sequencing library construction protocols need be standardized to improve the NGS data quality of FFPE samples. Secondly, many samples available for testing are small amount of material obtained from biopsies, which require optimized protocols that accommodate limited amount of DNA input (Kerick et al. 2011). Thirdly, some clinical specimens present low tumor content, which will influence the sensitivity of detection. As a result, uniformly high sequence coverage across all regions of interest and appropriate analysis algorithms are required.
In this study, we have developed and validated a NGS-based cancer genomic diagnosis test targeting 115 cancer-related and therapeutically relevant genes on multiple types of cancer and specimens. We have assessed the analytical sensitivity, specificity, and accuracy of the assay. We also developed NGS bioinformatics analysis pipeline for detecting base substitutions,indels, and gene copy number variations (CNVs), which can be efficiently validated by Sanger sequencing or real-time quantitative PCR (qPCR) method. Our study showed that NGS-based molecular diagnosis test is more sensitive in detecting genomic alterations in cancer, and supported a direct clinical use for this method to guide targeted therapy.
Materials and Methods
Ethical compliance
The patient information and clinical samples were obtained from the Ruijin Hospital. The sample collection and preparation protocol was approved by the Ruijin Hospital Ethics Committee (reference number: 2013-70).
DNA extraction
Four to eight 5–10 μm FFPE sections were obtained per case. FFPE sections were then scraped into microcentrifuge tubes. The tissues were deparaffinized with 1 mL xylene at 56°C for 10 min, washed with 1 mL 100% ethanol for 5 min at RT, and then dried at 37°C for 10 min. QIAamp DSP DNA FFPE tissue kit (Qiagen, Valencia, CA, USA) was used to extract the genomic DNA from FFPE samples and DNeasy Blood & Tissue kit (Qiagen) was used to extract genomic DNA from blood and body fluid with in-house modifications. DNA concentration was determined by Qubit dsDNA HS assay kit on the Qubit Fluorometer according to the manufacturing protocol (Life Technologies, Carlsbad, CA, USA). DNA quality (A260/280 and A260/230) was measured by Nanodrop-2000 (Thermo Fisher Scientific, Waltham, MA, USA).
Library preparation
Sequencing library was prepared by Illumina TruSeq DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturing protocol. In brief, genomic DNA sample was fragmented into 350 or 550 bp in AFA fiber snap-cap microTUBE using Covaris M220 (Covaris, Woburn, MA, USA). End repair and size selection were performed according to the fragment size, followed by 3′ end adenylation. Finally, multiple indexing adapters were ligated to the ends of the DNA fragments. Library concentration was determined using Qubit according to the manufacturing protocol. For low DNA input samples, PCR-free library was further amplified with Illumina p5 (AATGATACGGCGACCACCGA) and p7 (CAAGCAGAAGA-CGGCATACGA) primers in NEB Next High-Fidelity 2XPCR Master Mix (NEB, Ipswich, MA, USA).
Hybrid capture and sequencing
Different libraries with unique indexes were pooled together with desirable ratio to up to 2 μg of total library input. A quantity of 5 μg human cot-1 DNA (Life Technologies) and 1 nmol of each xGen Universal blocking oligos (p5 or p7; IDT, Coralville, IA, USA) were added as blocking reagents. TruSight Cancer Panel Probes (Illumina) and customized xGen lockdown probes (IDT) were used for targeted enrichment, which collectively targets 115 cancer-related genes (Table S1). A quantity of 10 μL 2 × Hybridization buffer (0.5 mol/L Sodium phosphate buffer, pH 7.0, 1% SDS, 2 mmol/L EDTA, 2 × SSC and 4 × Denhardt's solution) was added to make the total reaction volume of 20 μL. The hybridization mix was denatured on a thermal cycler at 95°C for 5 min, and then incubated 30 cycles of 1 min duration, starting at 94°C, then decreasing 1°C per cycle with final incubation at 65°C for 16–24 h. Dynabeads M-270(50 μL); (Life Technologies) was washed with Bind and Wash buffer (10 mmol/L Tris-HCl, pH7.5, 2 mol/L NaCl, 1 mmol/L EDTA and 0.1% Tween-20). Hybridization reaction was added to Dynabeads M-270, and incubated for 30 min at RT with rotation. Beads were then washed at 65°C with Wash buffer I (1 × SSC/0.1% SDS) for 5 min, Wash buffer II (0.1 × SSC/0.1% SDS) for 5 min twice, and at RT with Wash buffer II for 5 min, and finally, Wash buffer III (0.2 × SSC) for 30 sec. Captured libraries were eluted from beads by boiling beads in DNase/RNase-free water at 98°C for 10 min, followed by postcapture amplification with Illumina p5 and p7 primers in NEB Next High-Fidelity 2 × PCR Master Mix. Postcapture amplified library was purified and quantified by qPCR using KAPA Library Quantification kit (KAPA Biosystems, Boston, MA, USA). Library fragment size was determined by Agilent Technologies 2100 Bioanalyzer using a High Sensitivity DNA chip (Agilent Technologies, Santa Clara, CA, USA). Capture-enriched library was sequenced on Illumina MiSeq NGS platform (Illumina) according to its instruction.
Sequence data processing
Trimmomatic (Bolger et al. 2014) was used for FASTQ file quality control. Leading/trailing low quality (below quality 15) or N bases were removed. Reads from each sample were mapped to reference sequence hg19 (Human Genome version 19) using Burrows–Wheeler Aligner (BWA) (Li and Durbin 2009) with modified parameters. SNPs/indels were identified using modified Haplotype Caller in Genome Analysis Toolkit (GATK) (DePristo et al. 2011). Enrichment efficiency was determined based on the percentage of reads that map to the targeted regions with 150 bp padding. (CNVs) were detected using ADTEx (Amarasinghe et al. 2013) with default parameters. In brief, CNVs were identified using tested sample and normal human hapmap DNA NA18535 average read depths at each captured region (exonic region). Proposed discrete wavelet transform (DWT) was used to reduce intrinsic noise. The copy number gains/losses of each targeted region are performed by a Hidden Markov Model (HMM).
Validation of SNPs/Indels and CNVs
SNPs/Indels were validated by Sanger sequencing. 200–500 bp of targeted DNA area was amplified by PCR using 2× AccuStartTM II PCR SuperMix (Quanta BioSciences, Gaithersburg, MD, USA). PCR products were purified and sequenced by ABI 3730xl DNA Analyzer.
For CNVs validation, qPCR primers were designed for targeted exons of test genes and ZNF80 (reference gene) by Primer-blast (Table S2). Normal human hapmap genomic DNA NA18535 was used as normal control sample. qPCR reactions was performed in triplicates using 2× SYBR Select Master Mix (Life Technology).
Results
Extraction of DNA from clinical specimens
In this study, we chose 14 samples from 10 different patients with different specimens and cancer types (Table 1). These samples included blood, tumor FFPE, and body fluid from the patients of lung cancer, colon cancer, rectal cancer, breast cancer, and neuroectodermal tumor crossing different genders and age ranges. Four cancer patients (patient 7–10) had both tumor/body fluid and matching blood samples.

Genomic DNA from blood samples or body fluid was extracted using DNeasy Blood & Tissue kit with good quantity and quality. However, DNA extraction from FFPE samples remains a challenge. To optimize the extraction condition, we have explored several different methods and established the protocol using xylene for deparaffinization followed by extracting DNA with QIAamp DSP DNA FFPE tissue kit. In general, genomic DNA extracted from FFPE samples had certain level of fragmentation. As shown in Figure S1, FFPE DNA sample 1–2 showed a moderate fragmentation, while sample 3 showed a severe fragmentation with DNA fragments ranging from 100 to 1000 bp. We also observed that DNA quantification with Qubit dsDNA HS assay was more accurate compared with Nanodrop analysis. Using our optimized extraction protocol, the FFPE-derived tumor DNA samples were all of quantity and quality sufficient for constructing NGS libraries on Illumina sequencing platform.
Targeted next-generation sequencing for cancer-related genes
In this study, we have developed an NGS-based cancer genomic diagnostic test (Fig. 1) targeting 115 cancer-related and therapeutically relevant genes (Table S1). Fragmented DNA underwent whole-genome sequencing library construction. Size distribution of the constructed libraries was analyzed by Agilent Bioanalyzer (Fig. S2A). We note that the average library insert size of FFPE genomic DNA was smaller than blood counterparts, due to the fragmented nature of FFPE DNA. Regions or genes of interest were then capture enriched by biotin-labeled DNA probes through hybridization, and amplified postenrichment. Using the Illumina Miseq platform, the hybrid-capture-enriched libraries were sequenced to high uniform depth. Library preparation and target enrichment protocols have been optimized to assure even coverage, low PCR duplicates, and robust performance for different type of samples.
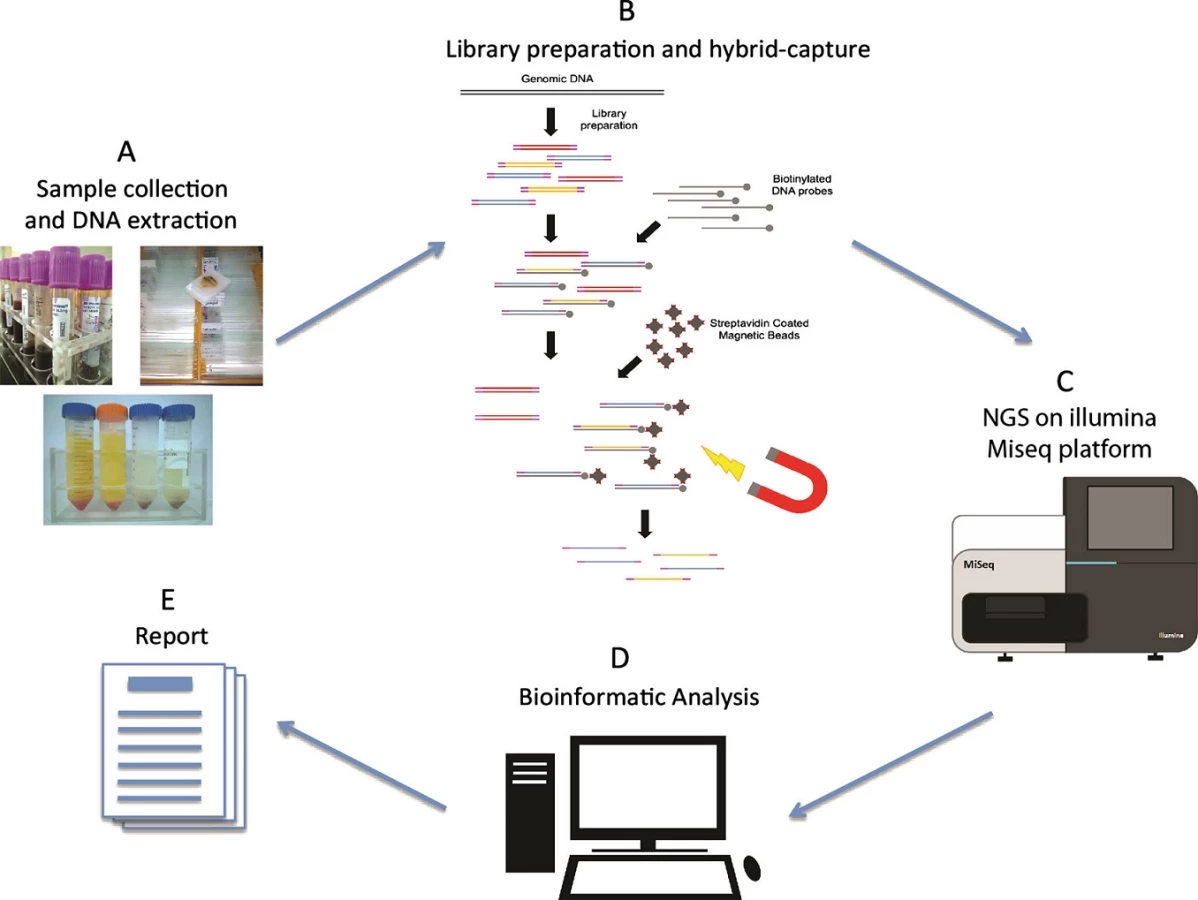
Sequencing data was analyzed by our self-developed bioinformatics analysis pipeline for accurately detecting multiple classes of genomic alterations, including base substitutions (SNVs), indels, and gene (CNVs) (See 'Materials and Methods'). The target capture and sequencing performance were summarized in Table 2. Blood samples were sequenced at 50–100× mean coverage, while tumor samples were sequenced up to 200–500× mean coverage depending on its tumor content within the samples in order to identify low abundance gene mutations. With our optimized DNA extraction and library preparation protocols for the poor quantity or quality samples, the uniformity of coverage at regions of interest (percentage of coverage >0.2× mean coverage) was able to reach 93% for all types of samples together with significantly reduced PCR duplicates. Our protocol dramatically improved the coverage depth with the similar amount of sequencing data, resulting in >92% of target bases being spanned by at least 50 sequencing reads for tumor samples. The on-target rate of all the sequencing reads was able to reach 75–88% on our 115 genes target panel.

Identification and validation of SNP and indels in targeted genes
SNPs and indels were identified using Haplotype Caller in GATK (DePristo et al. 2011). The known germline variants captured in dbSNP and in the 1000 genomes project were removed from all the SNPs called, thus showing the list of private germline variants and somatic mutations. By comparing with tumor-matching blood sample control, the private germline variants were further removed from somatic mutations (Fig. 2). The examples for SNPs and indels detected by NGS were shown in Figure 3. To validate the accuracy of base substitution/indels detection, Sanger sequencing was used for validating the mutations contained in the PCR amplified DNA fragments. In total, 28 base substitution/indels detected by NGS pipeline were tested (Table S3 and Fig. 3), within which 27 could be confirmed by Sanger validation. Studies have shown that the value of the mutant allele frequency, beyond which it is not detectable with confidence, is around 15% for the point mutations, and around 10% for the frame shift mutation (Chen et al. 2014). The mutation RECQL4 (c.448T>A), which could not be validated by Sanger sequencing, had a low frequency as 9% (46 out of 511 reads), suggesting that Sanger sequencing is less capable of detecting low-frequency mutation. The cut-off value of mutant frequency to be reported was set at 5% for tumor samples and 10% for blood samples, with at least 5 reads for mutant allele.


Identification and validation of CNV in targeted genes
CNVs were detected using ADTEx (Amarasinghe et al. 2013) (See 'Materials and Methods'). The examples for copy number gain (ERBB2and KRAS) and loss (RB1) detected by NGS were shown in Figure 4. To validate the accuracy of CNVs detection, real-time qPCR was used for targeted exons of test genes and ZNF80 gene (reference gene) (Fig. 5). Normal human hapmap genomic DNA NA18535 was used as normal control sample. The results showed that the copy number change identified by NGS was validated by the qPCR results.


Clinical implications for target therapy
From our NGS-based cancer genes genomic testing, functional genetic mutations were detected in these patient samples (Table S4). The detected actionable mutations along with the direct clinical implications were shown in the Table 3. Here, we found, in ERBB2 gene (EGFRfamily member, also known as HER2), the c.2329G>T missense mutation caused amino acid substitution (p.Val777Leu) which resulted in excessive activation of downstream signaling pathways in breast cancer, lung cancer, and other tumors (Greulich et al. 2012; Bose et al.2013). Tumor cells with activating mutations in ERBB2 gene respond well to ERBB2 inhibitors such as Trastuzumab (Table 3) (Bose et al.2013).

The EGFR is another EGFR family member involved in the pathogenesis and progression of different carcinomas (Normanno et al. 2006). In our study, we detected exon 19 deletion or exon 21 L858R point mutation in a NSCLC patient. These mutations increased EGFR kinase activity and resulted in hyperactivation of downstream prosurvival signaling pathways (Ladanyi and Pao 2008). In the treatment of mutatedEGFR lung cancer, the first generation of tyrosine kinase inhibitors (TKIs) is commonly used, however, the efficacy of TKIs is limited due to the emergence of drug-resistant secondary mutation T790M (Table 3), which increases ATP affinity at the ATP-binding pocket and confers drug resistance (Yun et al. 2008). Thus, the irreversible inhibitors, such as Afatinib and Lapatinib, are capable of overcoming this resistance through covalent binding.
Inactivation of TP53 occurs in more than half (~60%) of the cancers, and is a sign of poor prognosis in many types of cancer (Olivier et al.2010; Muller and Vousden 2013). We found that c.472C>T mutation hotspot in TP53 caused premature termination of codon formation, resulting in a truncated p53 that promoted tumor development and drug resistance to platinum treatment (Brachova et al. 2015). Additionally, we detected c.3310G>C polymorphism (rs17655) in ERCC5 (also known as XPG), which is involved in platinum-based drug-induced DNA damage repair (Saldivar et al. 2007). The mutated ERCC5 (p.Asp1140His, Table 3) lost the ability to repair DNA damage caused by platinum-based drug treatment, resulting in increased toxicity (Zhu et al. 2012; He et al. 2013). These results indicate that the actionable mutations detected by our NGS protocol are capable of providing more accurate information for treating cancer patients, in contrast this valuable information may be missed in the regular Sanger sequencing, particularly when the sample contains limited tumor cells or the gene shows low-mutating frequency.
Discussion
Formalin fixation and paraffin embedding (FFPE) is a standard method for long-term preservation of most archived pathological specimens. FFPE tissue is an excellent source of DNA, but its extraction remains a challenge. Formaldehyde, the effective component of formalin, leads to the generation of cross-linking between nucleic acids and proteins (Gilbert et al. 2007), and causes nucleic acids to fragment because of fixation process conditions, such as extremely low pH (<1). Cross-linking not only causes problems in DNA extraction, but blocks PCR amplification. Considerable effort has been made to optimize methods for extracting high-quality DNA from FFPE samples. Shi et al. (2004) suggested that heating FFPE samples at a higher temperature in 0.1 mol/L NaOH solution highly increased the efficiency of DNA extraction. In our protocol, we have adopted the step for heating protein K digested FFPE DNA samples at 90° for 1 h, which greatly improved the productivity of FFPE derived DNA.
Poor quality and low amount of DNA also greatly influences the library preparation efficiency. Using Illumina TruSeq DNA PCR-Free Sample Preparation Kit, sequencing library could be successfully generated from as little as 25 ng genomic DNA. Limited PCR amplification cycles were applied to low DNA input samples in order to increase the amount of library for later enrichment with minimum increase on PCR duplicates using NEB Next High-Fidelity PCR master mix, which is specially optimized for the robust, high-fidelity amplification of NGS libraries even with GC-rich amplicons. On the other hand, libraries generated from poor quality DNA tented to have low PCR efficiency. Therefore, comprehensive pooling guideline needed to be applied in order to compensate the different PCR efficiency for each sample during postcapture PCR amplification.
The percentage of tumor content greatly influences the sensitivity of mutation identification, especially by traditional Sanger sequencing method. In our study, an EGFR p.Thr790Met mutant with 13% frequency was detected by NGS analysis in one patient's body fluid, which could be barely detected by Sanger validation. It is very easy to be missed when doing de novo testing for this mutant using Sanger method since it is very close to the detecting limit. As a result, important drug-resistant information can be missed, and dramatically influences patient treatment decision. Therefore, our NGS-based cancer genes genomic testing is a sensitive and efficient method to detect low abundant mutations. On the other hand, traditional clinical testing can only detect very limited markers, which will lose the whole picture of cancer genome. For example, in one patient with EGFR activating mutation, we also detected KRAS amplification, which will not be tested at the same time by traditional clinical testing, but will cause EGFR-TKI resistance. As many more cancer-related and therapeutically relevant genes have been discovered, additional target genes need to be added to our current panel. To this end, IDT xGEN lockdown probes greatly offered us the flexibility of expanding the current panel with additional customized DNA probes.
In summary, we have developed and validated an NGS-based cancer genomic diagnosis test targeting 115 cancer-related and therapeutically relevant genes on multiple types of cancer and specimens including difficult FFPE DNA samples. Using our self-developed NGS data bioinformatics analysis pipeline, we were able to detect base substitutions, indels and gene CNVs. Our test possesses high analytical sensitivity, specificity and accuracy, supported a direct clinical use for this method to guide targeted therapy.
Supporting Information
Table S1. Gene targeted in hybridization capture.
Table S2. qPCR primer sequences.
Table S3. Summary of SNVs/Indels validation by Sanger sequencing.
Table S4. Mutation identified in all samples.
Figure S1. Integrity of genomic DNA extract from blood and FFPE samples.
Figure S2. Size distribution of sequencing library.
Acknowledgment
This work is supported by the research grant of Science & Technology Commission of Shanghai (grant no: 14XD1402800).
Conflict of Interest
Xue Wu, Chao Zhang, Xiaofang Huang, Zhili Chang, and Yang Shao are the employees or shareholders of Geneseeq Technology Inc.
Funding Information
This work is supported by the research grant of Science & Technology Commission of Shanghai (grant no: 14XD1402800).
[Corrections added after initial online publication on January 10, 2016: the affiliations of the following authors have been changed: Jing Xie, Xiongxiong Lu, XinjingWang, ChenleiWen, Xiaomei Tang, Minmin Shi, Qian Zhan, Hao Chen, Xiaxing Deng, Chenghong Peng, Yuan Fang and Baiyong Shen.]
† These authors contributed equally to this work.
Received: 4 November 2015
Revised: 10 December 2015
Accepted: 12 December 2015
Version of Record online: 10 January 2016
* Correspondence:
Baiyong Shen and Yuan Fang
Research Institute of Pancreatic Disease, Ruijin Hospital, 197 Ruijin Er Road, Shanghai 200025, China
Tel: +86-21-34187470
Fax: 86-21-64373909
E-mails: shenby@shsmu.edu.cn and yuan.fang@shsmu.edu.cn
Yang Shao
Geneseeq Technology Inc., Suite 300, MaRS Centre, South Tower, 101 College Street, Toronto, ON, M5G1L7, Canada.
Tel: +1-647-255-1075
Fax: 1-647-255-1076
E-mail: yang.shao@geneseeq.com
Zdroje
1. Amarasinghe, K. C., J. Li, and S. K. Halgamuge. 2013. CoNVEX: copy number variation estimation in exome sequencing data using HMM. BMC Bioinf. 14(Suppl. 2):S2.
2. Barretina, J., G. Caponigro, N. Stransky, K. Venkatesan, A. A. Margolin, S. Kim, et al. 2012. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483 : 603–607.
3. Bolger, A. M., M. Lohse, and B. Usadel. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 : 2114–2120.
4. Bose, R., S. M. Kavuri, A. C. Searleman, W. Shen, D. Shen, D. C. Koboldt, et al. 2013. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 3 : 224–237.
5. Brachova, P., S. R. Mueting, M. J. Carlson, M. J. Goodheart, A. M. Button, S. L. Mott, et al. 2015. TP53 oncomorphic mutations predict resistance to platinum and taxanebased standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 46 : 607–618.
6. Camidge, D. R., W. Pao, and L. V. Sequist. 2014. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat. Rev. Clin. Oncol. 11 : 473–481.
7. Cancer Genome Atlas N. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490 : 61–70.
8. Cancer Genome Atlas Research N. 2012. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489 : 519–525.
9. Chen, Z., K. Moran, J. Richards-Yutz, E. Toorens, D. Gerhart, T. Ganguly, et al. 2014. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum. Mutat. 35 : 384–391.
10. Chin, L., and J. W. Gray. 2008. Translating insights from the cancer genome into clinical practice. Nature 452 : 553–563.
11. Craig, D. W., J. A. O’Shaughnessy, J. A. Kiefer, J. Aldrich, S. Sinari, T. M. Moses, et al. 2013. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 12 : 104–116.
12. DePristo, M. A., E. Banks, R. Poplin, K. V. Garimella, J. R. Maguire, C. Hartl, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43 : 491–498.
13. Dias-Santagata, D., S. Akhavanfard, S. S. David, K. Vernovsky, G. Kuhlmann, S. L. Boisvert, et al. 2010. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol. Med. 2 : 146–158.
14. Diaz, L. A. Jr, R. T. Williams, J. Wu, I. Kinde, J. R. Hecht, J. Berlin, et al. 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486 : 537–540.
15. Frampton, G. M., A. Fichtenholtz, G. A. Otto, K. Wang, S. R. Downing, J. He, et al. 2013. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31 : 1023–1031.
16. Garnett, M. J., E. J. Edelman, S. J. Heidorn, C. D. Greenman, A. Dastur, K. W. Lau, et al. 2012. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483 : 570–575.
17. Gilbert, M. T., T. Haselkorn, M. Bunce, J. J. Sanchez, S. B. Lucas, L. D. Jewell, et al. 2007. The isolation of nucleic acids from fixed, paraffin-embedded tissues-which methods are useful when? PLoS One 2:e537.
18. Greulich, H., B. Kaplan, P. Mertins, T. H. Chen, K. E. Tanaka, C. H. Yun, et al. 2012. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc. Natl. Acad. Sci. USA 109 : 14476–14481.
19. Hadd, A. G., J. Houghton, A. Choudhary, S. Sah, L. Chen, A. C. Marko, et al. 2013. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffinembedded and fine-needle aspiration tumor specimens. J. Mol. Diagn. 15 : 234–247.
20. He, C., Z. Duan, P. Li, Q. Xu, and Y. Yuan. 2013. Role of ERCC5 promoter polymorphisms in response to platinum-based chemotherapy in patients with advanced non-small-cell lung cancer. Anticancer Drugs 24 : 300–305.
21. International Cancer Genome C; Hudson, TJ, Anderson, TJ, A Artez, AD Barker, C Bell, RR Bernabe, et al. 2010. International network of cancer genome projects. Nature 464 : 993–998.
22. Kerick, M., M. Isau, B. Timmermann, H. Sultmann, R. Herwig, S. Krobitsch, et al. 2011. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genomics 4 : 68.
23. Ladanyi, M., and W. Pao. 2008. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod. Pathol. 21(Suppl. 2):S16–S22.
24. Li, H., and R. Durbin. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 : 1754–1760.
25. Liang, W. S., D. W. Craig, J. Carpten, M. J. Borad, M. J. Demeure, G. J. Weiss, et al. 2012. Genome-wide characterization of pancreatic adenocarcinoma patients using next generation sequencing. PLoS One 7:e43192.
26. MacConaill, L. E., C. D. Campbell, S. M. Kehoe, A. J. Bass, C. Hatton, L. Niu, et al. 2009. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One 4:e7887.
27. Mardis, E. R. 2012. Genome sequencing and cancer. Curr. Opin. Genet. Dev. 22 : 245–250.
28. McCourt, C. M., D. Boyle, J. James, and M. Salto-Tellez. 2013. Immunohistochemistry in the era of personalised medicine. J. Clin. Pathol. 66 : 58–61.
29. Muller, P. A., and K. H. Vousden. 2013. p53 mutations in cancer. Nat. Cell Biol. 15 : 2–8.
30. Nik-Zainal, S., L. B. Alexandrov, D. C. Wedge, P. Van Loo, C. D. Greenman, K. Raine, et al. 2012a. Mutational processes molding the genomes of 21 breast cancers. Cell 149 : 979–993.
31. Nik-Zainal, S., P. Van Loo, D. C. Wedge, L. B. Alexandrov, C. D. Greenman, K. W. Lau, et al. 2012b. The life history of 21 breast cancers. Cell 149 : 994–1007.
32. Normanno, N., A. De Luca, C. Bianco, L. Strizzi, M. Mancino, M. R. Maiello, et al. 2006. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366 : 2–16.
33. Olivier, M., M. Hollstein, and P. Hainaut. 2010. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2:a001008.
34. Ross, J. S. 2011. Update on HER2 testing for breast and upper gastrointestinal tract cancers. Biomark. Med. 5 : 307–318.
35. Roychowdhury , S, Iyer, MK, DR Robinson, RJ Lonigro, YM Wu, X Cao, et al. 2011. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci. Transl. Med. 3 : 111ra121.
36. Saldivar, J. S., X. Wu, M. Follen, and D. Gershenson. 2007. Nucleotide excision repair pathway review I: implications in ovarian cancer and platinum sensitivity. Gynecol. Oncol. 107(1 Suppl. 1):S56–S71.
37. Shi, S. R., R. Datar, C. Liu, L. Wu, Z. Zhang, R. J. Cote, et al. 2004. DNA extraction from archival formalin-fixed, paraffin-embedded tissues: heat-induced retrieval in alkaline solution. Histochem. Cell Biol. 122 : 211–218.
38. Stegmeier, F., M. Warmuth, W. R. Sellers, and M. Dorsch. 2010. Targeted cancer therapies in the twenty-first century: lessons from imatinib. Clin. Pharmacol. Ther. 87 : 543–552.
39. Stephens, P. J., P. S. Tarpey, H. Davies, P. Van Loo, C. Greenman, D. C. Wedge, et al. 2012. The landscape of cancer genes and mutational processes in breast cancer. Nature 486 : 400–404.
40. Stratton, M. R., P. J. Campbell, and P. A. Futreal. 2009. The cancer genome. Nature 458 : 719–724.
41. Thomas, R. K., A. C. Baker, R. M. Debiasi, W. Winckler, T. Laframboise, W. M. Lin, et al. 2007. High-throughput oncogene mutation profiling in human cancer. Nat. Genet. 39 : 347–351.
42. Yun, C. H., K. E. Mengwasser, A. V. Toms, M. S. Woo, H. Greulich, K. K. Wong, et al. 2008. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 105 : 2070–2075.
43. Zhu, M. L., M. Wang, Z. G. Cao, J. He, T. Y. Shi, K. Q. Xia, et al. 2012. Association between the ERCC5 Asp1104His polymorphism and cancer risk: a meta-analysis. PLoS One 7:e36293.
Štítky
GenetikaČlánek vyšel v časopise
Molecular Genetics & Genomic Medicine

2016 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Capture-based next-generation sequencing reveals multiple actionable mutations in cancer patients failed in traditional testing
- Variable expressivity and co-occurrence of LDLR and LDLRAP1 mutations in familial hypercholesterolemia: failure of the dominant and recessive dichotomy
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy