
Dědičné arytmické syndromy v dětském věku
Inherited arrhythmic syndromes in children
Inherited arrhythmic syndromes, also known as cardiac channelopathies, are group of cardiac electrophysiological disorders characterized by a specific pattern on electrocardiogram and a disease specific risk for malignant ventricular arrhythmias. This group includes long QT syndrome (LQT), short QT syndrome (SQT), catecholaminerg polymorphic ventricular tachycardia (CPVT) and a sodium channelopathies (SCN5A) like Brugada syndrome, progressive cardiac conduction disease (PCCD) etc. Channelopathies manifest mostly in form of syncopes, epileptic seizures or sudden cardiac death. Most types of cardiac channelopathies are inherited in autosomal dominant pattern, autosomal recessive variants are rare and associated with a severe course of the disease. In some cases arrhythmic syndromes overlap arrhythmic cardiomyopathies, which are mainly caused by disorders in desmosomal proteins.
The genetic stratification allows in many cases installation of individualized therapy and presymptomatic treatment of mutation carriers, who are at risk of malignant arrhythmias. The proper clinical diagnosis, genetic analysis and long-term treatment demand multidisciplinary approach and close cooperation among pediatric and adult cardiologists, geneticist, molecular geneticist, psychologists, sport and forensic medicine professionals.
Keywords:
Arrhythmia – child – channelopathies – genetic analysis – ventricular tachycardia – dysritmia
Autoři:
T. Tavačová 1; J. Janoušek 1; A. Krebsová 2
Působiště autorů:
Dětské kardiocentrum 2. lékařské fakulty Univerzity Karlovy a Fakultní nemocnice Motol, Praha
1; Klinika kardiologie, Institut klinické a experimentální medicíny, Praha
2
Vyšlo v časopise:
Čes-slov Pediat 2020; 75 (1): 27-33.
Kategorie:
Sympozium: lékařská genetika
Souhrn
Primární dědičné arytmické syndromy patří do skupiny onemocnění, které způsobují poruchy elektrické aktivace srdečních buněk a tím predisponují ke vzniku život ohrožujících arytmií. Mezi dědičné arytmické syndromy patří syndrom dlouhého QT intervalu, krátkého QT intervalu, katecholaminergní polymorfní komorová tachykardie, či onemocnění sodíkového kanálu (gen SCN5A), která zahrnují např. syndrom bratří Brugadových, progresivní převodní poruchy, familiární sick sinus syndrom apod. Volně je k nim řazena i arytmogenní kardiomyopatie, která je však způsobena poruchou desmosomálních proteinů.
Abnormality elektrické aktivace srdečních buněk jsou dány ve většině případů geneticky podmíněnou poruchou funkce iontových kanálů kardiomyocytů, proto se někdy označují i jako kanálopatie a jejich dědičnost je nejčastěji autosomálně dominantní. Prvním příznakem těchto syndromů můžou být palpitace, synkopy, křeče, ale i náhlá srdečná smrt. Jejich příčinou je shodně u všech uvedených onemocnění maligní komorová tachy-arytmie vedoucí k mozkové hypoxii. Diagnostika těchto syndromů je založena na symptomech, typickém EKG obrazu a genetické stratifikaci, která přispívá k volbě terapie včetně specifických doporučení úpravy životního stylu. Nutná je úzká vzájemná mezioborová spolupráce pediatra, dětského kardiologa, dospělého kardiologa, klinického genetika, sportovního lékaře a v neposlední řadě i soudního lékaře a psychologa.
Klíčová slova:
arytmie – deti – poruchy srdečního rytmu – tachykardie – dysrytmie – genetická analýza
ÚVOD
Dědičné arytmické syndromy (ArS) představují skupinu onemocnění s narušenou elektrickou aktivací srdečních buněk (změny akčního potenciálu) a jsou spojeny s rizikem komorových arytmií a náhlé srdeční smrti. Nejčastěji jsou výrazem vrozené poruchy funkce minerálových transportních kanálů pro ionty sodíku, draslíku a kalcia. Proto se dědičné arytmické syndromy také nazývají srdečními „kanálopatiemi“. Diagnostika těchto syndromů je založena na symptomech, specifickém EKG obrazu a genetické stratifikaci, která přispívá k volbě terapie včetně specifických doporučení úpravy životního stylu [1].
Mezi ArS patří syndrom dlouhého QT intervalu (LQT), krátkého QT intervalu (SQT), katecholaminergní polymorfní komorová tachykardie (CPVT), či onemocnění sodíkového kanálu (gen SCN5A), která zahrnují např. syndrom bratří Brugadových, progresivní převodní poruchu, familiární sick sinus syndrom apod. Kombinaci fenotypických projevů mutací v genu SCN5A můžeme někdy nalézt i u jednoho pacienta, nebo jeho příbuzných. V tomto smyslu se hovoří o tzv. překrývajících se syndromech („overlap syndrom“). Volně se k dědičným arytmickým syndromům řadí i primární onemocnění srdečního svalu způsobené poruchou desmosomálních bílkovin, arytmogenní kardiomyopatie (AC).
Prvním příznakem všech zmíněných onemocnění mohou být palpitace, synkopy, bezvědomí někdy připomínající epileptický záchvat, ale i náhlá srdečná smrt bez morfologických změn srdečního svalu u dospělých (Sudden Arrhythmic Death Syndrome, SADS), dětí i kojenců (Sudden Infant Death Syndrome, SIDS).
Dědičnost výše uvedených onemocnění je především autosomálně dominantní, kdy dědičná vloha se dědí s 50% pravděpodobností, bez závislosti na pohlaví. Naštěstí, na základě neúplné penetrance a expresivity, mají ne všichni nosiči dědičné vlohy klinické onemocnění, nebo onemocní s různou intenzitou (obr. 1).
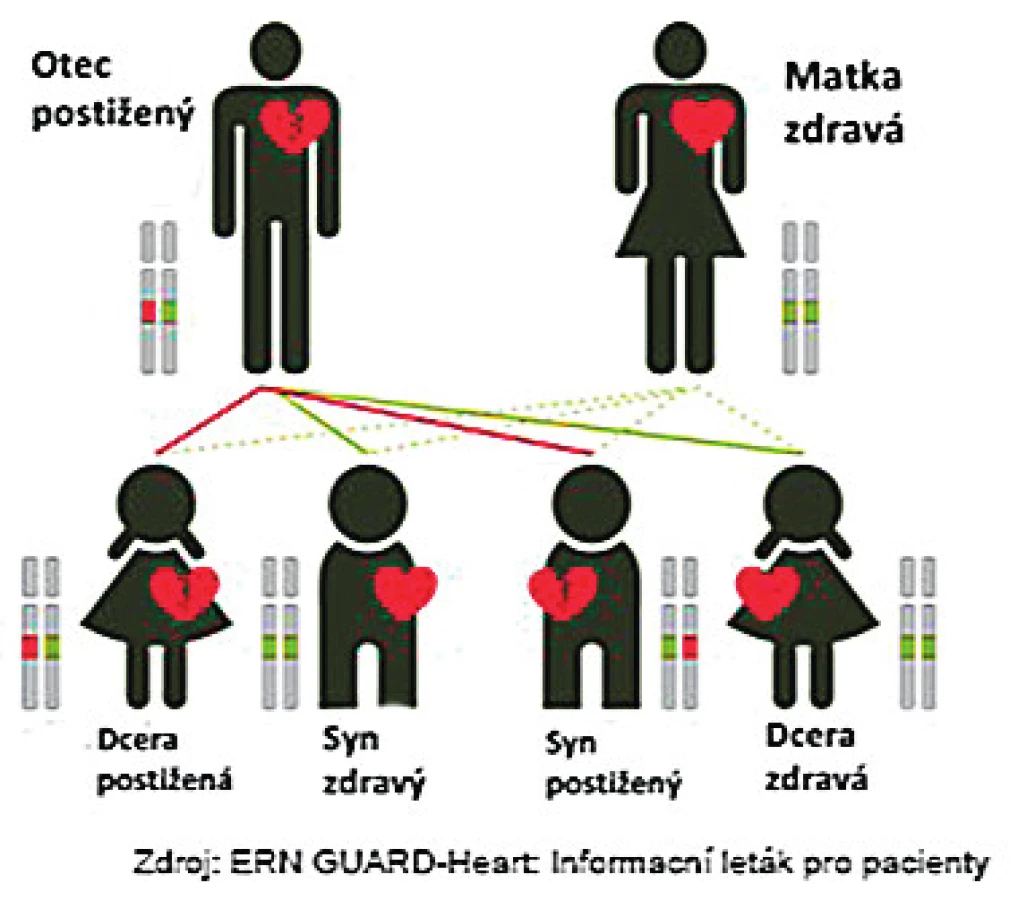
Zásadní roli v péči o pacienty s dědičným arytmickým syndromem hraje tzv. kaskádový rodinný screening sloužící k detekci onemocnění u asymptomatických příbuzných probanda s klinickým onemocněním a tedy i pro primární prevenci významných poruch srdečního rytmu a náhlého úmrtí.
V případě všech zmíněných onemocnění se jedná o monogenně dědičná onemocnění, která jsou způsobena variantami v různých genech. Přelom v genetické diagnostice přinesla metoda sekvenování nové generace (next generation sequencing – NGS), která umožňuje získat sekvenci vícero genů najednou až celé kódující i nekódující sekvence dědičné molekuly DNA. Nález příčinné varianty DNA se podaří u cca 50–60 % rodin. Negativní výsledek molekulárně genetického vyšetření nevylučuje přítomnost dědičného onemocnění, pouze odráží naše současné hranice diagnostických možností a znalostí.
Diagnostika a terapie dědičných arytmických syndromů u dětí a jejich rodin je založena na multidisciplinární spolupráci pediatra, dětského i dospělého kardiologa/arytmologa, sportovního lékaře, klinického a molekulárního biologa a v neposlední řadě i soudního lékaře nebo psychologa [2].
Aktuálně se péče o dětské pacienty soustřeďuje v ČR do Dětského kardiocentra FN Motol a Oddělení dětské kardiologie FDN Brno, kde je současně zajištěno napojení na další výše uvedené odbornosti k zajištění péče o všechny rodinné příslušníky v riziku.
SYNDROM DLOUHÉHO QT INTERVALU
Syndrom dlouhého QT intervalu (LQTS) je onemocnění definované klinicky abnormálním prodloužením korigovaného QT intervalu k tepové frekvenci nejčastěji za využití Bazettovy formule QTc = QT/√RR, kde QT je délka QT intervalu a RR hodnota délky jednoho srdečního cyklu, obě v sekundách (>460 ms u dětí, >450 ms u dospělých mužů a >470 u dospělých žen ve vícero EKG) (tab. 1). Abnormální prodloužení korigovaného QT intervalu predisponuje ke vzniku život ohrožujících arytmií, nejčastěji polymorfní komorové tachykardie typu Torsades de Pointes (TdP) (obr. 2). Prevalance LQTS je asi 1 : 2000 [3].


Podle genového defektu v současné době rozeznáváme 17 typů LQT. Nicméně asi 70 % pacientů s diagnózou LQTS má LQT typ 1, 2 nebo 3 se specifickým obrazem v EKG (obr. 3). Pro LQT 1 jsou zodpovědné DNA varianty v genu pro draslíkový kanál typu KCNQ1, pro LQT2 v genu pro draslíkový kanál typu KCNH2 a pro LQT3 v genu pro sodíkový kanál SCN5A.

Pro LQT typu 1 byla popsána i autosomálně recesivní forma onemocnění s přítomností patogenních variant KCNQ1 na obou alelách (syndrom Jervell Lange-Nielsen). Tato forma je mnohem vzácnější, má závažnější průběh a je často asociována s neurosenzorickým typem hluchoty.
V dětském věku a v rané dospělosti se LQTS může manifestovat synkopou, ke které přispějí fyzické nebo psychické zátěže spojené s nárůstem sympatické stimulace srdce. Tak je tomu například při sportu (u LQT1 typicky plavání), u emočních situací nebo při náhlých hlasitých zvucích (typicky např. zvonění u LQT2). U LQT 3 vznikají arytmie při bradykardii ve spánku.
Základem diagnostiky je opakovaný nález prodloužení QTc intervalu. Nicméně až jedna třetina pacientů má normální korigovaný QTc interval v klidovém EKG. Mnohem citlivějším vyšetřením k určení prodloužení QT intervalu je zátěžové vyšetření – ergometrie, kde typickým nálezem u LQTS je porucha schopnosti adekvátně zkrátit QT interval ve vyšších frekvencích (při zátěži). Hodnota QTc >/ = 480 ms ve 4. minutě zotavení po zátěži patří mezi diagnostická kritéria. Během Holterovy monitorace EKG se mohou zachytit změny morfologie repolarizační fáze a vztah těchto abnormalit k běžné denní zátěži pacienta. Lze také využít provokační test s adrenalinem k posouzení schopnosti adekvátně zkrátit QT interval při farmakologicky navozené zátěží (diagnostické pro LQT1). V praxi se diagnostika LQTS opírá o Schwartzova kritéria, na základě kterých se přiřadí každému ze symptomů určitá bodová hodnota a výsledný součet pak určí pravděpodobnost diagnózy (tab. 2).

Terapie spočívá v podávání neselektivních betablokátorů (nejlépe nadolol) jak u symptomatických pacientů, tak u asymptomatických nosičů příčinné varianty DNA [4]. Dále se doporučuje vyhýbat se lékům, které mohou prodlužovat QTc interval, jejichž seznam je uveden na www.crediblemeds.com. Je třeba se vyvarovat situací s hypokalémií např. při průjmových onemocněních. U pacientů s LQT3 jsou kromě betablokátorů indikovány i blokátory sodíkového kanálu (např. mexiletin). U pacientů po srdeční zástavě či neúčinné medikaci je nutné zvážit implantaci kardioverter-defibrilátoru (ICD) a/nebo levostrannou sympatickou srdeční denervaci (left sympathetic cardiac denervation, LSCD), kdy se přerušuje nervové spojení vylučující adrenalin v nervových zakončeních myokardu [3, 4].
SYNDROM KRÁTKÉHO QTc INTERVALU
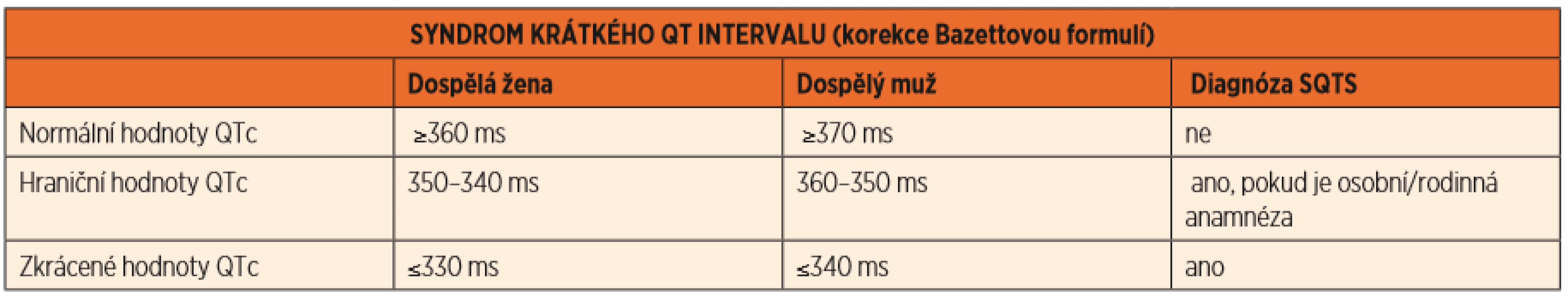
Syndrom krátkého QTc intervalu je velmi vzácné, maligní onemocnění, které je v klidovém EKG charakterizováno zkrácením QTc intervalu <330 ms u dospělého muže a <340 ms u dospělé ženy, v případě prodělané srdeční zástavy již <360 ms (tab. 3).
Toto onemocnění je spojeno často s časnou fibrilací síní, či srdeční zástavou v důsledku komorové fibrilace. Geneticky je podmíněno geny kódujícími draslíkové, vápníkové i sodíkové kanály. Terapie je opět genotypově vázaná a představuje neselektivní betablokátory (nadolol) a blokátory sodíkového kanálu, nejlépe chinidin. V případě prodělané zástavy srdce či jiných rizikových faktorů je implantován ICD [7].
KATECHOLAMINERGNÍ POLYMORFNÍ KOMOROVÁ TACHYKARDIE
Katecholaminergní polymorfní komorová tachykardie (CPVT) je vzácné onemocnění (1 : 10 000), které je charakteristické vznikem život ohrožujících komorových tachykardií v průběhu fyzické nebo psychické zátěže. Klidové EKG je zcela normální.
CPVT je způsobeno poruchou cyklování kalcia mezi sarkoplazmatickým retikulem a cytosolem myokardiální buňky, při které je cytosol přetížen kalciem. U cca 60 % pacientů je CPVT způsobeno dědičnou poruchou funkce ryanodinového receptoru. Vzácněji se popisují příčinné varianty v genu CASQ2, které mohou být jak v autosomálně dominantní, tak i recesivní formě.
Klinická diagnostika CPVT je založena na výskytu typických síňových a komorových arytmií při fyzické nebo farmakologicky navozené zátěži při podávání isoprenalinu (obr. 4).
4A. Klidová EKG křivka.
4B. Zátěžové komorové extrasystoly.
4C. Síňová tachykardie (černá šipka) a bidirekční komorová tachykardie (červená šipka).
4D. Komorová fibrilace.

Terapie CPVT spočívá opět v podávání betablokátorů, nejlépe neselektivních v kombinaci s blokátory sodíkového kanálu (flecainid). Implantace ICD je u tohoto onemocnění velmi sporná, protože adekvátní výboje mohou u pacientů vést k dalšímu vyplavení katecholaminů, které způsobí opětovné komorové arytmie. Obdobně jako u LQT, i u CPVT je terapií volby u rezistentních forem kardiální denervace. Obecně se pacientům doporučuje vyhýbat se sportovním aktivitám [8].
DĚDIČNÉ ARYTMICKÉ SYNDROMY SODÍKOVÉHO KANÁLU SCN5A
Iontový kanál SCN5A je komplexní transmembránový protein. Změny v něm mohou způsobit celou řadu dědičných arytmických syndromů. Mezi tyto patří např. progresivní porucha vedení vzruchu (Progressive Cardiac Conduction Disease, PCCD), tedy forma sinoatriální a atrioventrikulární blokády, dále tzv. multifokální ektopie z blízkosti Purkyněho buněk (Multifocal Ectopic Purkinje-Related Premature Contractions – MEPPC), syndrom bratří Brugadových apod. Některé varianty jsou považovány za molekulární příčinu arytmogenní kardiomyopatie či dilatační kardiomyopatie, čímž fenotypicky dědičné kanálopatie přechází do spektra dědičných kardiomyopatií. Protože kombinaci zmíněných onemocnění můžeme nalézt u jednoho pacienta nebo u jeho příbuzných, hovoří se dnes o tzv. překrývajících se syndromech, tzv. „overlap syndromes“[9].
SYNDROM BRUGADOVÝCH
Brugadových syndrom (BrS) je vzácné dědičné onemocnění, které je charakterizováno typickými změnami EKG ve smyslu sedlovité elevace ST úseku následováné negativní T vlnou. Tyto změny mohou být jen tranzientní, vyvolané typicky horečkou, objemným jídlem a alkoholovým excesem či specifickými léky (např. ajmalin), nebo trvalé (obr. 5). Syndromem Brugadových trpí asi 1 z 2000 lidí v jihovýchodní Asii, ve střední Evropě je toto onemocnění velmi vzácné (1 : 10 000).
5A. Nativní EKG křivka
5B. Po provokaci ajmalinem: vznik Brugada obrazu typu I (šipky)

Ve většině případů se diagnostika BrS opírá o typický nález na EKG spontánně, nebo po jeho vyvolání specifickými léky (ajmalinový test). Diagnóza BrS je stanovena jenom v případě, je-li na klidovém EKG zaznamenána jasná morfologie 1. typu. Pokud pacient nemá žádné symptomy a na EKG křivce je nalezen jenom typ 2 nebo 3 i po ajmalinovém testu, diagnózu BrS nelze potvrdit ani vyloučit (obr. 6).

U 25 % rodin s diagnózou syndromu Brugadových bývá nalezena kauzální mutace v genu SCN5A. U ostatních 75 % rodin se předpokládá komplexnější původ onemocnění, s největší pravděpodobností se bude jednat o vícečetné mutace v různých genech s významným podílem faktorů životního prostředí [10].
Většina jedinců s asymptomatickým BrS nevyžaduje terapii. Důležitá je úprava životního stylu ve smyslu vyhýbání se lékům kontraindikovaným u BrS (www.brugadadrugs.org) a vyhýbání se nadměrnému požití alkoholu a objemných jídel. Sportování není kontraindikováno, ale má-li pacient symptomy v průběhu zátěže, je doporučeno omezit účast na sportovních aktivitách. Zejména u dětských pacientů s tímto syndromem je důležité časně snížit teplotu antipyretiky za hospitalizace a trvalé monitorace EKG, protože právě febrilní stavy predisponují ke vzniku život ohrožujících arytmií. Očkování by mělo probíhat také za hospitalizace a pečlivé monitorace tělesné teploty. U pacientů po srdeční zástavě či dokumentaci setrvalých komorových arytmií je indikována sekundárně preventivní implantace ICD. V případě, že je implantace ICD nemožná nebo kontraindikována, jedinou farmakologickou možností terapie je chinidin.
ARYTMOGENNÍ KARDIOMYOPATIE
Arytmogenní kardiomyopatie (AC) je onemocnění srdečního svalu, které postihuje pravou i levou komoru v kombinaci či jednotlivě. Často prvním a posledním příznakem onemocnění mohou být komorové arytmie. Onemocnění je vzácné, s prevalencí 1 : 5000, má obecně velmi nízkou penetranci, nicméně intenzivní sportovní aktivita vede k progresi tohoto onemocnění a je vedoucí příčinou náhlého úmrtí při sportu.
Geneticky je onemocnění způsobeno hlavně DNA variantami v genech kódujících desmosomální proteiny. Nejčastěji se jedná o geny pro plakophilin 2 (PKP2), desmoplakin (DSP) nebo desmoglein (DSG2). Obzvláště maligní formu kardiomyopatie způsobují DNA varianty v genu pro lamin A/C (LMNA/C), kdy komorovým arytmiím často předchází symptomatická AV blokáda. Poruchy funkce desmosomálních proteinů v typickém případě vedou k časné přestavbě srdečního svalu v tukovou tkáň a tím ke vzniku arytmií a srdečního selhání.
Diagnostika se opírá o typické změny de - a repolarizace v klidovém EKG (obr. 7), nebo o nález pozdních potenciálů v signálově průměrovaném EKG (Signal Averaged ECG, SAECG). Echokardiografické vyšetření může prokázat poruchy kinetiky a dilataci ve vtokové a výtokové části pravé komory či redukci systolické funkce obou komor. Časně je možno myokardiální změny detekovat magnetickou rezonancí s nálezem tukové infiltrace a oblastí pozdního sycení v myokardu (obr. 8) svědčícího pro fibrotizaci. Ke stanovení diagnózy se může využít i katetrizační endomyokardiální biopsie, která ale s ohledem na převážně epikardiální postižení při AC může být velmi neinformativní.


Terapie AC spočívá především v podávání betablokátorů, implantaci ICD v případě resucitované srdeční zástavy a jednoznačném vynechání sportovních aktivit. V případě arytmických bouří je možné provést terapeutickou radiofrekvenční ablaci hlavně z epikardiálního přístupu, která, je-li provedena v expertních centrech, je velmi dobře a dlouhodobě účinná. V ojedinělých případech může být nutná transplantace srdce z důvodu pokročilého srdečního selhání [11].
MUDr. Terézia Tavačová
Dětské kardiocentrum FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: terezia.tavacova@fnmotol.cz
Zdroje
1. Priori SG, Blomström-Lundqvist C, et al. 2015 European Society of Cardiology Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co-chairs. Eur Heart J 2015; 36 (41): 2757–2762.
2. Wilde AAM, Amin A. Channelopathies, genetic testing and risk stratification. Int J Cardiol 2017; 237 : 53–55.
3. Priori SG, Wilde AAM, Horie M, et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. Heart Rhythm 2013; 10 (12): 1932–1963.
4. Ahn J, et al. Effectiveness of beta-blockers depending on the genotype of congenital long-QT syndrome: A meta-analysis. PLoS One 2017; 12 (10): 1–13.
5. Priori SG, Blomström-Lundqvist C, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Eur Heart J 2015; 36 (41): 2793–2867.
6. Mazzanti A, et al. Interplay between genetic substrate, QTc duration, and arrhythmia risk in patients with Long QT Syndrome. J Am Coll Cardiol 2018; 71 (15): 1663–1671.
7. Campuzano O, Sarquella-Brugada G, Cesar S, Arbelo E. Recent advances in short QT Syndrome. Front Cardiovasc Med 2018; 5 : 1–7.
8. Roston TM, et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: Findings from an international multicentre registry. Europace 2018; 20 (3): 541–547.
9. Li W, et al. SCN5A Variants : Association with cardiac disorders. Front Physiol 2018; 9 : 1–13.
10. Walsh R, Wilde AAM. SCN5A variants in Brugada syndrome: True, true false, or false true. J Cardiovasc Electrophysiol 2018;30 (1): 128–131.
11. Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2017; 376 (1): 61–72.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2020 Číslo 1
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- OZP pečuje o zdraví svých pojištěnců již 30 let. V čem je lepší a jaké výhody nabízí?
- Cytomegalovirové infekce u novorozenců a dětí
- Cytomegaloviróza a spalničky u dětí
Nejčtenější v tomto čísle
- Nikdy nie je neskoro – herpetická encefalitída vyvolaná vírusom HSV1
- Dědičné arytmické syndromy v dětském věku
- Genetické příčiny syndromu náhlého úmrtí kojence. Co přineslo sekvenování nové generace
- Rozšířené testování nosičství recesivních chorob (CarrierTest) v klinické praxi
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy