
Talasémie a hemoglobinové varianty u dětí
Thalassemias and hemoglobin variants in children
Hemoglobinopathies (thalassemias and structural hemoglobin variants) represent a group of inhereted microcytic anemias that are rarely diagnosed in our country. In most cases, these are individual patients or families, often with a different ethnic origin. Clinical manifestation can be heterogenic - from mild microcytic hypochromic anemia to severe, lifelong, transfusion-dependent anemia with multiorgan involvement. The aim of this work is to present a group of patients diagnosed with some forms of hemoglobinopathy together with characteristics of individual mutation.
Methods:
32 children were examed for suspected hemoglobinopathy between 2007–2017. Complete blood count, iron metabolism parameters and special hematological tests were evaluated. Mutations in HBA and HBB genes were confirmed by molecular genetic testing.
Results:
We diagnosed eight α-thalassemia carriers, one patient with hemoglobin H disease, eighteen patients with β-thalassemia minor, one patient with β-thalassemia major and four patients with hemoglobin variants (HbE, Hb Monroe, Hb Sydney and sickle cell disease).
Conclusion:
Hemoglobinopathies can be exactly diagnosed by molecular genetic testing – MLPA and Sanger sequencing. As a result of global migration, new mutations of globin genes can be imported in Czech population genofond.
Key words:
hemoglobinopathies, thalassemias, structural hemoglobin variants
Autoři:
L. Sulovská 1; M. Divoká 2; Z. Novák 1; J. Hak 3; M. Orviská 2; D. Pospíšilová 1
Působiště autorů:
Dětská klinika, Fakultní nemocnice a Lékařská fakulta Univerzity Palackého, Olomouc
1; Hemato-onkologická klinika, Fakultní nemocnice a Lékařská fakulta Univerzity Palackého, Olomouc
2; Dětská klinika, Fakultní nemocnice Hradec Králové
3
Vyšlo v časopise:
Čes-slov Pediat 2017; 72 (8): 464-470.
Věnováno panu profesorovi Hrodkovi, zakladateli moderní dětské hematologie v České republice
Souhrn
Hemoglobinopatie (talasémie a hemoglobinové varianty) reprezentují skupinu vrozených mikrocytárních anémií, které jsou raritně diagnostikované i na našem území. Ve většině případů se jedná o jednotlivé pacienty nebo rodiny, často s odlišným etnickým původem. Klinické projevy onemocnění jsou rozdílné – od asymptomatického nosičství mutace až po těžké anémie s nutností pravidelné aplikace erytrocytárních koncentrátů. Cílem této práce je představit soubor pacientů, u kterých byla diagnostikovaná některá z forem hemoglobinopatie spolu s charakteristikou jednotlivých mutací.
Metody:
V letech 2007–2017 bylo vyšetřeno 32 dětí pro podezření na hemoglobinopatii. U pacientů byl vyšetřen kompletní krevní obraz, parametry metabolismu železa a speciální hematologická vyšetření. Jednotlivé mutace v HBA a HBB genech byly prokázané pomocí molekulárně genetických analýz.
Výsledky:
V našem souboru jsme detekovali u osmi pacientů tiché nosičství α-talasémie, u jedné pacientky nemoc hemoglobinu H, osmnáct pacientů s mutací β-globinového genu vedoucí k β-talasémii minor, jednu pacientku s β-talasémií major a čtyři pacienty s hemoglobinovými variantami – HbE, Hb Monroe, Hb Sydney a srpkovitá anémie (HbS).
Závěr:
Hemoglobinopatie lze v dnešní době velmi přesně diagnostikovat pomocí molekulárně genetických vyšetření – MLPA a Sangerova sekvenování. V důsledku celosvětové migrace lze očekávat importování dalších mutovaných alel globinových řetězců do genofondu české populace.
Klíčová slova:
hemoglobinopatie, talasémie, strukturní hemoglobinové varianty
ÚVOD
Hemoglobinopatie představují heterogenní skupinu monogenně dědičných poruch červené krevní řady. Molekulární podstatou onemocnění je porucha syntézy globinové složky hemoglobinu (Hb) vedoucí ke snížení nebo úplné zástavě syntézy některého typu globinového řetězce (talasémie) nebo k syntéze abnormálního globinového řetězce (hemoglobinové varianty). Syntéza některých abnormálních globinových řetězců může být kombinována s redukcí jejich tvorby a mohou tak vznikat hemoglobinové varianty s nerovnováhou α - a β-řetězců s talasemickým klinickým obrazem (tzv. talasemický hemoglobin).
Talasémie jsou způsobeny kvantitativní poruchou syntézy jednoho typu globinového řetězce; nejčastěji se vyskytují α-talasémie (α0-talasémie s naprostou absencí α-globinu nebo α+-talasémie se sníženou syntézou α-globinu z mutované alely) a β-talasémie (analogicky β0 a β+-talasémie). Geny pro jednotlivé globinové řetězce se nachází na chromozomu 16 v HBA lokusu pro α-globin a na 11. chromozomu v lokusu HBB pro β-globin. V diploidním lidském genomu jsou na každém chromozomu (maternálním a paternálním) geny pro α-globin ve dvou kopiích (α1 a α2) a pro β-globin v jedné kopii. Klinická manifestace, diagnostika a léčba jsou detailněji popsány v článku „Talasemické syndromy“ (s. 457–463).
Strukturální Hb varianty jsou způsobeny kvalitativní poruchou syntézy globinového řetězce - dochází ke změně pořadí aminokyselin a vzniku odlišného polypeptidového řetězce. Dosud bylo popsáno více než 1000 různých Hb variant [1].
Na molekulární úrovní se strukturální Hb varianty projevují:
- změnou fyzikálních vlastností Hb (polymerizace HbS v deoxygenovaném stavu nebo tvorba krystalů u HbC);
- vznikem nestabilního Hb (precipitace Hb a vznik Heinzových tělísek, např. Hb Haná nebo Hb Hradec Králové [2, 3];
- změnou afinity Hb ke kyslíku – snížení afinity má za následek periferní cyanózu – Hb Kansas [4], zatímco zvýšená afinita ke kyslíku je kompenzovaná erytrocytózou – Hb Olomouc [5, 6];
- oxidací hemového železa – změna pořadí aminokyselin v místě vazby hemové kapsy vede k oxidaci železa (Fe) z Fe2+ na Fe3+ za vzniku methemoglobinu.
Klinická významnost těchto změn je heterogenní – zahrnuje asymptomatické nosičství mutace, různě vyjádřený stupeň hemolýzy v důsledku přítomnosti precipitovaného Hb až po těžké anémie s nutností transfuze erytrocytárních koncentrátů. Klinické příznaky závisí na typu dědičnosti. Z hlediska formální genetiky vykazují tzv. endemické hemoglobiny (jako jsou HbS, HbC) recesivní dědičnost, zatímco nestabilní Hb varianty a varianty se změněnou afinitou Hb ke kyslíku mají většinou dominantní typ dědičnosti. Talasémie mají typicky recesivní typ dědičnosti.
Hemoglobinopatie se v České republice vyskytují u dětí rodičů pocházejících ze zemí s vysokou frekvencí recesivně dědičných mutovaných alel, kde je heterozygotní nosičství (platí pro talasémie, HbS) nebo homozygotní postižení (u HbC) protektivním faktorem před onemocněním malárií (Středomoří, Afrika aj.) nebo u dětí z rodin s de novo vzniklou mutací globinového řetězce (Hb Haná, Hb Olomouc, Hb Hradec Králové).
METODY
V průběhu deseti let bylo vyšetřeno 32 dětí z 28 nepříbuzných rodin pro podezření na talasémii nebo Hb variantu. Indikací k vyšetření byla mikrocytární hypochromní anémie (po vyloučení nedostatku železa) a u části pacientů také pozitivní rodinná anamnéza. Odběr krevních vzorků byl realizován klasickou venepunkcí. Vyšetření parametrů krevního obrazu bylo provedeno na přístroji Sysmex XE-500 analyzer (Sysmex) podle pokynů výrobce. Hladina plazmatického Fe byla stanovena fotometricky na přístroji Cobas 8000 (Hitachi, Tokyo, Japan), na stejném přístroji byla imunoturbidimetricky vyšetřena i hladina solubilního transferinového receptoru (sTfR). Ke stanovení hladiny feritinu byla použita chemiluminiscenční metoda a analyzátor Architect i2000SR (Abbott Laboratories, Illinois, USA).
Hladina HbA2 byla stanovena chromatograficky na koloně a hladina HbF alkalickou denaturací. Elektroforéza hemoglobinu byla provedena v polyakrylamidovém gelu (PAGE). Při podezření na nestabilní Hb variantu byly provedeny testy stability (izopropanolový test, test na Heinzova tělíska – obr. 1).

K detekci jednotlivých typů mutací v genech HBA a HBB bylo využito pro bodové mutace Sangerovo sekvenování na genetickém analyzátoru ABI PRISM TM 3100 (Applied Biosystems, USA) a vyšetření MLPA (Multiplex Ligation Probe Amplification; MRC-Holland, Holandsko) pro deleční typy mutací.
VÝSLEDKY
α-talasémie
V našem souboru jsme detekovali deleční formu α-talasémie u 9 dětí z 9 nepříbuzných rodin (tab. 1), průměrný věk v době diagnózy byl 9 let (věkové rozmezí 1–16 let). V krevním obraze jsme u těchto pacientů zaznamenali mikrocytózu a hypochromii erytrocytů s kompenzatorním zvýšením počtu erytrocytů (medián: ERY 5,77 x 1012, MCV 63,5 fl; MCH 21,6 pg). Počet retikulocytů byl ve fyziologickém rozmezí. Hladina plazmatického Fe byla u šesti pacientů ve fyziologickém rozmezí, u dvou byla snížená. U tří dětí jsme zaznamenali snížení hladiny Hb, feritinu a zvýšení sTfR, protože se v jejich případě jednalo o kombinaci talasemického nosičství se sideropenií, byla u nich zahájena feroterapie.

U všech pacientů byla normální hladina HbA2, hladina HbF a standardní Hb spektrum na elektroforéze.
Na molekulární úrovni jsme u pěti dětí detekovali nejčastější typ α-talasemické delece dlouhé 3,7 kb, která zahrnuje část α2 i α1 globinových genů (v lokusu tak vzniká z původních dvou α-globinových genů jeden funkční fúzní-α3.7 gen) (obr. 2). Tato delece je typická pro různé populace, nejvíce je zastoupena v populaci indické, africké a středozemní a v populacích zemí Dálného východu. Dědeček jedné pacientky pocházel z Angoly, dědeček druhého pacienta pocházel z oblasti Dálného východu a rodiče třetího pacienta pochází z Pákistánu, u dvou dětí se nepodařilo zjistit etnický původ. Ve všech případech se jednalo o heterozygotní postižení, které označujeme jako tzv. „tiché nosičství“ α-talasémie.
U jednoho pacienta jsme detekovali deleci označovanou jako –SEA, která zahrnuje celé geny α2 i α1, ale i další pseudogeny na HBA lokusu (obr. 2). Pro heterozygoty je typická mírná mikrocytární hypochromní anémie a tato delece je častá v populaci jihovýchodní Asie, náš pacient je původem z Vietnamu.

Další dítě neslo deleci označovanou jako –MED typickou pro středozemní oblast a zahrnující opět oba α-globinové geny na jednom chromozomu (obr. 2). Dědeček této pacientky pocházel z Řecka.
U jednoho dětského pacienta byla zjištěna delece celého HBA lokusu včetně oblasti označované HS-40 (obr. 2), která je regulační oblastí celého HBA lokusu a je zodpovědná za expresi α-globinových genů. Tyto typy delecí bývají diagnostikovány vzácně v různých populacích. Rodina této pacientky nemá informace o jiném etnickém původu svých předků.
U poslední pacientky jsme diagnostikovali nemoc hemoglobinu H, která je způsobena delecí tří funkčních genů pro α-globinové řetězce. Sedmnáctiletá dívka byla odeslaná ke specializovanému hematologickému vyšetření pro symptomatickou mikrocytární hypochromní anémii. Matka pacientky byla vyšetřována a opakovaně neúspěšně léčena preparáty železa pro mikrocytární anémii. Otec dívky byl po splenektomii, indikace není matce dívky známá. Prarodiče dívky nikdy netrpěli žádným hematologickým onemocněním, matka matky pocházela z Itálie.
Při laboratorním vyšetření byla potvrzena výrazná mikrocytóza 59,3 fl (N: 82–98 fl) a hypochromie 17,5 pg (N: 28–34 pg) erytrocytů a snížená hladina hemoglobinu 87 g/l (N: 120–160 g/l). Celkový počet erytrocytů byl v normě. Distribuční šíře erytrocytů byla zvýšená 24,6 % (N: 10–15,2 %). Při mikroskopickém zhodnocení nátěru periferní krve byly zachyceny výrazné morfologické odchylky tvaru erytrocytů – terčovité erytrocyty, poikilocyty, eliptocyty, anulocyty, ojediněle s Howellovými–Jollyho tělísky. Počet retikulocytů byl mírně zvýšený, neodpovídal však tíží anémie (0,029; N: 0,005–0,025). Celkový počet leukocytů, trombocytů a manuální diferenciální rozpočet leukocytů byly v normě. Markery hemolýzy nebyly zvýšené. Součástí základního vyšetření byly parametry metabolismu železa: hladina plazmatického železa a feritinu byly v normě. Nadhraniční hodnoty dosahovala celková vazebná kapacita pro železo (72,5 μmol/l; N: 24,2–70,1 μmol/l) a výrazně zvýšené byly solubilní transferinový receptor (10,4 mg/l; N: 1,9–4,4 mg/l) a hladina hepcidinu 120 ng/ml (střední hodnota zdravé populace 25,86 ng/ml; [7]). V rámci diferenciální diagnostiky vrozených mikrocytárních anémií byla doplněna elektroforéza hemoglobinu v polyakrylamidovém gelu (PAGE) s nálezem standardního hemoglobinového spektra (HbH není na proteinové elektroforéze patrný, protože je nestabilní). Hladina HbA2 stanovená chromatograficky byla na spodní hranici normy (1,3 %), hladina HbF vyšetřená pomocí alkalické denaturace byla v normě (0,5 %). Barvení periferní krve briliant-kresylovou modří prokázalo přítomnost H inkluzí v erytrocytech, které vznikají precipitací volných β-globinových řetězců (obr. 3).

Molekulárně genetickým vyšetřením MLPA byly detekované dvě různé deleční mutace v HBA lokusu, které vedly k inaktivací tří α-globinových genů a nemoci hemoglobinu H (delece regulační oblasti HS-40 HBA lokusu na jednom chromozomu, na druhém chromozomu se jednalo o deleci zahrnující HBA geny -α3.7).
β-talasémie
V našem vyšetřovaném souboru jsme detekovali nosičství β-talasemické alely (heterozygotní postižení) u celkem 18 dětí ze 14 nepříbuzných rodin a β-talasémii major (dvojitá heterozygotní mutace) u jedné pacientky původem z Moldávie. Talasémie byla diagnostikovaná v průměrném věku 5 let (věkové rozmezí 1–13 let).
Všichni pacienti s β-talasémií minor měli v krevním obraze patrnou mikrocytární hypochromní anémii (medián: Hb 105 g/l; MCV 59 fl; MCH 19 pg). U všech pacientů byl zvýšený sTfR (medián: 5,4 mg/l) při normální hladině feritinu a plazmatického Fe. Všichni pacienti měli zvýšenou hladinu HbA2 a HbF a standardní Hb spektrum na elektroforéze. Molekulárně genetická analýza pomocí metody Sangerova sekvenování odhalila u všech pacientů přítomnost bodové nebo posunové mutace v genu pro β-globin (HBB). U jednoho pacienta byla pomocí metody MLPA jako příčinná mutace nosičství β-talasémie určena 619 bp dlouhá delece v genu HBB. Označení jednotlivých mutací zjištěných v našem souboru dětských pacientů, jejich dopad na fenotyp a jejich etnický původ shrnuje tabulka 2. Většina β-talasemických alel zjištěných na našem území pochází z oblasti Středomoří, jak již bylo dříve popsáno [8]. Předci našich pacientů pocházeli z Řecka, Bulharska, Turecka, Itálie, Egypta a v jednom případě z Ukrajiny.

Pacientka s β-talasémií major měla v krevním obraze výraznou anémii (Hb 68 g/l; ERY 2,76 x 1012, MCV 67,1 fl). Hladina HbA2 byla v normě, měla ovšem vysokou hladinu HbF (72 %), což bylo patrné také na elektroforéze, která ukazovala v podstatě pouze přítomnost HbF. Pomocí molekulárně genetického vyšetření byly detekovány dvě bodové mutace v HBB genu CD 8 (-AA) a CD 39 (C->T), které vedly u pacientky k závažnému fenotypu β-talasémie major. U pacientky byla ihned po stanovení diagnózy zahájena pravidelná hemoterapie erytrocytárními koncentráty. Od čtyř let je současně léčena chelatačními preparáty pro přetížení železem.
Strukturní hemoglobinové varianty
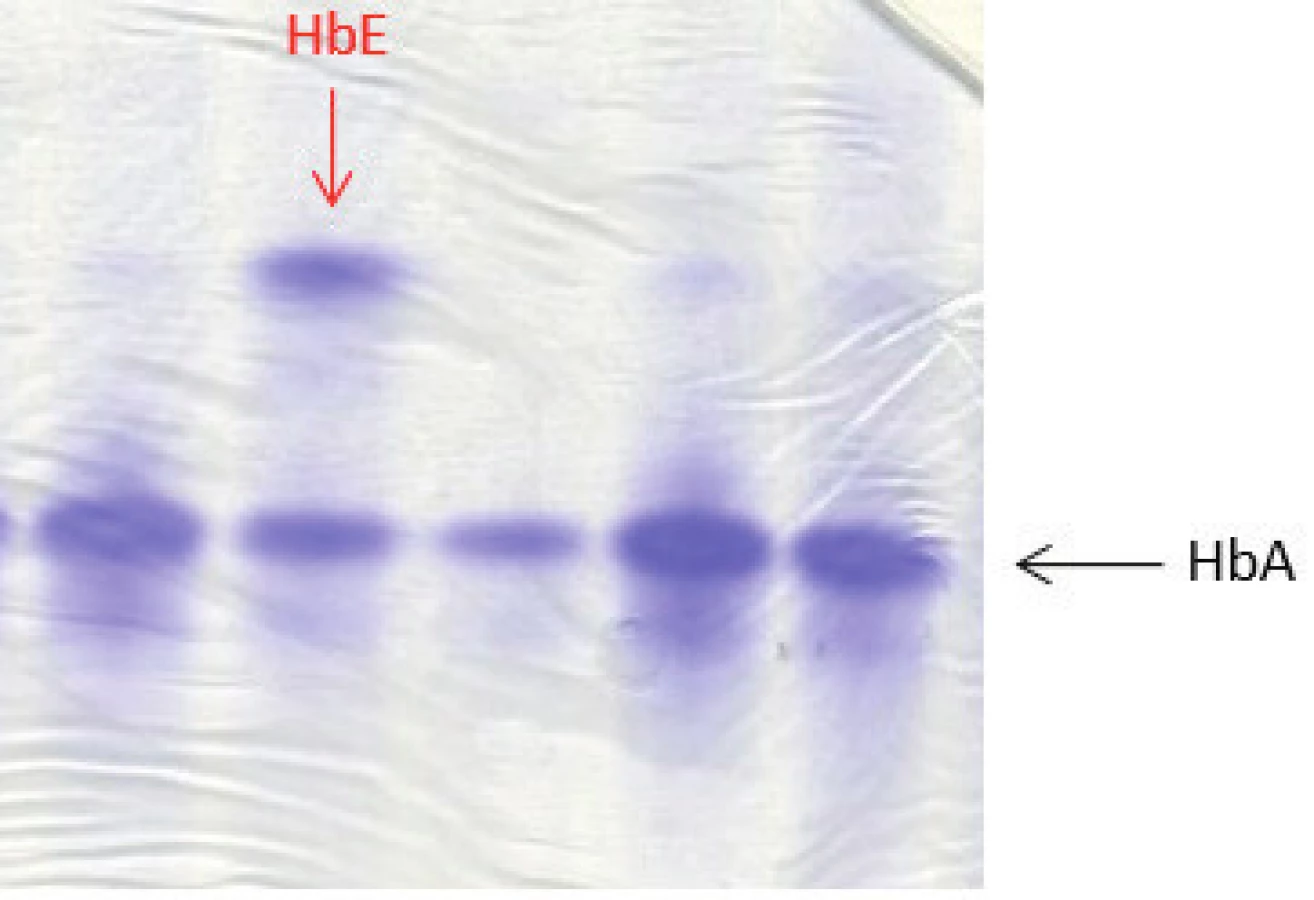
U 4 dětí ze 4 nepříbuzných rodin jsme detekovali některou formu Hb varianty (tab. 3). Jeden pacient byl nosič alely pro HbE β26 (Glu→Lys), který patří mezi tzv. talasemické hemoglobiny. Většina heterozygotů je bezpříznakových nebo s mírnou mikrocytární anémií stejně jako náš pacient (Hb 124 g/l; ERY 5,55 x 1012, MCV 66,1 fl) [9]. HbE lze dobře detekovat na proteinové elektroforéze, vykazuje jen velmi mírnou nestabilitu (obr. 4). Nosičství alely pro HbE jsme potvrdili pomocí sekvenační analýzy DNA, kdy byla stanovena příčinná mutace v HBB genu.

Hb Sydney β67 (Val→Ala) patří mezi nestabilní Hb varianty. Projevuje se hemolytickou anémií s nálezem Heinzových tělísek. Naše pacientka měla makrocytární anémii (Hb 103 g/l; ERY 3,67 x 1012, MCV 91,8 fl), měla pozitivní testy Hb stability (izopropanolový test, test na Heinzova tělíska), na elektroforéze byl nález odpovídající nestabilní hemoglobinopatii. Genetická analýza potvrdila nosičství alely pro Hb Sydney.
Pacientka s Hb Monroe (β30 Arg→Thr) měla indický původ. Jednalo se o heterozygotní nosičství, které se projevuje mírnou mikrocytární hypochromní anémií (Hb 103 g/l; ERY 5,86 x 1012, MCV 53,4 fl) se zvýšenou hladinou HbA2 (talasemický fenotyp). Hb Monroe je nestabilní, proto je nezachytitelný na proteinové elektroforéze. Mutace byla potvrzena sekvenační analýzou HBB genu, kde byla detekována mutace v CD 30 (AGG→ACG).
U posledního pacienta z našeho souboru byla diagnostikována srpkovitá anémie – homozygot pro HbS β6 (Glu→Val). Měl výraznou anémii (Hb 81 g/l; ERY 3,10 x 1012, MCV 74,5 fl). Jednalo se o imigranta z Afriky. Na elektroforéze Hb byly patrné dominantní frakce HbS a minoritní frakce HbF, frakce HbA chyběla (obr. 5). Pacienti se srpkovitou anémií mají hladinu HbS obvykle mezi 70–90 %, hladinu HbF mezi 5–15 %. Molekulárně genetická analýza potvrdila homozygotní mutaci v CD 6 HBB genu.

DISKUSE
Talasémie a hemoglobinové varianty se raritně vyskytují i v České republice a bývají nejčastější příčinou vrozené mikrocytární anémie. Průměrný věk diagnostikovaných pacientů v našem souboru spadá do školního věku v případě α-talasemického nosičství a do předškolního věku u β-talasémie minor, pravděpodobně v důsledku závažnějšího fenotypového projevu u β-talasémie minor. Obvykle je uváděno, že pacienti s tichým nosičstvím α-talasémie mají parametry krevního obrazu ve fyziologickém rozmezí, v našem souboru jsme u všech pacientů zaznamenali mikrocytózu a hypochromii erytrocytů a u dvou třetin pacientů i kompenzatorní erytrocytózu. Výskyt těchto odchylek v krevním obraze je indikací k podrobnějšímu vyšetření parametrů metabolismu železa a po vyloučení sideropenie následně i specializovanému hematologickému vyšetření. Všichni pacienti s β-talasémií minor měli zvýšenou hladinu sTfR, která je odrazem erytropoetického úsilí kostní dřeně, na rozdíl od pacientů s nosičstvím α-talasemické mutace (s výjimkou pacientů s koexistující sideropenií). Tyto výsledky dokumentují odlišný dopad β-talasemických mutací na fyziologii erytropoézy.
Přítomnost nespárovaných řetězců v časných stadiích erytropoézy má za následek zánik erytroidních prekurzorů ve dřeni a výsledný počet erytrocytů uvolněných do cirkulace pak neodpovídá počtu erytroidních prekurzorů vstupujících do diferenciace (neefektivní erytropoéza). Volné α-řetězce jsou „toxičtější“ pro progenitorové buňky červené krevní řady v kostní dřeni než volné β-řetězce u α-talasémie, více jich podléhá apoptóze a neefektivní erytropoéza je znatelnější u pacientů s β-talasémií než u pacientů s α-talasémií. U pacientů s nemocí HbH pozorujeme nižší míru neefektivní erytropoézy než u β-talasémie intermedia, právě v důsledku schopnosti β-řetězců agregovat do tetramerů. Molekula HbH je v pozdějších stadiích erytropoézy nestabilní, proto nemusí být HbH při elektroforéze Hb detekovatelný, jako tomu bylo i v případě naší pacientky. HbH zároveň vykazuje výrazný posun disociační křivky doleva, což má za následek snížení extrakce kyslíku tkáněmi vedoucí k tkáňové hypoxii [10].
Jones et al. a Guimares et al. v nedávno publikovaných studiích poukazují na alteraci osy erytropoéza – homeostáza Fe v důsledku převažujících erytropoetických signálů nad signály o stavu zásob Fe [11, 12]. Klíčovou molekulou v regulaci příjmu a utilizace Fe v organismu je hepcidin a právě jeho neadekvátní suprese u pacientů s talasemickým nosičstvím by mohla vést k akumulaci Fe v parenchymatózních orgánech. Pozvolná akumulace Fe v organismu se může podobně jako u hemochromatózy typu I projevit až později v dospělém věku orgánovým postižením ve smyslu rozvoje diabetu a jiných endokrinopatií, hepatopatie nebo kardiálního postižení. Metabolismus železa je proto nutné i u talasémie minor pečlivě monitorovat.
ZÁVĚR
Většina hemoglobinopatií diagnostikovaných na našem území byla importována z oblastí Středomoří, Afriky, Blízkého i Dálného východu a Asie. V následujících letech a desetiletích lze očekávat další obohacení genofondu české populace o nové druhy mutovaných alel globinových genů v důsledku celosvětové migrace a globalizace. Zpřesnění diagnostiky je dáno zejména používáním moderních molekulárně genetických postupů – MLPA a Sangerova sekvenování.
MUDr. Lucie Sulovská, Ph.D.
Dětská klinika
FN a LF UP
I.P. Pavlova 6
775 20 Olomouc
Zdroje
1. Divoký V, Indrák K, Mojzíková R. Hemoglobinopatie: talasémie a strukturní Hb varianty. In: Pospíšilová Š, Dvořáková D, Mayer J (Eds). Molekulární hematologie. Praha: Galén, 2013 : 270–283.
2. Divoký V, Luhový M, Divoká M, et al. Hemoglobin Haná or alpha 2 beta 2 63 (E7) His-Asn: a new unstable hemoglobin variant with a paradoxically different clinical manifestations in smokers and non-smokers in the same family. Vnitr Lek 1997; 43 (5): 267–272. Czech PubMed PMID: 9601847.
3. Pospíšilová D, Divoký V, Čermák J, et al. Nestabilní hemoglobinové varianty u dětí v České a Slovenské populaci. Čes-slov Pediat 2007; 62 (9): 519–526.
4. Steinberk SM, Mahoney DH, Raby BA. Genetic disorders of hemoglobin oxygen affinity. www.uptodate.com. Last update: Oct 2017.
5. Indrák K, Divoký V, Kynčlová E, et al. Hemoglobin Sydney alfa2beta2 (E11) Val-Ala a hemoglobin Olomouc alfa2beta2 86 (F2) Ala-Asp v českých rodinách. Přínos sekvenační analýzy DNA pro zpřesnění diagnostiky hemoglobinopatií. Vnitr Lék 1998; 44 : 347–349.
6. Divoky V, Walczyskova S, Pospisilova D, et al. Rare forms of hereditary anaemia in the Czech and Slovak populations – β - and δβ-thalassaemia and unstable haemoglobin variants. Vnitr Lek 2005; 51 : 886–893.
7. Sulovska L, Holub D, Zidova Z, et al. Characterization of iron metabolism and erythropoiesis in erythrocyte membrane defects and thalassemia traits. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2016; 160 (2): 231–237.
8. Divoká M, Partschová M, Pospíšilová D, et al. Alfa-talasémie u 45 českých rodin a 37 rodin cizinců žijících v České republice: přehled literatury a molekulárně-genetická diagnostika. Transfuze Hematol dnes 2016; 22 (3): 201–209.
9. Indrák K, Brabec V, Divoký V, et al. Structural variants in hemoglobin occurring in the Czech Republic. Vnitr Lek 1995; 41 (1): 13–20.
10. Benz EJ, Schrier SL Landaw SA. Clinical manifestation and diagnosis of the talassemias. www.uptodate.com. Last updated: Oct 2017.
11. Jones E, Pasricha SR, Allen A, et al. Hepcidin is suppressed by erythropoiesis in hemoglobin E β-thalassemia and β-thalassemia trait. Blood 2015; 125 (5): 873–880.
12. Guimarães JS, Cominal JG, Silva-Pinto AC, et al. Altered erythropoiesis and iron metabolism in carriers of thalassemia. Eur J Haematol 2015; 94 (6): 511–518.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2017 Číslo 8
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Cytomegalovirové infekce u novorozenců a dětí
Nejčtenější v tomto čísle
- Talasemické syndromy
- Chronický zánět středního ucha v dětském věku
- Mozgový absces – zriedkavá, ale závažná infekcia v detskom veku
- Talasémie a hemoglobinové varianty u dětí
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy