
Etiologie dětských ALL a AML, molekulární genetika a minimální reziduální nemoc
Aetiology of childhood ALL and AML, molecular genetics and minimal residual disease
Childhood acute leukaemias are heterogeneous group of diseases. Besides the basic classification into acute lymphoblastic leukaemias (ALL) and acute myeloid leukaemias (AML), the heterogeneity is mainly a consequence of variety of primary genetic aberrations. These aberrations result in various biological background, variable response to treatment and variable prognosis of different leukaemia subtypes. In ALL, the most common primary aberrations with a very good prognosis are hyperdiploidy and TEL/AML1 fusion gene. On the other hand, patients with BCR/ABL fusion or MLL gene rearrangements have poor prognosis. In AML, the AML1/ETO, CBFB/MYH11 and PML/RARA fusions are considered favourable. Probably the most reliable prognostic factor in ALL is an early response to treatment measured as levels of minimal residual disease (MRD) in specific time-points during therapy. The role of MRD in AML is less clear; however, its monitoring becomes a part of the standard treatment protocols recently.
Key words:
ALL, AML, aetiology, molecular genetics, minimal residual disease
Autoři:
J. Zuna; M. Žaliová
Působiště autorů:
CLIP, Klinika dětské hematologie a onkologie 2. LF UK a FN Motol, Praha, vedoucí pracoviště prof. MUDr. Jan Starý, DrSc.
Vyšlo v časopise:
Čes-slov Pediat 2015; 70 (2): 70-84.
Kategorie:
Dětské akutní leukémie
Souhrn
Dětské akutní leukémie jsou heterogenní skupinou onemocnění. Kromě základního rozdělení na akutní lymfoblastické leukémie (ALL) a akutní myeloidní leukémie (AML) je heterogenita dána především rozmanitostí primárních genetických aberací, které s sebou nesou rozdílný biologický základ, rozdílnou odpověď na léčbu i rozdílnou prognózu jednotlivých podtypů. Nejčastějšími primárními aberacemi s velmi dobrou prognózou jsou u ALL hyperdiploidie a fúzní gen TEL/AML1, naopak špatnou prognózu mají pacienti s fúzí BCR/ABL či s přestavbami genu MLL. U AML jsou za příznivé aberace považovány fúze AML1/ETO, CBFB/MYH11 a PML/RARA. Zřejmě nejspolehlivějším ukazatelem prognózy u ALL je časná odpověď na terapii, stanovená jako hladiny minimální reziduální nemoci (MRN) v předem stanovených časových bodech léčby. Role MRN u AML je méně jasná, v poslední době se však i zde stává její vyšetřování součástí léčebných protokolů.
Klíčová slova:
ALL, AML, etiologie, molekulární genetika, minimální reziduální nemoc
Akutní leukémie se tradičně dělí na akutní lymfoblastické leukémie (ALL) a akutní myeloidní leukémie (AML). Poměr výskytu těchto podtypů je v dětském věku asi 5 : 1, ALL jsou tak s velkým odstupem nejčastější dětskou malignitou a i díky tomu jsou v řadě ohledů lépe prozkoumány než AML. Proto je i v následujících kapitolách o něco více prostoru věnováno ALL.
Etiologie akutních leukémií
Akutní leukémie dětského věku jsou v naprosté většině případů de novo onemocněními způsobenými novými, nedědičnými aberacemi DNA v nezralých hematopoetických buňkách. Jak je podrobněji popsáno dále, jedná se ve skutečnosti o velmi heterogenní skupinu onemocnění, proto i etiologie může být v různých případech velmi odlišná. Následující kapitola tak shrnuje základní obecné mechanismy a zmiňuje jen některá specifika jednotlivých podtypů.
Jen velmi vzácně jsou dětské leukémie spojeny s dědičnými syndromy charakterizovanými vyšším rizikem vzniku malignit (syndrom Li-Fraumeni, Fanconiho anémie, ataxia telangiectasia, Wiskottův-Aldrichův syndrom, Bloomův syndrom) [1]. Nejčastějším vrozeným syndromem s významně vyšším rizikem vzniku leukémie je Downův syndrom (asi 20x vyšší riziko vzniku akutní lymfoblastické leukémie (ALL) a až 500x vyšší riziko vzniku akutní megakaryoblastické leukémie (AMKL)); i tak však leukémie dětí s Downovým syndromem tvoří pouze asi 1–2 % všech dětských leukémií [2].
Riziko vzniku de novo aberací je ovlivněno kombinací několika hlavních okolností: expozice rizikovým faktorům, vrozené náchylnosti – a náhody [3]. Dosud jediným spolehlivě potvrzeným vnějším rizikovým faktorem způsobujícím leukémie je ionizující záření [4]. Zvýšený výskyt leukémií byl prokázán v místech zasažených zářením z atomových bomb v Japonsku v roce 1945 (Hirošima, Nagasaki) [5] a, i když v daleko menší míře, také u dětí matek, které v těhotenství prodělaly rentgenovou pelvimetrii, tedy měření pánevních rozměrů [6]. Řada studií se zabývala významem dalších potenciálních rizikových faktorů pro vznik dětské leukémie (vystavení rizikovým faktorům během těhotenství – pesticidy, chemikálie zejména benzenového základu, kouření, užívání alkoholu či léků, infekce, dlouhodobý pobyt dětí v blízkosti elektromagnetických polí či jaderných elektráren, čistota ovzduší a mnoho dalších), výsledky však jsou při provedení meta-analýz z velkého počtu studií vesměs hraniční, nepříliš přesvědčivé, protichůdné či nereprodukovatelné [4].
Náchylnost ke vzniku leukémie mohou ovlivňovat vrozené genetické variace. Celogenomové asociační studie (genome-wide association studies, GWAS, tedy vyšetření vrozených genetických variant, nejčastěji jednonukleotidových polymorfismů, u velkého počtu pacientů a zdravých kontrol za účelem nalezení asociace genotypu s příslušnou nemocí) odhalily, že varianty v genech ARID5B, IKZF1, CEBPE, PIP4K2A, GATA3 a CDKN2A/CDKN2B mají vliv na riziko vzniku dětské ALL [7–9]. Všechny tyto geny hrají důležitou roli v průběhu vývoje lymfoidní řady, regulaci buněčného cyklu či v kontrole nádorové progrese a variabilita způsobující změny jejich funkce či interakčních schopností tak skutečně může hrát v leukemogenezi důležitou úlohu.
Podle současných představ o původu leukémií jsou k vyvolání onemocnění zapotřebí nejméně dva zásahy do DNA nezralé krvetvorné buňky [4]. Po prvním zásahu vzniká preleukemický klon buněk, které mohou mít jistou výhodu v přežívání a určitou funkční abnormalitu, nejsou však ještě schopny vyvolat klinicky aktivní onemocnění. Může v nich však docházet k dalším genetickým aberacím (a mohou například oslabením genetické stability//schopnosti detekovat a opravovat vzniklá poškození DNA riziko vzniku dalších aberací samy významně zvyšovat) a některý nebo některé z těchto dalších zásahů již mohou vést ke kompletní zástavě vyzrávání buňky a současně k potlačení mechanismů programované buněčné smrti (apoptózy). To pak způsobí hromadění těchto nezralých a funkčně nezpůsobilých buněk na úkor normální fyziologické hematopoézy a následně vede ke klinickým projevům leukémie.
Vysoká konkordance (shodný výskyt) kojeneckých akutních leukémií u jednovaječných dvojčat vedla k odhalení skutečnosti, že k prvnímu zásahu může docházet již před narozením, v průběhu intrauterinního vývoje [10, 11]. Pokud v krvetvorbě jednoho z dvojčat vznikne preleukemický klon, mohou některé jeho buňky cestou placentárních anastomóz přejít do oběhu jeho sourozence a tam jsou v případě jednovaječných dvojčat přijaty jako vlastní. Po narození pak s různě dlouhou latencí může v některé z těchto preleukemických buněk dojít k další genetické aberaci, „druhému zásahu“, a stane se z ní buňka leukemická. Výsledkem jsou pak konkordantní leukémie nesoucí shodnou základní genetickou aberaci (například přestavbu genu MLL nebo fúzi TEL/AML1, viz dále), ale různé aberace sekundární (například různá poškození dalších genů důležitých pro lymfocytární vývoj). Konkordance leukémie u jednovaječných monochoriálních dvojčat je nejvyšší v kojeneckém věku, kdy se blíží až 100 % (onemocní-li jedno z dětí v prvním roce života, je téměř jisté, že leukémie bude diagnostikována i u jeho jednovaječného dvojčete), pak však relativně rychle klesá a v předškolním věku už pravděpodobně není vyšší než 10 % [12].
Po důkazu, že konkordantní leukémie u jednovaječných dvojčat bývají iniciovány již prenatálně, bylo metodami „backtrackingu“, tedy zpětného hledání leukemogenních aberací v novorozenecké krvi, prokázáno, že prenatální původ je společný pro podstatnou část (pravděpodobně významnou většinu) dětských akutních lymfoblastických leukémií (ALL) [13–15], zatímco u akutních myeloidních leukémií (AML) je intrauterinní vznik zřejmě méně častý [16] (přinejmenším pro některé subtypy však platí podobně jako pro ALL – kromě kojeneckých AML s přestavbami genu MLL jsou to například leukémie s fúzními geny AML1//ETO [17] či CBFB/MYH11 [18], viz dále). Jako novorozenecký materiál pacientů, kteří v dětském věku onemocněli leukémií, slouží v těchto „backtrackingových“ studiích takzvané Guthrieho kartičky, tedy archivované zaschlé kapky krve odebírané těsně po narození na filtrační papír k diagnostickému screeningu vrozených metabolických vad. Na těchto kartičkách zůstává uchována novorozenecká DNA a je možno v ní zpětně hledat změny způsobené prvním, prenatálním zásahem [19].
Jak je již zmíněno výše, po prenatálním zásahu je k vyvolání leukémie pravděpodobně vždy nutný ještě nejméně jeden další zásah, typicky postanatální. Tato skutečnost, dále prudký pokles konkordance leukémie u dvojčat s přibývajícím věkem a také případy, kdy dvojčata sice ve vyšším věku skutečně onemocní konkordantní leukémií, ale v rozdílném věku (nejdelší vzájemná latence byla dosud popsána u dvojčat, která onemocněla jedno v pěti a druhé až ve čtrnácti letech) [20] vedou logicky k otázce, jestli prenatálně vzniklý preleukemický klon vždy nevyhnutelně dříve či později skončí klinicky diagnostikovanou leukémií, či zda je běžné, že po určité době preleukemické buňky z krvetvorby vymizí, aniž by byla kterákoliv z nich postižena dalším kritickým genetickým zásahem.
Odpověď na tuto otázku přinesly studie zaměřené na detekci preleukemických změn v pupečníkové krvi zdravých novorozenců. Cílené hledání buněk s fúzními geny TEL/AML1 (vyvolávajícím B-buněčné ALL, viz dále) a AML1//ETO (vyvolávajícím AML) v novorozenecké krvetvorbě ukázalo, že velmi malé množství buněk s některou z těchto aberací lze nalézt asi u 1 %, respektive 1 ‰ zdravých dětí, což je přibližně stokrát více, než jaká je výsledná incidence TEL/AML1, respektive AML1/ETO-pozitivních dětských leukémií [21, 22]. Jinými slovy, pouze asi 1 % dětí s preleukemickým klonem vzniklým před narozením skutečně onemocní leukémií – u zbylých 99 % se tento klon nikdy klinicky neprojeví.
Věkový průběh vzniku leukémií ukazuje, že ke druhému zásahu (předcházejícímu klinickou diagnózu leukémie zpravidla nejspíše o několik týdnů) dochází u ALL typicky v předškolním věku [1, 23]. Významný vrchol incidence mezi třetím a šestým rokem života je patrný na datech všech vyspělých států. Tento dominantní vrchol je i jedním z důležitých vodítek pro vytváření teorií, čím je bezprostředně vyvolán rozhodující druhý (či další) zásah a tím samotná ALL. V současné době zřejmě nejpřesvědčivější a obecně nejakceptovanější je takzvaná „infekční teorie“ [4, 24]. Podle ní je pro správnou funkci imunitního systému důležité, aby byl od časného věku stimulován běžnými infekcemi. Pokud tato stimulace chybí, kontakt s běžnými infekčními agens v pozdějším (předškolním) věku může vyvolat neadekvátní reakci imunitního systému a jejím důsledkem pak může být i vznik závažného onemocnění – leukémie, ale pravděpodobně i například dětské cukrovky (diabetes mellitus 1. typu), alergií, průduškového astmatu. Nepřímé důkazy podporující tuto hypotézu se opírají například o nižší výskyt ALL u dětí navštěvujících v časném věku kolektivní zařízení (jesle, mateřské školy), u dětí, které již mají v rodině starší sourozence, v socioekonomicky slabších společnostech (i v ČR byl popsán nárůst v počtu těchto předškolních leukémií mezi roky 1980–1999, tedy v době zřetelného posunu našeho životního standardu na vyšší socioekonomickou úroveň [25]). Všechny tyto skutečnosti (přítomnost dalších dětí v bezprostřední blízkosti či nižší socioekonomický standard a s ním obvykle i méně striktní hygienické nároky v širším smyslu) zvyšují v raném věku kontakt s běžnými infekcemi a tím přirozeně stimulují imunitní systém k adekvátnímu a správnému vyzrávání [1, 4].
U AML výrazný vrchol incidence v dětském věku zcela chybí – častější jsou v kojeneckém věku, pak jejich výskyt klesá na nejnižší hodnoty, zvyšuje se dále přibližně rovnoměrně s věkem až do dospělosti a vrcholu dosahuje ve věku nad 60 let. To nasvědčuje spíše postupné, náhodné kumulaci genetických změn s věkem než konkrétnímu věkově specifickému mechanismu vzniku AML (obr. 1).

Studie z poslední doby potvrzují, že preleukemické buňky u AML vznikají prvním zásahem ve stadiu časných hematopoetických kmenových a progenitorových buněk a že tyto buňky mají ještě zachovanou diferenciaci a jsou schopné dát vznik různým liniím [26–28]. Kromě fúzních genů AML1/ETO a CBFB/MYH11, které v případě dětských AML mohou vznikat často (nebo dokonce vždy) prenatálně, mohou být preleukemickými změnami například mutace genů ovlivňujících buněčnou metylaci (DNMT3A, IDH2, TET2). Tyto mutace vedou ke klonální expanzi postižených buněk a další zásah (například mutace genu NPM1) již může vyvolat AML [28].
Preleukemické buňky – jak u ALL, tak u AML – mohou uniknout léčbě cílené primárně na plně maligní klon, mohou tak přežívat i po ukončení terapie a být potenciálním rizikem pro návrat onemocnění v případě dalšího zásahu. Přítomnost buněk s primární aberací byla u pacientů s AML dokumentována před vznikem onemocnění či v období remise (buňky s fúzním genem AML1/ETO či s mutací DNMT3A přítomné v dlouhodobé remisi po léčbě, buňky s přestavbou genu MLL přítomné několik let před vznikem leukémie) [28–32]. U ALL nebývají preleukemické buňky po terapii detekovatelné (nebo je použitými metodami zpravidla nedokážeme odlišit od buněk leukemických) – o jejich přítomnosti však nepřímo svědčí „relapsy“ onemocnění vyvolané buňkami se stejnou primární aberací (fúzí TEL/AML1), avšak s odlišným druhým zásahem (delecí netranslokované alely genu TEL) [33, 34]. Existenci a dlouhou životnost preleukemických buněk u ALL pak kromě metod backtrackingu dokládají i případy přítomnosti fúze TEL/AML1 či hyperdiploidních buněk u zdravých jednovaječných dvojčat pacientů s příslušným typem ALL [35, 36]. Genetická patogeneze ALL z B-prekurzorů je znázorněna na obrázku 2.

Určitou výjimku z pohledu etiologie dětských leukémií tvoří sekundární AML. Takto označujeme jednak leukémie vznikající progresí jiného hematologického onemocnění (myelodysplastického syndromu nebo jiné myeloproliferace) a jednak leukémie vyvolané předchozí radio - či chemoterapií jiného maligního onemocnění. Tyto léčbou indukované sekundární AML jsou způsobeny dvěma hlavními typy cytostatik – alkylačními látkami (mechlorethamin, chlorambucil, cyklofosfamid, busulfan, ...) a inhibitory topoizomerázy II (etoposid a částečně i antracykliny (doxorubicin, daunorubicin)). Sekundární leukémie způsobené těmito dvěma typy cytostatik se liší – zatímco pro AML vyvolané alkylačními látkami je typická delší latence od terapie (5–7 let) a delece dlouhých ramének chromozomu 5 a/nebo 7, AML způsobené inhibicí topoizomerázy II jsou diagnostikovány krátce po chemoterapii (1–2 roky) a jsou charakterizované přestavbami genu MLL [37, 38].
Molekulární genetika akutních leukémií
Akutní leukémie jsou i v rámci hlavních diagnóz – ALL a AML – ve skutečnosti heterogenní skupinou onemocnění. Zatímco u AML je tato heterogenita zřejmá již z morfologických nálezů, u ALL se postupně odkrývala a odkrývá až s rozvojem cytogenetické a molekulárně genetické diagnostiky. V posledních letech naše představy o heterogenitě a komplexnosti dětských leukémií významně posunují moderní, zejména celogenomové a „single-cell“ metody (celogenomové vyšetření DNA pomocí SNP-arrays, sekvenování nové generace, molekulární vyšetření jednotlivých izolovaných buněk leukemického klonu a další). Díky těmto technikám se čím dál více ukazuje, že při diagnóze leukémie, jak ALL, tak AML, je v kostní dřeni zpravidla přítomno několik různých leukemických subklonů, které nesou stejnou základní aberaci, ale v dalších změnách se liší [39, 40]. Změn, kterými se leukemické buňky liší od buněk normálních, může být celá řada (podle rozlišovací schopnosti a citlivosti použité detekční techniky), ale biologický význam pro vznik či progresi leukémie má zřejmě jen několik málo z nich („drivers“). Zbylé aberace jsou pravděpodobně pouze časem naakumulované změny, ke kterým dochází přirozeně v průběhu zrání a stárnutí buněk a na onemocnění nejspíše nemají žádný vliv („passengers“). Odlišení jedněch od druhých není vždy snadné, aberace považovaná původně za vedlejší nález může s postupem znalostí o buněčné biologii a s dalšími publikovanými daty mít v procesu leukemogeneze důležitou roli. Obecně za „driver“ mutace považujeme změny, které nacházíme u stejného typu leukémie opakovaně (rekurentní aberace), a změny, které postihují významným způsobem geny účastnící se lymfoidního/myeloidního vývoje, regulace buněčného cyklu, tumor-supresorové geny a podobně. „Driver“ vlastnosti příslušné aberace u daného typu leukémie by však vždy měly být potvrzeny ve funkční studii.
S heterogenitou leukemické populace při diagnóze onemocnění souvisí i fakt, že asi u poloviny pacientů s relapsem onemocnění není leukemický klon při relapsu totožný s dominantním klonem popsaným při původní diagnóze [40]. Často je odvozen od některého z minoritních diagnostických klonů, jak je možno zjistit nejspíše při backtrackingu aberací z relapsu v původním diagnostickém materiálu. Jde-li o skutečný relaps (a ne o zcela novou, léčbou vyvolanou sekundární leukémii) [41], je základní (preleukemická) aberace vždy zachována, sekundární změny však mohou být různé – setkáváme se se ztrátami některých původních aberací i s objevením se nových. Často se v relapsu objevují aberace spojené s vyšší agresivitou či rezistencí buněk na chemoterapii a tyto změny považujeme za důsledek selekčního tlaku v průběhu intenzivní léčby.
V následujících odstavcích se krátce zmíníme o hlavních rekurentních genetických aberacích u ALL a AML.
Akutní lymfoblastické leukémie
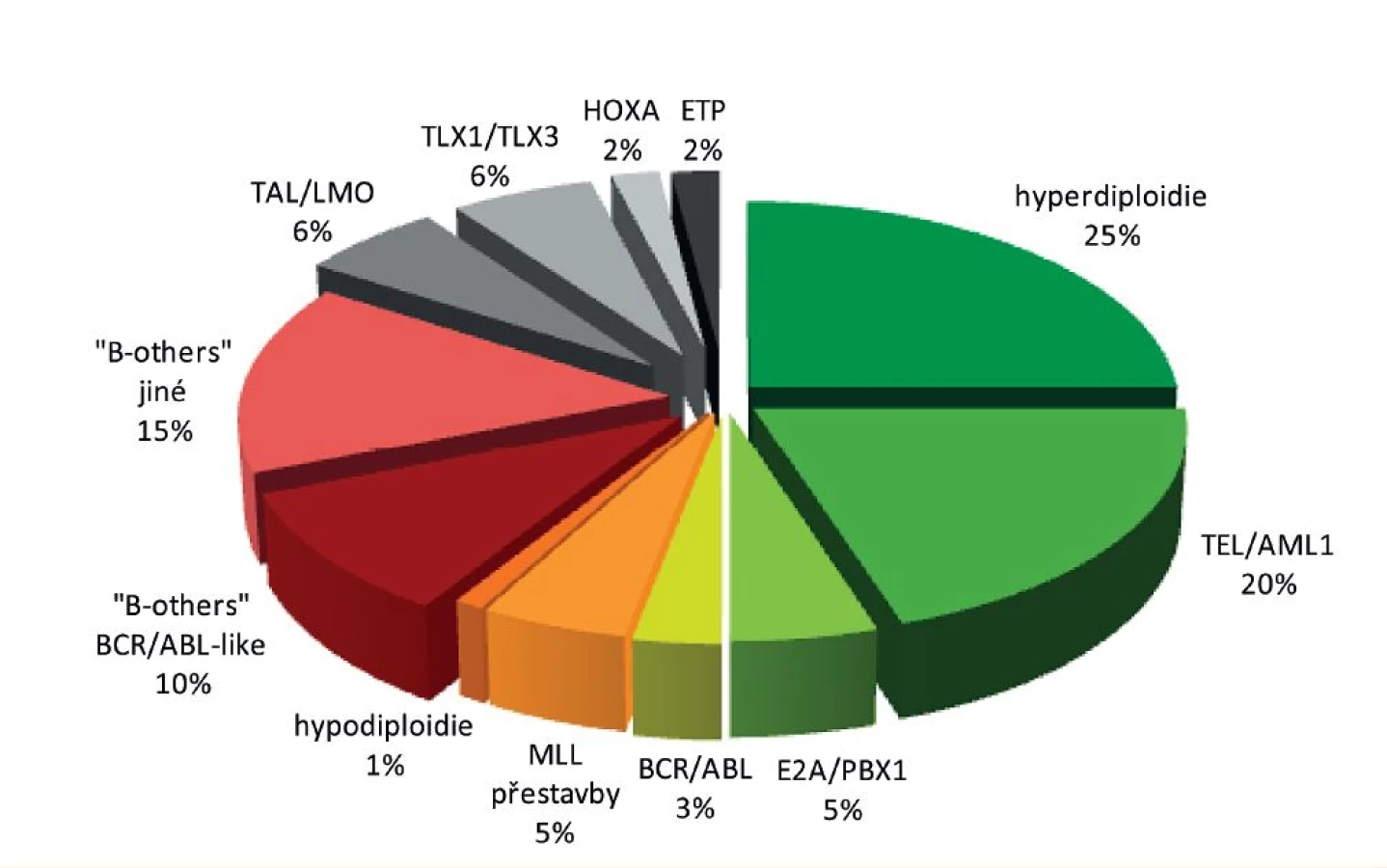
Nejčastějšími genetickými abnormalitami u dětských ALL jsou hyperdiploidie (tedy zmnožení počtu chromozomů nad 50) a přítomnost fúzního genu TEL/AML1 – každá z těchto změn se vyskytuje asi u čtvrtiny ALL z B-prekurzorových buněk. Fúzní gen TEL/AML1 (ETV6/RUNX1) je produktem translokace t(12;21)(p13;q22). Tato translokace na běžném karyotypu není patrná, a proto i přes svoji vysokou incidenci u dětských ALL unikala až do roku 1995 pozornosti [42]. Jak je již uvedeno v kapitole o etiologii, bylo prokázáno, že tato aberace vzniká často – a možná vždy – prenatálně [11]. Je typická pro dětské předškolní ALL a v dospělém věku je velmi vzácná. Mechanismus, jakým fúzní produkt TEL/AML1 přispívá k leukemogenezi, není stále zcela jasný. Zdá se, že preleukemické lymfoidní buňky s touto aberací reagují odlišně na infekční stimulaci, získávají v takové situaci výhodu ve smyslu přežívání a zvyšují pravděpodobnost dalšího zásahu [43]. Souvislost s aberantní imunitní reakcí nepřímo podporuje i skutečnost, že právě TEL//AML1-pozitivní leukémie tvoří hlavní část výrazného vrcholu incidence v předškolním věku (přisuzovaného právě infekční etiologii) [23]. Jestli je fúzní gen nutný i k udržení již vzniklé leukémie, není přesvědčivě prokázáno [44]. Z klinického hlediska je důležité, že se jedná o leukémie s velmi dobrou prognózou. I když se v této skupině objevují relapsy, jsou zpravidla pozdní a obvykle dobře odpovídají na další chemoterapii [45–48]. Jak je zmíněno výše, některé z těchto relapsů mohou být i „novými“ leukémiemi vzniklými novým druhým zásahem (často pravděpodobně delecí netranslokované alely TEL) do přežívající preleukemické populace [34].
Dalším typickým nálezem u dětských ALL jsou změny ploidie, tedy počtu chromozomů. Jedná se především o hyperdiploidie, které nacházíme přibližně stejně často jako fúzní gen TEL/AML1. O hyperdiploidii (správně „vysoké hyperdiploidii“) hovoříme při počtu chromozomů nad 50 a pod 68. Nadbytečné chromozomy nejsou u ALL rozloženy náhodně – u všech nebo téměř všech hyperdiploidních ALL je zmnožen chromozom 21 a nejčastěji ho nacházíme dokonce ve čtyřech kopiích. Dalšími chromozomy zmnoženými u většiny případů jsou chromozomy 4, 6, 10, 14, 17, 18 a X [49, 50]. Hyperdiploidie pravděpodobně nevzniká postupným hromaděním jednotlivých chromozomů, ale dochází k ní v jedné mitóze. Vzniklý karyotyp je pak zřejmě relativně stabilní – od preleukémie (neboť hyperdiploidie u dětských ALL je považována za první zásah, ke kterému dochází prenatálně, jak potvrzují backtrackingové studie [13, 15]) až do leukémie, která bývá nejčastěji opět diagnostikována v předškolním věku. Stejně jako TEL/AML1 i hyperdiploidní ALL jsou spojovány s dobrou prognózou. Týká se to zejména hyperdiploidií s větším počtem chromozomů a DNA indexem (určujícím množství DNA v leukemické buňce v poměru k buňce zdravé) nad 1,16. Za prognosticky zvláště příznivé jsou považovány hyperdiploidní ALL s trisomií chromozomů 4, 10 a 17 („triple-trisomy“), ale zřejmě jde ve skutečnosti jen o jinou definici leukémií s vyšším počtem přídatných chromozomů, neboť tyto dvě skupiny se v podstatě překrývají [51].
Hypodiploidie leukemických buněk, tedy počet chromozomů nižší než 44, je naopak považována za významně negativní prognostický ukazatel a v léčebných protokolech bývá sama o sobě příčinou k zařazení pacienta do skupiny nejvyššího rizika. Je naštěstí relativně vzácná – tvoří asi 1 % dětských ALL. Podle počtu chromozomů se dále dělí na „téměř haploidní“ s 24–31 chromozomy, nízce hypodiploidní (32–39 chromozomů) a vysoce hypodiploidní (40–43 chromozomů) případy. V některých případech se hypodiploidní genom může následně reduplikovat a působí pak na první pohled jako hyperdiploidní. Tuto „maskovanou hypodiploidii“ spolehlivě odhalí genetické vyšetření například pomocí SNP-array, které prokáže stejný původ duplikovaných chromozomů (uniparentální disomii). Díky celogenomovým metodám bylo nedávno prokázáno, že vysoké procento (90 %) pacientů s nízce hypodiploidní ALL nese mutace genu TP53 a téměř polovina z nich nejen v leukemických, ale i v normálních buňkách. Tato data nasvědčují tomu, že nízce hypodiploidní ALL jsou často ve skutečností prezentací syndromu Li-Fraumeni [52].
Leukémie s přestavbami genu MLL (KMT2A) ležícím na chromozomálním lokusu 11q23 se vyskytují jak u ALL, tak u AML. U ALL tvoří asi 3–5 % všech případů a jsou specificky spojeny s kojeneckým věkem, zejména s leukémiemi diagnostikovanými do půl roku od narození, u starších dětí je nacházíme relativně vzácně. Gen MLL je zřejmě nejpromiskuitnějším genem, co se týče počtu translokačních partnerů – dosud bylo popsáno přes 120 různých fúzí genu MLL, z toho asi 80 fúzních partnerů bylo charakterizováno na molekulární úrovni. Nejčastějšími partnery genu MLL jsou u dětských ALL geny AF4 (AFF1), AF9 (MLLT3) a ENL (MLLT1). Obecně jsou ALL s přestavbou genu MLL považovány za prognosticky velmi nepříznivé a ve většině protokolů jsou indikací k nejintenzivnější terapii [53, 54].
Fúzní gen BCR/ABL (t(9;22)(q34;q11)) s Philadelphským (Ph) chromozomem, první cytogenetickou abnormalitou specificky spojenou s konkrétní malignitou [55, 56], je typickým znakem chronické myeloidní leukémie (CML), vyskytuje se však i u ALL. Jeho incidence u ALL stoupá s věkem, takže zatímco u dětí tvoří tyto leukémie asi jen 3 % (a vídáme je více u větších dětí), u starších dospělých je to až třetina všech ALL. Fúzní gen se podle místa zlomu v BCR genu objevuje ve dvou hlavních variantách – pro CML je typická varianta Major-BCR/ABL (M-BCR/ABL), která dává vznik proteinu označovanému p210, u ALL vídáme častěji variantu minor-BCR/ABL (m-BCR/ABL) s výsledným proteinem p190 [57].
Porovnání hladin transkriptu BCR/ABL a hladin leukemických buněk měřených pomocí přestaveb imunoreceptorových genů (viz dále v kapitole o reziduální nemoci) odhalila časté odchylky, způsobené přinejmenším v některých případech přítomností BCR/ABL přestavby i v neleukemických buňkách včetně buněk myeloidních [58]. To otevírá otázku, jestli některé případy diagnostikované jako BCR/ABL-pozitivní ALL (s minor i Major přestavbou) nejsou ve skutečnosti lymfoidními blastickými zvraty chronické myeloidní leukémie.
Prognóza pacientů s fúzním genem BCR/ABL se významně zlepšila po objevu specifického inhibitoru kinázové domény genu ABL (imatinib mesylát) a jeho uvedení na trh v roce 2001. Po úspěšném použití u pacientů s CML byly vytvořeny i protokoly pro léčbu dětských ALL, a i když hledání optimálního terapeutického schématu stále trvá, posun v prognóze pacientů je již dnes významný [59, 60].
ALL s fúzním genem E2A/PBX1 (TCF3/PBX1) způsobeným translokací t(1;19)(q23;p13) jsou ukázkou toho, jak se s intenzivní léčbou může původně negativní prognostický ukazatel změnit na znak prognosticky zcela neutrální – zatímco ve starších protokolech v osmdesátých letech minulého století měly tyto ALL významně horší prognózu než jiné podtypy, na moderní terapii reagují velmi dobře a jejich léčebné výsledky jsou srovnatelné s jinými podskupinami dětských ALL [61]. ALL s translokací t(1;19) jsou spojené spíše s vyšším věkem (nad 10 let) a tvoří asi 5 % dětských ALL [62].
Významnou skupinou mezi ALL z B-prekurzorů jsou leukémie, které nenesou žádnou z výše popsaných specifických změn. Podle toho se označují jako „B-others“ a tvoří téměř čtvrtinu dětských ALL. Moderní celogenomové metody však u leukémií v posledních letech odhalily řadu nových rekurentních aberací a tak se postupně odhaluje genetické pozadí ALL i u této skupiny. Nápadné je, že velká část těchto aberací má za následek alteraci lymfoidního vývoje – jedná se například o mutace, delece, fokální amplifikace či nově objevené translokace genů PAX5, IKZF1 (IKAROS) či EBF1, kódujících transkripční regulátory časné lymfoidní maturace [3]. Gen PAX5 je postižen asi u čtvrtiny ALL, gen IKZF1 méně často (asi 10–15 % ALL) [63], ale je specificky spojen s určitými subtypy ALL (je alterován až u 80 % BCR/ABL-pozitivních a o něco méně i u „BCR/ABL-like“ (viz dále) ALL) [64]. Podle řady studií jsou delece genu IKZF1 spojeny se špatnou prognózou jak u BCR/ABL-pozitivních, tak negativních pacientů a uvažovalo se o zařazení této aberace do stratifikačního schématu léčby ALL [65–67]. V poslední době se však ukazuje, že i tato skupina je heterogenní a celkové souhrnné výsledky není možné generalizovat. Dnes je například zřejmé, že současná intragenová delece genu ERG, vyskytující se asi u 15 % pacientů s IKZF1 delecí, nepříznivý prognostický vliv IKZF1 ruší a pacienti se současným výskytem obou aberací mají prognózu dobrou [68]. Je tedy zřejmé, že pro případné zařazení do algoritmu rizikové stratifikace bude nutné pacienty s delecí IKZF1 dále přesněji charakterizovat.
Další častou aberací této skupiny ALL jsou přestavby cytokinového receptoru CRLF2 [69]. Jedná se o aktivační přestavby způsobené jednak translokací CRLF2 ke genu těžkého imunoglobulinového řetězce (IGH) a jednak fokální delecí krátkého úseku bezprostředně před kódující oblastí CRLF2, která vede k expresi fúzního produktu P2RY8/CRLF2 (sekvenční mutace CRLF2 jsou méně časté) [70]. Přestavby tohoto genu nacházíme asi u 8 % dětských ALL a asi u 50 % ALL u dětí s Downovým syndromem. Asi v polovině případů jsou asociovány s aktivačními mutacemi v kinázových genech JAK1 nebo JAK2 – u ALL se však jedná o jiné mutace, než které nacházíme v JAK genech u maligních myeloproliferací [71].
Studie profilování celogenomové exprese ukázaly, že 10–15 % dětských ALL z B-řady má expresní profil velmi podobný BCR/ABL-pozitivním ALL, přestože tento fúzní gen nenesou. Tato skupina tak byla nazvána „BCR/ABL-like“ či „Ph-like“ [66, 72]. Tyto ALL jsou ve skutečnosti velmi těžko přesně definovatelné, neboť expresní profily, které je charakterizují, jsou v různých studiích definovány různě a jejich průnik je překvapivě malý. Navzdory tomu se v poslední době dostává tato skupina do popředí zájmu dětských hematoonkologů, neboť ať už definovaná jakkoliv, byla podle prvotních studií spojena se špatnou prognózou. Poslední publikovaná americká práce tento trend nepotvrdila a ukazuje, že v současných protokolech založených na stratifikaci podle reziduální nemoci mohou tyto ALL svůj negativní prognostický vliv pozbýt [73]. Na jednoznačné zhodnocení bude však zřejmě ještě nutno potvrdit tato data v dalších studiích. Kromě delecí genu IKZF1 zmíněných výše jsou nejčastější genetickou aberací této skupiny přestavby genu CRLF2 (až u poloviny případů) a mutace genů JAK [74]. Celogenomové sekvenování posledních několika málo let ukazuje, že zbylé případy nesou celou řadu různých přestaveb, delecí a mutací, které vedou k aktivaci cytokinové a kinázové signalizace (ABL, EPOR, IL7R, JAK2, PDGFRB a další). To je velmi důležité pro jejich diagnostiku a terapii, neboť při včasném odhalení konkrétní aberace je velké části z nich možno nabídnout cílenou terapii specifickými kinázovými inhibitory [75].
Všechny dosud popsané aberace se u ALL vyskytují téměř výhradně u leukémií vycházejících z B-buněčných prekurzorů a u ALL z T-řady jsou vzácné (přestože výjimky existují). T-ALL tvoří asi 15–20 % dětských ALL. Podle genetického profilu je možné je rozdělit do čtyř podskupin charakterizovaných dysregulací (způsobenou nejčastěji přestavbami s T-buněčným receptorem) genů TAL/LMO, TLX1, TLX3, respektive HOXA (sem patří i translokace CALM/AF10, MLL/ENL a SET/NUP214) [76]. Další skupinou jsou velmi nezralé T-ALL nazývané ETP („early T-cell precursor ALL“) popsané nedávno jako podskupina se špatnou prognózou [77], podle posledních studií však není tento efekt jednoznačný [78]. Nejčastějšími genetickými aberacemi u T-ALL, které se obě vyskytují u více než poloviny případů, napříč jednotlivými výše popsanými podskupinami, jsou aberace genů CDKN2A/B a NOTCH1. První způsobuje inaktivaci inhibitorů buněčného cyklu p15 a p16, druhá aktivaci transkripčního faktoru NOTCH1 [76].
Původně horší léčebné výsledky T-buněčných ALL se v posledních letech začínají blížit výsledkům ALL z B-prekurzorů. T-ALL mají obvykle pomalejší odpověď na iniciální léčbu, ale tato skutečnost nemá tak významný vliv na prognózu jako u B-prekurzorových ALL [79]. Rozložení nejvýznamnějších genetických aberací u dětské ALL je znázorněno na obrázku 3 a prognóza dětské ALL podle hlavních genetických podskupin na obrázku 4.

Akutní myeloidní leukémie
Jak ukazují výsledky celogenomového profilování pomocí SNP-arrays a celogenomového sekvenování (genom AML byl mimochodem v roce 2008 vůbec prvním osekvenovaným nádorovým genomem [80]), ke vzniku AML je ve srovnání s ALL či jinými malignitami zřejmě zapotřebí méně aberací [63, 81]. Rozhodně to však neznamená, že je tím pádem svět genetických aberací u AML omezenější než u ALL. Není tomu tak, AML jsou přinejmenším stejně (ne-li více) geneticky heterogenní malignitou. S ohledem na dvouzásahovou teorii vzniku leukémií bývají genetické aberace AML tradičně děleny do dvou tříd: třídy I, zahrnující aberace poskytující proliferační výhodu (například mutace genů FLT3, NRAS, KRAS, KIT, CBL, PTPN11 či JAK2), a třídy II, kam patří aberace narušující buněčnou diferenciaci (například fúzní geny AML1/ETO, CBFB/MYH11, PML//RARA, přestavby genu MLL, mutace genů CEBPA nebo AML1) [82]. Toto třídění však není dokonalé a některé aberace, například ty s ne zcela objasněnou rolí v leukemogenezi (mutace genů DNMT3A, IDH1, IDH2, TET2 popsané teprve nedávno) či takové, jejichž biologické následky vybočují z relativně úzce definovaných výše popsaných tříd nebo jsou pleiotropní (například mutace genů NPM1 či WT1), zůstávají nezařazené [83].
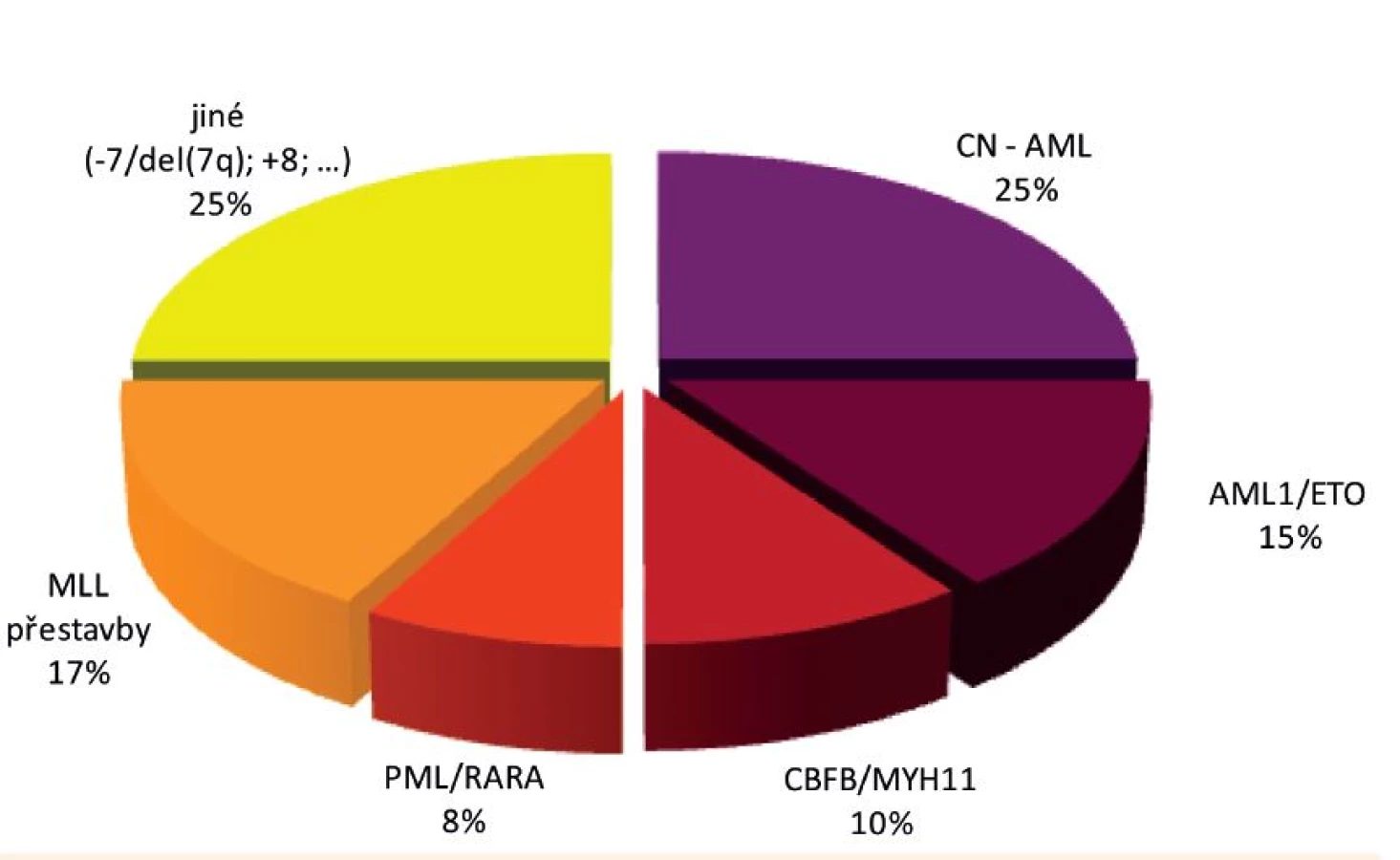
Největší genetickou podskupinu dětských AML (přibližně pětinu až čtvrtinu všech případů) představují tak zvané „Core binding factor“ (CBF) AML, kam řadíme leukémie s fúzními geny AML1/ETO a CBFB/MYH11. U obou těchto aberací je postižena alfa (AML1 neboli CBFA2) či beta (CBFB) podjednotka významného hematopoetického heterodimerního transkripčního faktoru CBF, následkem čehož dochází k poruše buněčné diferenciace, ale i k deregulaci apoptózy, proliferace a sebeobnovy [84]. Je zajímavé (a dosud nevysvětlené), že na rozdíl od těchto dvou AML aberací se třetí aberace postihující CBF – fúzní gen TEL/AML1 popsaný výše – vyskytuje výhradně u ALL.
Fúzní gen AML1/ETO (RUNX1/RUNX1T1) vzniká následkem translokace t(8;21)(q21;q22) a vyskytuje se u 12–14 % dětských AML, typicky u akutní myeloblastické leukémie s granulocytární diferenciací (AML-M2) [85]. CBFB/MYH11 vzniká následkem inverze či translokace inv(16)/t(16;16)(p13;q22) u 8–10 % dětských AML, nejčastěji u akutní mye-lomonocytární leukémie, kde bývá typicky doprovázen eozinofilií v kostní dřeni (AML-M4Eo) [86, 87]. CBF AML mají v dětském stejně jako v dospělém věku dobrou prognózu, některé práce však popisují horší léčebné výsledky AML1/ETO-pozitivních AML nesoucích současně mutace genu KIT (nejčastější spolupřítomná/kooperující aberace CBF AML) [88–90].
Další aberací pojící se s dobrou prognózou je fúzní gen PML/RARA. Nacházíme jej u naprosté většiny (asi u 95 %) akutních promyelocytárních leukémií (AML-M3, APL), což představuje asi 6–10 % všech dětských AML. Gen RARA kóduje jaderný receptor, který tlumí genovou expresi, avšak pouze v nepřítomnosti svého přirozeného ligandu – kyseliny retinové. Fúzí s genem PML se RARA mění v trvalý represor, jehož tlumivý efekt na genovou expresi není ovlivnitelný přítomností fyziologických hladin kyseliny retinové. Tím dochází k deregulaci buněčné diferenciace, apoptózy a sebeobnovy [84]. Represivní funkci PML/RARA lze farmakologicky inhibovat pomocí all-trans-retinové kyseliny (ATRA) v dávce navozující suprafyziologickou hladinu lignadu. Léčba APL pomocí ATRA představuje první do praxe uvedenou úspěšnou molekulárně cílenou léčbu leukémií [91]. Nejčastější kooperující aberací u PML//RARA-pozitivních AML je interní tandemová duplikace genu FLT3 (FLT3/ITD). Zatím není k dispozici dostatečné množství údajů pro posouzení jejího vlivu na jinak příznivou prognózu dětských APL.
Jak je již zmíněno výše, MLL přestavby se vyskytují u ALL i AML. U AML jsou kromě kojeneckých leukémií spojeny specificky i se sekundárními AML vzniklými následkem předchozí léčby inhibitory topoizomerázy II (etoposid, antracykliny) (viz kapitola o etiologii leukémií). Kromě těchto dvou kategorií se u AML setkáváme s MLL-fúzními geny u akutních monocytárních/monoblastických leukémií, tedy u AML podtypu M5. Spektrum fúzních partnerů genu MLL je i tady bohaté, nejčastější jsou AF9 (MLLT3), AF10 (MLLT10), AF6 (MLLT4), ELL a ENL (MLLT1) [53].
Obecně i tady jsou AML s MLL přestavbou spojeny spíše s horší prognózou, rozdíly oproti ostatním AML však nejsou velké. Některé studie ukazují, že léčebné výsledky závisí i na translokačním partnerovi genu MLL a že například AML-M5 s fúzí MLL/AF9 mají spíše lepší prognózu, zatímco MLL/AF6 či MLL/AF10 naopak horší. Výjimkou jsou AML s translokací t(1;11)(q21;q23) a fúzním genem MLL/AF1Q (MLLT11), které mají prognózu velmi dobrou – jsou však relativně vzácné [92].
Genetické přestavby, na základě kterých vznikají uvedené fúzní geny, jsou běžně detekovatelné rutinními cytogenetickými postupy. Přibližně čtvrtinu dětských AML však představují tak zvané cytogeneticky normální AML (CN AML), tedy AML bez cytogeneticky diagnostikované aberace. Pomocí molekulárně genetických metod u nich však můžeme detekovat různorodé mutace, mezi které patří například mutace transkripčních faktorů (CEBPA, WT1, AML1 (například u syndromu dědičné poruchy destiček s predispozicí k AML – FPD/AML), GATA1 (například u akutní megakaryoblastické leukémie – AMKL)), mutace genu NPM1, mutace genů postihující kinázovou signalizaci (vyskytující se typicky napříč podskupinami; KIT, FLT3, NRAS, KRAS, PTPN11) a mutace genů postihující epigenetickou regulaci (DNMT3A, IDH1, IDH2, TET2). Nejčastějšími z těchto změn jsou mutace NRAS (asi 20 % AML, nemají prognostický význam), interní tandemové duplikace genu FLT3 (FLT3/ITD; asi 10 % AML – případy s vysokým poměrem mutované alely k alele normální (>0,4) jsou spojovány s nepříznivou prognózou), mutace genu NPM1 (5–10 % AML, příznivý vliv na prognózu), mutace genu CEBPA (5 % AML, rovněž prognosticky příznivé), mutace genů WT1 a KIT [88, 93].
Jak je již uvedeno výše, heterogenita a rozmanitost aberací rekurentně nacházených u dětských AML je však ještě podstatně větší a s postupem celogenomových metod v čele se sekvenováním nové generace dále narůstá. Rozložení nejvýznamnějších genetických aberací u dětské AML je znázorněno na obrázku 5.
Minimální reziduální nemoc
Pojem minimální reziduální nemoc (MRN) definujeme jako množství buněk leukemického klonu pod rozlišovací schopností klasické morfologie, které v průběhu léčby ještě zůstávají v kostní dřeni (případně v periferní krvi) pacienta. Citlivá detekce těchto buněk (v běžné praxi se zpravidla jedná o schopnost zachytit jednu leukemickou buňku mezi deseti tisíci až sto tisíci buňkami normálními, hovoříme tak o citlivosti detekce 10-4 až 10-5) zejména v časných fázích terapie se stala dosud nejpřesnějším samostatným nezávislým ukazatelem prognózy dětské ALL. Tuto vysokou citlivost umožňují v současné době v zásadě tři hlavní přístupy ke sledování MRN – metody průtokové cytometrie, metody založené na polymerázové řetězové reakci (polymerase chain reaction, PCR) a v poslední době i metody sekvenování nové generace (NGS – next generation sequencing) neboli masivně paralelní sekvenování.
Techniky a cíle
Metody průtokové cytometrie využívají k detekci leukemických buněk toho, že exprese některých antigenů (zejména povrchových, ale i intracelulárních) je u leukemických buněk jiná než u jejich fyziologických protějšků. Dochází k aberantní expresi (přítomnost antigenu, který na buňkách dané hematopoetické řady normálně přítomen není), či k asynchronii (přítomnost antigenu z dané hematopoetické řady, avšak ve vývojovém stadiu, ve kterém se normálně neobjevuje). Podrobné vyšetření imunofenotypu při diagnóze leukémie, kdy abnormální buňky tvoří zpravidla většinu (často téměř stoprocentní) vyšetřovaného materiálu (nejčastěji kostní dřeně), umožní nalézt téměř ve všech případech unikátní kombinaci antigenů specifickou pro leukemické blasty. Tuto kombinaci pak označujeme jako LAIP (leukemia-associated aberrant immunophenotype), v průběhu léčby pak hledáme buňky tomuto imunofenotypu odpovídající a jejich množství určuje hladinu MRN – při analýze je však nutno brát v úvahu i fakt, že exprese některých antigenů se v průběhu terapie může do určité míry měnit [94, 95].
Výhodou průtokové cytometrie je rychlost (výsledek vyšetření může být k dispozici do několika hodin po odběru) a široká aplikovatelnost. Navíc kromě hladiny MRN získáváme při tomto vyšetření i celkový obraz o stavu a počtu ostatních buněk ve vzorku, nejčastěji v kostní dřeni. Na druhou stranu je tato metoda detekce MRN zatím méně standardizovaná než metody založené na PCR (přestože v posledních letech se i zde standardizace významně posouvá [96, 97]) a její citlivost je většinou o něco nižší [98]. Citlivost je závislá především na konkrétním imunofenotypu leukemických buněk (jak specifická je kombinace antigenů, odlišující leukemické buňky od normálních) a na počtu parametrů, které můžeme na buňce současně sledovat. U tří až čtyřbarevné detekce se pohybuje kolem 10-3–10-4, citlivost moderních přístrojů s možností detekce šesti až devíti barev je pak i více než o řád vyšší. Důležitým faktorem je ovšem samozřejmě i celkový počet vyšetřených buněk, k dosažení vysoké citlivosti je nutno analyzovat odpovídající množství materiálu. Vyšetření a jeho citlivost a specificitu může ovlivňovat i povaha normálních, nemaligních buněk ve vyšetřovaném odběru – ve vzorcích odebíraných ve fázích léčby, kdy dochází k významné regeneraci kostní dřeně, je obvykle náročnější spolehlivě oddělit leukemické buňky a zmnožené nezralé hematopoetické progenitory. Zodpovědná interpretace výsledků měření MRN vyžaduje zvláště u vícebarevných přístupů dostatečné zkušenosti [99, 100].
Pro metody založené na PCR (a stejně tak pro metody NGS) je nejdříve nutno nalézt sledovatelný „cíl“ – úsek DNA nebo RNA, který je specifický pouze pro leukemické buňky a v buňkách normálních se nevyskytuje. Takovým cílem může být genetická aberace, která je přímo příčinou samotné leukemické transformace – nejčastěji fúzní gen, ale i některé druhy duplikací, delecí, inzercí, mutací. Ojediněle můžeme ke sledování MRN použít i expresi normálního, nepřestavěného genu, který se však aberantně exprimuje pouze v leukemických buňkách, zatímco v normální hematopoéze je jeho exprese buď nulová, nebo významně (o několik řádů) nižší. Příkladem je gen WT1 aberantně exprimovaný u některých AML – informace o hladinách MRN je však v těchto případech méně spolehlivá a citlivost detekce významně nižší [101, 102].
Cíle přímo spojené s patogenezí leukémie můžeme sledovat na úrovni DNA i na úrovni exprese (RNA, respektive „komplementární DNA“ (cDNA), do které v praxi RNA z důvodu lepší stability přepisujeme) [103]. Na úrovni exprese nejčastěji sledujeme fúzní geny (u ALL například BCR/ABL, TEL/AML1, E2A/PBX1, u AML pak PML/RARA, AML1//ETO, CBFB/MYH11). Zatímco na DNA úrovni dochází při chromozomálních translokacích ke zlomům mezi geny téměř vždy v intronových oblastech, které jsou obvykle desítky až stovky tisíc bází dlouhé (a hledání přechodového místa je tak technicky náročné), sestřižená RNA bez intronů má zpravidla jen jednu nebo velmi omezené množství variant a k její detekci nám stačí dvojice nebo několik málo dvojic PCR primerů. S tím však souvisí i určité riziko – přechody na DNA úrovni jsou vzhledem k výše popsanému mechanismu vzniku vždy zcela specifické pro daného pacienta, ale na RNA (cDNA) úrovni má řada pacientů pro sledování MRN stejný cíl, což při neopatrné manipulaci s PCR produkty přináší rizika kontaminace mezi vzorky a vzniku falešně pozitivních výsledků. I proto (avšak nejen proto – viz dále) využíváme k monitorování MRN u pacientů s fúzními geny v některých (i když spíše ojedinělých) případech jako cíl genomický (intronový) přechod a PCR na DNA úrovni – i přes větší technickou náročnost hledání tohoto zlomu (například u ALL i AML, kde jedním z fúzních partnerů je gen MLL).
Aberace spojené přímo s patogenezí leukémie sledujeme (ať už na RNA nebo DNA úrovni) hlavně u AML – kromě zmíněných fúzních genů jsou to například ještě interní tandemové duplikace genu FLT3 (FLT3/ITD) či mutace v genu NPM1 [104, 105]. K detekci MRN u ALL dnes standardně používáme jiné cíle, které s vlastní leukemickou transformací přímo nesouvisí – geny pro imunoglobuliny (Ig) a T-buněčné receptory (TCR) [106, 107].
Tyto geny totiž prodělávají v průběhu fyziologického vyzrávání lymfocytů unikátní genové přestavby, v průběhu kterých se k sobě přiřazují vždy jeden z mnoha V („variable“), D („diversity“) a J („joining“) genových segmentů, mezi něž se v průběhu přestavby ještě vkládají náhodné („N“) nukleotidy. Tak vzniká teoreticky téměř nekonečné množství různých kombinací a sekvencí, čímž je zaručeno, že celý proces proběhne v každé lymfoidní buňce zcela unikátně a pravděpodobnost, že dva nezávisle vzniklé lymfocyty budou mít tuto finální přestavbu zcela shodnou, je minimální (tento mechanismus hraje důležitou roli ve fyziologické odpovědi imunitního systému a jeho rozmanitosti). Pokud se však některá z lymfoidních buněk s již vzniklou přestavbou stane základem leukemického klonu, všechny její „dceřiné“ buňky pak tuto přestavbu „dědí“ a ta je tak specifickým znakem celé leukemické populace [108, 109]. Schéma V-D-J přestavby imunoreceptorového genu je znázorněno na obrázku 6. I když z tohoto mechanismu existují vzácné výjimky, kvantifikace přestaveb Ig/TCR pro účely monitorování MRN je aplikovatelná u více než 90 % pacientů s ALL. Určitou výjimku tvoří kojenecké leukémie, často vznikající z velmi nezralých lymfoidních progenitorů, které ještě neprošly procesem fyziologické přestavby imunoreceptorů a nemají tedy v některých případech společný klonální znak ve formě Ig/TCR přestavby. Tady pak využíváme k monitorování MRN nejčastěji fúzní geny pro kojenecké leukémie typické, kde jedním partnerem je gen MLL, a sledujeme je obvykle na DNA úrovni [110].

Pro určení hladiny MRN kvantifikujeme v praxi kromě specifického cíle ještě kontrolní „housekeeping“ gen – tedy gen, jehož přítomnost/exprese by měla být ve všech buňkách (leukemických i neleukemických) stejná. Dosahujeme tím normalizace množství vstupního materiálu a finální („normalizovaný“) výsledek měření MRN tak vydáváme jako poměr množství specifického cíle a kontrolního genu, tedy jako poměr leukemických buněk ze všech buněk vyšetřovaného vzorku.
Monitorování na DNA úrovni je technicky jednodušší (odpadá krok přepisu RNA do komplementární cDNA) a současně lépe odráží skutečný počet buněk s danou aberací – kvantifikaci MRN na úrovni RNA může ovlivňovat různá hladina exprese daného cíle (a stejně tak kontrolního genu) v různých buňkách (například změny exprese v důsledku probíhající terapie).
V posledních několika letech se v praxi monitorování MRN začíná postupně uplatňovat metoda masivně paralelního sekvenování neboli NGS. Je založená na nových technologiích, které při určování pořadí nukleotidů dokáží detekovat výslednou sekvenci pro každou vstupní molekulu nukleové kyseliny zvlášť. Původním využitím této technologie bylo pokrytí co nejrozsáhlejších oblastí genomu pro účely celogenomového sekvenování, stejně tak je ale možno pokrýt sekvenací pouze konkrétní oblast vysokým počtem přečtených sekvencí – „readů“. Místo jedné výsledné sekvence reprezentující dominantní variantu přítomnou ve vzorku (jak je tomu u klasického sekvenování) jsou zde výsledkem až miliony sekvencí jednotlivých molekul. Tímto způsobem je možné dosáhnout vysoké citlivosti detekce MRN, tedy odhalení jedné nebo několika málo molekul s konkrétní přestavbou mezi statisíci až miliony molekul normálních. Jako cíle pro sledování MRN pomocí NGS se nejčastěji používají rovněž přestavby imunoreceptorových genů [111, 112], ale tuto technologii lze samozřejmě využít i pro citlivou detekci mutací a krátkých insercí/delecí [113].
NGS má pro sledování MRN u leukémií jistě obrovský potenciál, nicméně jako metoda stále není spolehlivě standardizovaná a přestože analýza výsledných sekvenačních dat může svádět k jednoduchým interpretacím, při pečlivém a zodpovědném přístupu je stále ještě složitá a komplikovaná řadou nevyjasněných otázek (kvantifikace výsledků, tedy hladin MRN, zejména například v případech aplastické kostní dřeně, různá „amplifikovatelnost“ různých typů přestaveb, relativně časté chyby polymerázy a čtení výsledné sekvence (zejména v místech homopolymerů), absence standardizace postupů kvantifikace pro klinické účely a podobně).
Klinický význam MRN u akutních leukémií
V osmdesátých a především pak v devadesátých letech minulého století se při hledání prognostických znaků a stratifikačních kritérií zejména pro dětské pacienty s ALL dostaly do popředí zájmu metody detekce MRN a hledání optimálních strategií a cílů pro citlivou detekci účinnosti léčby u jednotlivých pacientů. Původní představy, že průběžné vyšetřování hladin MRN umožní časnou detekci molekulárního a hematologického relapsu, byly na konci devadesátých let definitivně nahrazeny jinou strategií – ukázalo se, že pro prognózu pacientů je nejdůležitějším ukazatelem rychlost poklesu MRN v prvních fázích léčby, tedy časná odpověď na terapii [114]. MRN se tak u dětské ALL stala a dosud zůstává nejsilnějším nezávislým prognostickým ukazatelem přežití.
V současné době se na frontline protokolech ALL měří hladiny MRN obvykle na konci indukční léčby, tedy asi po jednom měsíci kombinované chemoterapie, a hladiny MRN nad 10-3 (tedy více než jedna leukemická buňka mezi tisícem buněk normálních) se zpravidla považují za prognosticky nepříznivé, zejména u ALL z B-prekurzorů. K tomuto odběru však v různých léčebných protokolech přistupují i další časové body (v protokolech BFM používaných v České republice je to odběr v polovině indukce (den 15, měřený průtokovou cytometrií) a odběr před zahájením konsolidace (po 12 týdnech léčby)). Výsledná riziková skupina určená podle reziduální nemoci je tak výsledkem kombinace hladin MRN v těchto časových bodech (a zohledněna je i buněčná linie, ze které leukémie vychází – B nebo T) [115]. Na finální stratifikaci do příslušné větve léčebného protokolu (jejímž cílem je nabídnout dostatečně intenzivní terapii pacientům se špatně reagující leukémií a naopak uchránit od pozdních následků chemo(radio-)terapie pacienty odpovídající na léčbu od začátku velmi dobře) mají pak vliv ještě některé další nálezy molekulární genetiky a cytogenetiky (přítomnost některých fúzních genů, ploidie leukemických buněk) a odpověď na týdenní prednisonovou předfázi (měřená počtem blastů v periferní krvi osmý den léčby).
Podobnou roli jako u pacientů léčených podle frontline protokolů má MRN i u pacientů po relapsu leukémie. I tady je časná odpověď na terapii silným prognostickým ukazatelem a rozhoduje (samozřejmě opět s dalšími faktory) o zařazení do příslušné větve protokolu a volbě porelapsové léčby, především o indikaci k transplantaci [116, 117].
Právě před a po transplantaci kmenových buněk krve-tvorby (HSCT – haematopoietic stem cell transplantation) má monitorování hladin MRN roli poněkud odlišnou. Ukazuje se, že hladina MRN bezprostředně před transplantací je u dětí s ALL důležitým prognostickým ukazatelem – pacienti, vstupující do procesu vlastní transplantace s vyšší, detekovatelnou hladinou MRN mají významně menší šanci na vyléčení než ti, kteří mají před transplantací hladiny MRN v kostní dřeni negativní nebo neměřitelné [118, 119]. Proto v posledních letech převládá snaha pacientům s detekovatelnou MRN před transplantací ještě přidat jeden intenzivní léčebný blok a pokusit se reziduální nemoc co nejvíce potlačit. Po HSCT pak hladiny MRN hrají důležitou roli při postupném vysazování imunosupresivní terapie – objeví-li se po transplantaci pozitivita MRN, snažíme se vysazování imunosuprese urychlit i za cenu vyššího rizika reakce štěpu proti hostiteli, abychom tak potencovali imunologické mechanismy boje proti zbylým leukemickým buňkám (reakci „štěpu proti leukémii“).
U AML je prognostický význam hladin MRN v průběhu léčby méně jasný, monitorování reziduální nemoci dlouho nebylo (s výjimkou akutních promyelocytárních leukémií s fúzním genem PML/RARA) součástí standardních léčebných protokolů a pro rizikovou stratifikaci pacientů nehrálo roli. Hodnocení prognostického významu MRN bylo navíc ztíženo nejednotnou metodikou studií (různé techniky detekce MRN, různá senzitivita metod, různé časové odběry pro hodnocení odpovědi) a také nestejnou dynamikou odpovědi u různých genetických podskupin (například CBF AML mohou při hodnocení MRN pomocí kvantifikace fúzních transkriptů (AML1/ETO či CBFB//MYH11) odpovídat relativně pomaleji, přesto si zachovávají dobrou prognózu) [120, 121]. V současné době však řada světových skupin začíná používat či připravuje protokoly, ve kterých již jsou hladiny MRN brány v úvahu a stávají se součástí stratifikačního algoritmu a rozhodování o indikaci k HSCT. Na rozdíl od ALL, kde je dnes standardní používanou metodikou kvantifikace MRN pomocí PCR, je u AML daleko častěji používána jako metoda volby průtoková cytometrie [100, 122, 123]. Důvodem je mimo jiné absence obecně použitelného cíle, jakým jsou u ALL Ig/TCR přestavby, neboť myeloidní buňky své imunoreceptorové geny standardně nepřestavují. I u AML však v současné době přibývá studií zaměřených na analýzu hladin MRN detekovaných PCR amplifikací specifických cílů. Jak je uvedeno výše, k monitorování léčebné odpovědi se u AML tradičně používají fúzní geny (kromě PML/RARA i AML1/ETO, CBFB/MYH11 či fúzní geny s genem MLL jako jedním z partnerů) i jiné aberace (FLT3/ITD, NPM1 mutace a další) a v některých případech je možno použít i expresi genu WT1.
Pro léčebnou rozvahu u pacientů před HSCT nehraje u AML hladina MRN takovou roli jako u ALL, i když tendence snížit leukemickou nálož před transplantací na minimum je jistě stejná. Po HSCT je pak i u pacientů s AML snaha o co nejrychlejší vysazování imunosuprese, ale konkrétní algoritmy úpravy terapie v závislosti na MRN jsou zatím spíše experimentální.
S podporou grantů IGA NT/12428-5 a NT/14350-3.
Doc. MUDr. Jan Zuna, Ph.D.
CLIP, Klinika dětské hematologie a onkologie
2. LF UK a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: jan.zuna@lfmotol.cuni.cz
Zdroje
1. Greaves M. Molecular genetics, natural history and the demise of child-hood leukaemia. Eur J Cancer 1999; 35 : 1941–1953.
2. Seewald L, Taub JW, Maloney KW, et al. Acute leukemias in children with Down syndrome. Mol Genet Metab 2012; 107 : 25–30.
3. Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet 2013; 381 : 1943–1955.
4. Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer 2006; 6 : 193–203.
5. Preston DL, Kusumi S, Tomonaga M, et al. Cancer incidence in atomic bomb survivors. Part III. Leukemia, lymphoma and multiple myeloma, 1950–1987. Radiat Res 1994; 137: S68–S97.
6. Doll R, Wakeford R. Risk of childhood cancer from fetal irradiation. Br J Radiol 1997; 70 : 130–139.
7. Bartram T, Burkhardt B, Wossmann W, et al. Childhood acute lympho-blastic leukemia-associated risk-loci IKZF1, ARID5B and CEBPE and risk of pediatric non-Hodgkin lymphoma: a report from the Berlin-Frankfurt--Munster Study Group. Leuk Lymphoma 2014 : 1–3.
8. Migliorini G, Fiege B, Hosking FJ, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood 2013; 122 : 3298–3307.
9. Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet 2013; 45 : 1494–1498.
10. Ford AM, Ridge SA, Cabrera ME, et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature 1993; 363 : 358–360.
11. Wiemels JL, Cazzaniga G, Daniotti M, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 1999; 354 : 1499–1503.
12. Greaves MF, Maia AT, Wiemels JL, et al. Leukemia in twins: lessons in natural history. Blood 2003; 102 : 2321–2333.
13. Gruhn B, Taub JW, Ge Y, et al. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia 2008; 22 : 1692–1697.
14. Smith MT, McHale CM, Wiemels JL, et al. Molecular biomarkers for the study of childhood leukemia. Toxicol Appl Pharmacol 2005; 206 : 237–245.
15. Taub JW, Konrad MA, Ge Y, et al. High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood 2002; 99 : 2992–2996.
16. Burjanivova T, Madzo J, Muzikova K, et al. Prenatal origin of childhood AML occurs less frequently than in childhood ALL. BMC Cancer 2006; 6 : 100.
17. Wiemels JL, Xiao Z, Buffler PA, et al. In utero origin of t(8;21) AML1--ETO translocations in childhood acute myeloid leukemia. Blood 2002; 99 : 3801–3805.
18. McHale CM, Wiemels JL, Zhang L, et al. Prenatal origin of child-hood acute myeloid leukemias harboring chromosomal rearrangements t(15;17) and inv(16). Blood 2003; 101 : 4640–4641.
19. Wiemels J, Kang M, Greaves M. Backtracking of leukemic clones to birth. Methods Mol Biol 2009; 538 : 7–27.
20. Wiemels JL, Ford AM, Van Wering ER, et al. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood 1999; 94 : 1057–1062.
21. Mori H, Colman SM, Xiao Z, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A 2002; 99 : 8242–8247.
22. Zuna J, Madzo J, Krejci O, et al. ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood 2011; 117 : 368–369; author reply 370–361.
23. Trka J, Zuna J, Hrusak O, et al. Impact of TEL/AML1-positive patients on age distribution of childhood acute lymphoblastic leukemia in the Czech Republic. Pediatric Hematology Working Group in the Czech Republic. Leukemia 1998; 12 : 996–997.
24. Greaves MF, Alexander FE. An infectious etiology for common acute lymphoblastic leukemia in childhood? Leukemia 1993; 7 : 349–360.
25. Hrusak O, Trka J, Zuna J, et al. Acute lymphoblastic leukemia incidence during socioeconomic transition: selective increase in children from 1 to 4 years. Leukemia 2002; 16 : 720–725.
26. Pandolfi A, Barreyro L, Steidl U. Concise review: preleukemic stem cells: molecular biology and clinical implications of the precursors to leukemia stem cells. Stem Cells Transl Med 2013; 2 : 143–150.
27. Shlush LI, Minden MD. Preleukemia: the normal side of cancer. Curr Opin Hematol 2015, in press.
28. Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic hae-matopoietic stem cells in acute leukaemia. Nature 2014; 506 : 328–333.
29. Kusec R, Laczika K, Knobl P, et al. AML1/ETO fusion mRNA can be detected in remission blood samples of all patients with t(8;21) acute myeloid leukemia after chemotherapy or autologous bone marrow transplantation. Leukemia 1994; 8 : 735–739.
30. Miyamoto T, Nagafuji K, Akashi K, et al. Persistence of multipotent progenitors expressing AML1/ETO transcripts in long-term remission patients with t(8;21) acute myelogenous leukemia. Blood 1996; 87 : 4789–4796.
31. Preudhomme C, Philippe N, Macintyre E, et al. Persistence of AML1//ETO fusion mRNA in t(8;21) acute myeloid leukemia (AML) in prolonged remission: is there a consensus? Leukemia 1996; 10 : 186–188.
32. Zuna J, Burjanivova T, Mejstrikova E, et al. Covert preleukemia driven by MLL gene fusion. Genes Chromosomes Cancer 2009; 48 : 98–107.
33. Ford AM, Fasching K, Panzer-Grumayer ER, et al. Origins of „late“ relapse in childhood acute lymphoblastic leukemia with TEL-AML1 fusion genes. Blood 2001; 98 : 558–564.
34. Zuna J, Ford AM, Peham M, et al. TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res 2004; 10 : 5355–5360.
35. Bateman CM, Alpar D, Ford AM, et al. Evolutionary trajectories of hyperdiploid ALL in monozygotic twins. Leukemia 2015; 29 : 58–65.
36. Hong D, Gupta R, Ancliff P, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science 2008; 319 : 336–339.
37. Felix CA. Leukemias related to treatment with DNA topoisomerase II inhibitors. Med Pediatr Oncol 2001; 36 : 525–535.
38. Joannides M, Grimwade D. Molecular biology of therapy-related leu-kaemias. Clin Transl Oncol 2010; 12 : 8–14.
39. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011; 469 : 356–361.
40. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012; 481 : 506–510.
41. Zuna J, Cave H, Eckert C, et al. Childhood secondary ALL after ALL treatment. Leukemia 2007; 21 : 1431–1435.
42. Romana SP, Mauchauffe M, Le Coniat M, et al. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood 1995; 85 : 3662–3670.
43. Ford AM, Palmi C, Bueno C, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J Clin Invest 2009; 119 : 826–836.
44. Zaliova M, Madzo J, Cario G, et al. Revealing the role of TEL/AML1 for leukemic cell survival by RNAi-mediated silencing. Leukemia 2011; 25 : 313–320.
45. Bhojwani D, Pei D, Sandlund JT, et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 2012; 26 : 265–270.
46. Loh ML, Goldwasser MA, Silverman LB, et al. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 2006; 107 : 4508–4513.
47. Zuna J, Hrusak O, Kalinova M, et al. TEL/AML1 positivity in child-hood ALL: average or better prognosis? Czech Paediatric Haematology Working Group. Leukemia 1999; 13 : 22–24.
48. Zuna J, Hrusak O, Kalinova M, et al. Significantly lower relapse rate for TEL/AML1-positive ALL. Leukemia 1999; 13 : 1633.
49. Paulsson K, Forestier E, Lilljebjorn H, et al. Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2010; 107 : 21719–21724.
50. Paulsson K, Johansson B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 2009; 48 : 637–660.
51. Paulsson K, Forestier E, Andersen MK, et al. High modal number and triple trisomies are highly correlated favorable factors in childhood B-cell precursor high hyperdiploid acute lymphoblastic leukemia treated according to the NOPHO ALL 1992/2000 protocols. Haematologica 2013; 98 : 1424–1432.
52. Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013; 45 : 242–252.
53. Meyer C, Hofmann J, Burmeister T, et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013; 27 : 2165–2176.
54. Meyer C, Kowarz E, Hofmann J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009; 23 : 1490–1499.
55. Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst 1960; 25 : 85–109.
56. Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973; 243 : 290–293
57. Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood 1996; 88 : 2375–2384.
58. Zaliova M, Fronkova E, Krejcikova K, et al. Quantification of fusion transcript reveals a subgroup with distinct biological properties and predicts relapse in BCR/ABL-positive ALL: implications for residual disease monitoring. Leukemia 2009; 23 : 944–951.
59. Biondi A, Schrappe M, De Lorenzo P, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome--positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 2012; 13 : 936–945.
60. Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children‘s oncology group study. J Clin Oncol 2009; 27 : 5175–5181.
61. Schultz KR, Pullen DJ, Sather HN, et al. Risk - and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children‘s Cancer Group (CCG). Blood 2007; 109 : 926–935.
62. Andersen MK, Autio K, Barbany G, et al. Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 2011; 155 : 235–243.
63. Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446 : 758–764.
64. Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008; 453 : 110–114.
65. Dorge P, Meissner B, Zimmermann M, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 2013; 98 : 428–432.
66. Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009; 360 : 470–480.
67. van der Veer A, Zaliova M, Mottadelli F, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 2014; 123 : 1691–1698.
68. Zaliova M, Zimmermannova O, Dorge P, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 2014; 28 : 182–185.
69. Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor - and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 2009; 41 : 1243–1246.
70. Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2010; 107 : 252–257.
71. Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic//Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010; 115 : 5312–5321.
72. Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009; 10 : 125–134.
73. Roberts KG, Pei D, Campana D, et al. Outcomes of children with BCR--ABL1-like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol 2014; 32 : 3012–3020.
74. Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest 2012; 122 : 3407–3415.
75. Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 2012; 22 : 153–166.
76. Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol 2010; 23 : 307–318.
77. Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 2009; 10 : 147–156.
78. Patrick K, Wade R, Goulden N, et al. Outcome for children and young people with early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol 2014; 166 : 421–424.
79. Patrick K, Vora A. Update on biology and treatment of T-cell acute lymphoblastic leukaemia. Curr Opin Pediatr 2015; 27 : 44–49.
80. Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008; 456 : 66–72.
81. Radtke I, Mullighan CG, Ishii M, et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc Natl Acad Sci U S A 2009; 106 : 12944–12949.
82. Renneville A, Roumier C, Biggio V, et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia 2008; 22 : 915–931.
83. Takahashi S. Current findings for recurring mutations in acute myeloid leukemia. J Hematol Oncol 2011; 4 : 36.
84. Steffen B, Muller-Tidow C, Schwable J, et al. The molecular pathogenesis of acute myeloid leukemia. Crit Rev Oncol Hematol 2005; 56 : 195–221.
85. Downing JR. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. Br J Haematol 1999; 106 : 296–308.
86. Larson RA, Williams SF, Le Beau MM, et al. Acute myelomonocytic leukemia with abnormal eosinophils and inv(16) or t(16;16) has a favorable prognosis. Blood 1986; 68 : 1242–1249.
87. Liu P, Tarle SA, Hajra A, et al. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science 1993; 261 : 1041–1044.
88. Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 2012; 120 : 3187–3205.
89. Pollard JA, Alonzo TA, Gerbing RB, et al. Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 2010; 115 : 2372–2379.
90. Shimada A, Taki T, Tabuchi K, et al. KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood 2006; 107 : 1806–1809.
91. Huang ME, Ye YC, Chen SR, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988; 72 : 567–572.
92. Balgobind BV, Zwaan CM, Pieters R, et al. The heterogeneity of pediatric MLL-rearranged acute myeloid leukemia. Leukemia 2011; 25 : 1239–1248.
93. Martelli MP, Sportoletti P, Tiacci E, et al. Mutational landscape of AML with normal cytogenetics: biological and clinical implications. Blood Rev 2013; 27 : 13–22.
94. Campana D. Minimal residual disease monitoring in childhood acute lymphoblastic leukemia. Curr Opin Hematol 2012; 19 : 313–318.
95. Gaipa G, Basso G, Biondi A, et al. Detection of minimal residual disease in pediatric acute lymphoblastic leukemia. Cytometry B Clin Cytom 2013; 84 : 359–369.
96. Kalina T, Flores-Montero J, Lecrevisse Q, et al. Quality assessment program for EuroFlow protocols: Summary results of four-year (2010--2013) quality assurance rounds. Cytometry A 2014.
97. Kalina T, Flores-Montero J, van der Velden VH, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 2012; 26 : 1986–2010.
98. Inaba H, Coustan-Smith E, Cao X, et al. Comparative analysis of different approaches to measure treatment response in acute myeloid leukemia. J Clin Oncol 2012; 30 : 3625–3632.
99. Szczepanski T, van der Velden VH, van Dongen JJ. Flow-cytometric immunophenotyping of normal and malignant lymphocytes. Clin Chem Lab Med 2006; 44 : 775–796.
100. van der Velden VH, van der Sluijs-Geling A, Gibson BE, et al. Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia 2010; 24 : 1599–1606.
101. Trka J, Kalinova M, Hrusak O, et al. Real-time quantitative PCR detection of WT1 gene expression in children with AML: prognostic significance, correlation with disease status and residual disease detection by flow cytometry. Leukemia 2002; 16 : 1381–1389.
102. Willasch AM, Gruhn B, Coliva T, et al. Standardization of WT1 mRNA quantitation for minimal residual disease monitoring in childhood AML and implications of WT1 gene mutations: a European multicenter study. Leukemia 2009; 23 : 1472–1479.
103. Szczepanski T, Harrison CJ, van Dongen JJ. Genetic aberrations in paediatric acute leukaemias and implications for management of patients. Lancet Oncol 2010; 11 : 880–889.
104. Schiller J, Praulich I, Krings Rocha C, et al. Patient-specific analysis of FLT3 internal tandem duplications for the prognostication and monitoring of acute myeloid leukemia. Eur J Haematol 2012; 89 : 53–62.
105. Schnittger S, Kern W, Tschulik C, et al. Minimal residual disease levels assessed by NPM1 mutation-specific RQ-PCR provide important prognostic information in AML. Blood 2009; 114 : 2220–2231.
106. Szczepanski T, Flohr T, van der Velden VH, et al. Molecular monitoring of residual disease using antigen receptor genes in childhood acute lymphoblastic leukaemia. Best Pract Res Clin Haematol 2002; 15 : 37–57.
107. van der Velden VH, Cazzaniga G, Schrauder A, et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia 2007; 21 : 604–611.
108. van Dongen JJ, Wolvers-Tettero IL. Analysis of immunoglobulin and T cell receptor genes. Part II: Possibilities and limitations in the diagnosis and management of lymphoproliferative diseases and related disorders. Clin Chim Acta 1991; 198 : 93–174.
109. van Dongen JJ, Wolvers-Tettero IL. Analysis of immunoglobulin and T cell receptor genes. Part I: Basic and technical aspects. Clin Chim Acta 1991; 198 : 1–91.
110. van der Velden VH, Corral L, Valsecchi MG, et al. Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia 2009; 23 : 1073–1079.
111. Logan AC, Vashi N, Faham M, et al. Immunoglobulin and T cell receptor gene high-throughput sequencing quantifies minimal residual disease in acute lymphoblastic leukemia and predicts post-transplantation relapse and survival. Biol Blood Marrow Transplant 2014; 20 : 1307–1313.
112. Wu D, Emerson RO, Sherwood A, et al. Detection of minimal residual disease in B lymphoblastic leukemia by high-throughput sequencing of IGH. Clin Cancer Res 2014; 20 : 4540–4548.
113. Thol F, Kolking B, Damm F, et al. Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with FLT3-ITD or NPM1 mutations. Genes Chromosomes Cancer 2012; 51 : 689–695.
114. van Dongen JJ, Seriu T, Panzer-Grumayer ER, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 1998; 352 : 1731–1738.
115. Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010; 115 : 3206–3214.
116. Eckert C, Henze G, Seeger K, et al. Use of allogeneic hematopoietic stem-cell transplantation based on minimal residual disease response improves outcomes for children with relapsed acute lymphoblastic leukemia in the intermediate-risk group. J Clin Oncol 2013; 31 : 2736–2742.
117. Eckert C, von Stackelberg A, Seeger K, et al. Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia - long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 2013; 49 : 1346–1355.
118. Bader P, Kreyenberg H, Henze GH, et al. Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J Clin Oncol 2009; 27 : 377–384.
119. Krejci O, van der Velden VH, Bader P, et al. Level of minimal residual disease prior to haematopoietic stem cell transplantation predicts prognosis in paediatric patients with acute lymphoblastic leukaemia: a report of the Pre-BMT MRD Study Group. Bone Marrow Transplant 2003; 32 : 849–851.
120. Stentoft J, Hokland P, Ostergaard M, et al. Minimal residual core binding factor AMLs by real time quantitative PCR--initial response to chemotherapy predicts event free survival and close monitoring of peripheral blood unravels the kinetics of relapse. Leuk Res 2006; 30 : 389–395.
121. Zhang L, Cao Z, Ruan M, et al. Monitoring the AML1/ETO fusion transcript to predict outcome in childhood acute myeloid leukemia. Pediatr Blood Cancer 2014; 61 : 1761–1766.
122. Loken MR, Alonzo TA, Pardo L, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children‘s Oncology Group. Blood 2012; 120 : 1581–1588.
123. Rubnitz JE, Inaba H, Dahl G, et al. Minimal residual disease-directed thera py for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 2010; 11 : 543–552.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2015 Číslo 2
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Cytomegalovirové infekce u novorozenců a dětí
Nejčtenější v tomto čísle
- Akutní myeloidní leukémie v dětském věku
- Akútna lymfoblastová leukémia
- Leukémie u dětí ve 21. století
- Imunofenotypizace a jiné využití průtokové cytometrie u akutních leukémií
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy