
Nádory měkkých tkání očima molekulárního patologa
Soft tissue tumors - the view of the molecular biologist
Soft tissue tumors (SSTs) constitute a broad spectrum of neoplasms with diverse biological properties. Rare or unusual types are often difficult to classify. Recent studies show, that a significant subset of SSTs including many types of sarcomas are associated with specific genetic changes such as chromosomal translocations producing chimeric genes, which play a role in the pathogenesis of SSTs. Because SSTs represent a diagnostically challenging group of tumors, molecular-genetic techniques (FISH or PCR) are useful as supplementary and/or confirmatory diagnostic tools. In the present paper we demonstrate the usefulness of a combined diagnostic approach using the tools of classical histopathology and immunohistochemistry together with the molecular diagnostic approach in selected nosologic entites.
Keywords:
soft tissue tumors – translocation – RT-PCR
Autoři:
Lenka Krsková; Marcela Mrhalová; Markéta Kalinová; Vít Campr; Linda Čapková; Marek Grega; Jaromír Háček; Ludmila Hornofová; Mária Chadimová; Renata Chmelová; Daniela Kodetová; Josef Zámečník; Roman Kodet
Působiště autorů:
Ústav patologie a molekulární medicíny, 2. LF UK a FN Motol
Vyšlo v časopise:
Čes.-slov. Patol., 50, 2014, No. 3, p. 132-140
Kategorie:
Přehledový článek
Souhrn
Nádory měkkých tkání jsou z histopatologického pohledu extrémně širokou paletou nádorů, jejichž klasifikace je často obtížná a vyžaduje specializaci. V posledních desetiletích výzkumu těchto nádorů se ukázalo, že část nádorů měkkých tkání je spolu s morfologickými kritérii definovaná specifickými chromozomálními změnami, zejména translokacemi. Produktem translokací jsou fúzní geny a ty ve funkčním stavu hrají významnou roli v patogenezi těchto nádorů. S výhodou lze v diferenciálně diagnostické rozvaze nádorů měkkých tkání využít detekci chromozomálních zlomů pomocí fluorescenční in situ hybridizace (FISH) a metod na principu PCR detekujících specifické fúzní transkripty. V práci uvádíme příklady některých jednotek a jejich molekulární vymezení.
Klíčová slova:
Nádory měkkých tkání – translokace – RT-PCR
Nádory měkkých tkání představují široké spektrum nádorů vycházejících z mezenchymálních, především pojivových tkání. Většina z těchto nádorů vzniká v hlubokých měkkých tkáních, v podkoží a někdy i v kůži. V některých situacích je původ nádorů měkkých tkání spojen s buněčnou diferenciací, která v nádorem postiženém orgánu původní není: příkladem je rabdomyosarkom močového měchýře, prostaty, vedlejších dutin nosních a další lokalit v těle bez přítomnosti příčně pruhované svaloviny (1).
Podle současné klasifikace WHO jsou měkkotkáňové maligní nádory (sarkomy) děleny do více než 40 nozologických jednotek, jejichž zařazení se odvíjí od tkáně, kterou nejvíce připomínají (2). Mezi patology přetrvává tendence dále v již ustálené klasifikaci sarkomů rozlišovat nové podjednotky a již beztak široké spektrum dále dělit podle nově morfologicky definovaných detailů, popř. zjištěných genetických změn. Protože však jde o nádory s relativně nízkou incidencí, mají jak kliničtí lékaři, tak patologové mimo vysoce specializovaná pracoviště jen málo příležitostí získat dostatek zkušeností s celým tímto spektrem nádorů a jejich variant, a to jak z hlediska diferenciálně diagnostického, tak především prognostického a terapeutického.
Diferenciální diagnostika může být i dnes, přes možnost využití moderních imunohistochemických technik, obtížná, zvláště u méně diferencovaných podtypů kulatobuněčných či vřetenobuněčných nádorů díky jejich podobnému histopatologickému obrazu (3). Konkrétní diagnóza je však nutná vzhledem k variabilnímu biologickému chování měkkotkáňových nádorů od zcela benigního průběhu přes nádory lokálně invazivní se schopností lokálních recidiv, až po nádory s vysoce agresívním průběhem a časným metastatickým rozsevem (2). S výhodou v diferenciálně diagnostické rozvaze využíváme imunohistochemické metody. Na ně pak navazuje aplikace dalších přístupů, zejména detekce chromozomálních zlomů pomocí fluorescenční in situ hybridizace (FISH) a metod založených na principu PCR detekující specifické fúzní transkripty – produkty chromozomálních translokací. Aplikace těchto metodik pomáhá k ucelení diagnózy příslušné jednotky a mnohdy ji potvrzuje. Detekci fúzních transkriptů lze také s výhodou využít pro sledování infiltrace okrajů excize nádorovými buňkami a u některých jednotek také ke sledování dynamiky minimální diseminované nemoci (MDN) v kostní dřeni popř. v periferní krvi pomocí PCR v reálném čase.
Prognostickými faktory sarkomů měkkých tkání podle klinicko-patologického hodnocení jsou stadium, histopatologický grade, histotyp, velikost nádoru, jeho lokalizace a rozsah a kvalita primární resekce. Velikost nádoru ovlivňuje především riziko lokálních recidiv, zatímco histologický grade koreluje s časovým intervalem do výskytu metastáz a s celkovým přežíváním nemocných. V poslední době se s rozvojem molekulárně-genetických metod do popředí dostává i určení translokace a fúzního transkriptu, popř. přesného určení místa zlomu (rozštěpení genu), které mohou mít rovněž prognostický význam (např. negativní prognostický znak t(2;13) s fúzním genem PAX3-FOXO1 u alveolárních rabdomyosarkomů).
V následujícím textu se věnujeme měkkotkáňovým nádorům, u kterých jsou známé specifické genetické změny. K některým jednotkám jsme vybrali krátká kazuistická sdělení, na kterých dokládáme možnosti současné kombinované morfologické a molekulární diagnostiky.
Nádory z fibroblastů
Fibrosarkom
Fibrosarkom je ve skutečnosti nádorem vzácným, ačkoli dříve takto byla označována velká část vřetenobuněčných sarkomů. Fibrosarkom postihuje především starší pacienty. Jeho molekulární podstatu ve většině případů zatím neznáme. Existuje však i raně dětská varianta fibrosarkomu – tzv. infantilní fibrosarkom vykazující translokaci t(12;15)(p13;q25) s fúzním genem ETV6 - NTRK3.
Pod pojmem fibrosarkom se dále skrývá řada variant, jako jsou sklerotizující epiteloidní fibrosarkom, adultní fibrosarkom, myxofibrosarkom. Tzv. low-grade fibromyxoidní sarkom je charakterizován typickou chromozomální translokací t(7;16)(q33;p11) s fúzním genem FUS/CREB3L2. Charakteristické molekulární změny lze také identifikovat u fibrosarkomu vznikajícího na podkladě dermatofibrosarcoma protuberans (viz níže). I když tyto jednotky spojuje název „fibrosarkom“, jedná se o odlišné nádory. Dále se do této skupiny řadí myofibroblastický sarkom a zánětlivý myofibroblastický nádor s typickou přestavbou genu ALK v oblasti chromozómu 2p23. Mikroskopicky se jedná o nádory z fibroblastů, případně myofibroblastů s příměsí různě velkého množství extracelulárních komponent, zejména kolagenních vláken, které jsou produkovány nádorovými buňkami.
Infantilní fibrosarkom
Jedná se o silně buněčný nádor uspořádaný ve svazky vřetenitých buněk obvykle bez atypií, ale s nekrózami a s vysokou mitotickou aktivitou. Má intermediální maligní potenciál. Nádor má ve většině případů příznivý klinický průběh v závislosti na možnostech kompletní resekce. Histopatologickou diagnostikou této jednotky jsme se zabývali v samostatném sdělení již dříve (4).
Kazuistika 1
Vyšetřili jsme vzorek biopsie od 9měsíční dívky s nádorem předloktí, který dosahoval průměru 7 cm. Z histologického vyšetření bylo patrné, že jde o fascikulárně uspořádaný nádor z krátkých ovoidních a vřetenitých buněk (obr. 1) s imunohistochemickou pozitivitou vimentinu, proteinu bcl-2 a s fokální pozitivitou aktinu; negativní byl průkaz S100 proteinu, desminu, cytokeratinů, epiteliálního membránového antigenu a transkripčních faktorů MyoD1 a myogeninu. Pozitivita Ki-67 kolísala od 20 do 40 % nádorových buněk. Pomocí fluorescenční in situ hybridizace (FISH) jsme prokázali zlom v oblasti 12p13, kde je situován gen ETV6 (obr. 1). Pomocí metody RT-PCR jsme zjistili fúzní gen ETV6-NTRK, který vzniká translokací t(12;15). Nález jsme na základě všech uvedených vyšetření klasifikovali jako infantilní fibrosarkom.

Low-grade fibromyxoidní sarkom
Jde o nádor, který postihuje převážně dospělé v mladším věku. Roste infiltrativně v hlubokých měkkých tkáních a je typicky multinodulárně uspořádán. Okolo uzlíků myxoidního vzhledu nalézáme i celulární partie nádoru. Mohou být přítomny i mírné jaderné atypie. Nádor je cévnatý, cévy bývají vinuté až plexiformně uspořádané. Kromě myxoidních a buněčných oblastí mohou být přítomny hypocelulární silně kolagenizované okrsky a přibližně ve třetině případů tzv. obrovské kolagenní rozety s nádorovými buňkami uspořádanými kolem uzlíkovitě uspořádané kolagenizované tkáně. Nádor bývá fokálně pozitivní při průkazu epiteliálního membránového antigenu (EMA) a glykoproteinu MUC4. Kromě morfologického vzhledu je nádor charakterizován chromozomální translokací t(7;16)(q33;p11). Přes nízce proliferativní a zdánlivě neškodný vzhled má nádor lokálně recidivující a metastatický potenciál.
Kazuistika 2
Vyšetřili jsme vzorek nádoru od 60letého muže s nádorem zad. Šlo o laločnatě stavěný nádor rozměrů 110x95x45 mm, který byl na povrchu vazivově opouzdřený. Na řezu byl zastižen uzlovitý žlutobělavý nádor, který v jednom okraji přecházel do myxoidně prosáklé oblasti. Mikroskopicky byla nodularita procesu dobře patrná a nádor tvořily převážně vřetenité a místy hvězdicovité buňky, které byly uloženy v myxoidním stromatu (obr. 2) pozitivním v barvení alciánovou modří. Některé nádorové buňky měly velká hyperchromní atypická jádra s jadernými pseudoinkluzemi (obr. 3). Místy byl nádor buněčnější, tvořený fascikulárně uspořádanými vřetenitými nádorovými buňkami s protáhlými jádry. Lipoblastická diferenciace nebyla zastižena. Mitotická aktivita byla nízká. Ložiskově nádor nekrotizoval. Neprokázali jsme obrovské kolagenní rozety. V imunohistochemickém vyšetření byly nádorové buňky fokálně pozitivní v reakci s protilátkami proti S100 proteinu, CD34 a při průkazu EMA. Glykoprotein MUC4 jsme neměli k dispozici. Negativní byl průkaz hladkosvalového aktinu, desminu, h-kaldesmonu a směsi cytokeratinů. Ki-67 pozitivitu jsme odhadovali u 5 % nádorových buněk, včetně pozitivit u buněk s atypiemi. Na základě histologického nálezu jsme pro přítomné buněčné atypie a ložiska zvýšené buněčnosti zvažovali diagnózu high-grade myxofibrosarkomu, ale v úvahu přicházel také low-grade fibromyxoidní sarkom. Vzorek jsme vyšetřili pomocí metody RT-PCR a následné sekvenace. Identifikovali jsme fúzní gen FUS/CREB3L2. Prokázali jsme však variantní místo zlomu, a to fúzi části exonu 9 genu FUS s exonem 5 genu CREB3L2 (obr. 3). Toto místo zlomu nebylo zatím v literatuře popsáno. Běžně dochází k fúzi exonu 6 či 7 genu FUS s exonem 5 genu CREB3L2. Případ jsme na základě morfologické a zejména molekulární diagnostiky definitivně uzavřeli v rámci spektra obrazu tzv. nízkostupňového fibromyxoidního sarkomu. Biologický a klinický význam aberantní fúze exonů nelze při výskytu u jednoho nemocného posoudit.


Dermatofibrosarcoma protuberans (DFSP)
Jedná se o nádor, který je typicky lokalizován v dermis, ale často zasahuje do hlubších vrstev podkoží. Charakteristicky je tento nádor špatně ohraničen, je vysoce buněčný, typické je storiformní uspořádání svazků nádorových buněk. Dermatofibrosarcoma protuberans je nízce maligní nádor, který ale často recidivuje. Vzácně může metastazovat, obzvláště v tom případě, kdy se v nádoru nacházejí ložiska, která nelze odlišit od fibrosarkomu. Charakteristickým znakem tohoto onemocnění je reciproká translokace t(17;22)(q22;q13) a nadpočetný „ring” chromozóm, r(17;22). V obou případech dochází k fúzi genu pro kolagen typu 1A1 (Col1A1) a genu pro beta řetězec destičkového růstového faktoru (PDGFB). To vede k aktivaci exprese genu PDGFB a prostřednictvím autokrinní stimulace PDGF receptoru k nádorové transformaci buněk. PDGB receptor náleží do skupiny tyrozin kináz, které mohou být selektivně inhibovány specifickými nízkomolekulárními látkami typu imatinib mesylát. Poslední studie naznačují, že nádor u pacientů s metastatickým DFSP na léčbu tímto preparátem poměrně dobře reaguje (5).
Kazuistika 3
Vyšetřili jsme vzorek od patnáctiletého chlapce s kožní lézí v oblasti hrudníku. V dermis byl zastižen nádor tvořený protáhlými nádorovými buňkami uspořádanými do svazků, ložiskově bylo patrno i storiformní uspořádání nádorových buněk (obr. 4). Nádor infiltroval přilehlou subkutánní tukovou tkáň, nezasahoval do spodiny excize, zasahoval však těsně do blízkosti jednoho z laterálních okrajů excize. Imunohistochemicky jsme prokázali pozitivitu antigenu CD34 a vimentinu, Ki-67 pozitivita byla přibližně ve 3 % nádorových buněk. Morfologickou diagnózu jsme uzavřeli jako dermatofibrosarcoma protuberans. Vzorek tkáně byl následně vyšetřen pomocí metody RT-PCR s primery z genu PDGFB a primery pro exony 5, 8, 11, 15, 17, 20, 23, 26, 29, 32, 35, 38, 40, 43, 46, 49 genu Col1A1 (obr. 4). U pacienta jsme prokázali fúzní gen typický pro dermatofibrosarcoma protuberans - Col1A1/PDGFB s fúzí exonu 32 genu Col1A1 a exonu 2 genu PDGFB. Molekulární vyšetření t(17;22) je přínosné zvláště u pacientů s atypickým morfologickým obrazem, aberantním imunohistochemickým profilem, popř. u pacientů s diagnózou dermatofibrosarcoma protuberans v neobvyklém věku.

Nádory tukové tkáně
Liposarkom
Liposarkomy tvoří heterogenní skupinu nádorů s adipocytární diferenciací. Proto je blíže klasifikujeme do několika podtypů: atypický lipomatózní nádor (dříve dobře diferencovaný liposarkom) s typickou amplifikací genu MDM2 v oblasti chromozómu 12q15, dediferencovaný liposarkom, rovněž s amplifikací MDM2, pleomorfní liposarkom a myxoidní liposarkom. Kulatobuněčný liposarkom byl dříve považován za specifickou jednotku, ale dnes je zřejmé, že se jedná o dediferencovanou komponentu myxoidního liposarkomu (6). Myxoidní liposarkom je charakterizován cytogeneticky translokací t(12;16)(q13;p11) s fúzním genem FUS-DDIT3, případně translokací t(12;22)(q13;q12) a fúzním genem EWSR1-DDIT3. Prognóza pacientů s těmito nádory závisí především na určení daného histologického typu liposarkomu.
Kazuistika 4
K histopatologickému a molekulárnímu vyšetření jsme v roce 2007 obdrželi tkáň 32letého pacienta z oblasti distální částí stehna dorzálně a přilehlé podkolení jamky. Morfologické vyšetření ukázalo nádor o proměnlivé buněčnosti s myxoidním stromatem s početnými větvenými kapilárami, v němž byly drobnější většinou hvězdicovité nádorové buňky, některé s vakuolizovanou cytoplasmou (obr. 5). Fokálně byl nádor buněčnější se zastoupením kulatých buněk. Mitotická aktivita byla nízká. Ki-67 pozitivitu jsme odhadovali u 20 - 30 % nádorových buněk. Nález jsme uzavřeli jako myxoidní liposarkom s ložiskovou kulatobuněčnou složkou, nádor s intermediárním stupněm malignity. V roce 2009 pacient přichází s rezistencí v pravé axile, histologicky jsme nádor zařadili též jako myxoidní liposarkom. Nešlo však posoudit, zda se jedná o metachronně se vyskytující nový primární nádor či, zda jde o neobvykle lokalizovanou metastázu liposarkomu diagnostikovaného před dvěma roky. K dalšímu histologickému vyšetření byl pacient indikován v roce 2011 pro rezistenci v jizvě v oblasti axily, kde byla potvrzena recidiva myxoidního liposarkomu. V roce 2012 jsme po kardiochirurgické intervenci vyšetřili nádor exstirpovaný z komorového septa myokardu velikosti 40 x 25 x 20 mm. Nález měl histologicky obdobný charakter jako v předchozích vyšetřeních. Vyšetření pomocí FISH prokázalo zlom v oblasti 12q13 (gen DDIT3) a zlom v oblasti 16p11 (gen FUS). Molekulární vyšetření primárního nádoru, excize nádoru v axile i v odběru z myokardu prokázalo translokaci t(12;16) s fúzním genem FUS/DDIT3 (obr. 5). Ve všech vyšetřovaných vzorcích jsme prokázali stejné místo zlomu a to fúzi exonu 5 genu FUS s exonem 2 genu DDIT3. Tento nález podpořil naší úvahu, že v axile a v myokardu jde o metastatický myxoidní liposarkom z primárního místa ve stehně spíše, než o nádorovou multiplicitu.

Nádory vycházející z krevních nebo lymfatických cév
Epiteloidní hemangioendoteliom
Jedná se o nízce maligní sarkom. Mimo měkkých tkání se nádor také vyskytuje např. v plicích, v játrech a v kostech. Nezřídka se epiteloidní hemangioendoteliom nachází ve všech zmíněných lokalizacích najednou a potom je těžké určit, zda se jedná o metastázy či o multifokální proces. Jde o vaskulární nádor s metastatickým potenciálem, který je tvořen epiteloidními endoteliálními buňkami uspořádanými v krátkých pruzích a v hnízdech uložených v myxohyalinním stromatu. Biologické chování odpovídá nádoru s intermediálním stupněm malignity. Metastázy se popisují u 20 – 30 % osob. Tento typ nádoru je charakterizován translokací t(1;3)(p36;q25) s fúzním genem WWTR1/CAMTA1 (7).
Kazuistika 5
Vyšetřili jsme biopsii 60-leté pacientky s útvarem v levém nadklíčku rozměrů 20 – 25 mm. Z histologického pohledu byl nádor solidně trabekulární s trsovitým uspořádáním konglomerátů velkých polygonálních buněk s eozinofilní, místy světlou a místy vakuolizovanou cytoplazmou (obr. 6). Skupiny buněk byly uloženy ve fibrohyalinním vazivu a tvořily trámce nebo čepy, někdy se zřetelnými luminálními štěrbinami. Cytologicky byly místy patrné atypie nádorových buněk. V nádoru byly přítomny též regresivní změny s fokální nekrotizací. Z hlediska imunohistochemického byly nádorové buňky pozitivní v průkazu molekuly CD31 a faktoru VIII. Vzorek jsme dále vyšetřili pomocí RT-PCR a prokázali jsme fúzní gen WWTR1/CAMTA1 - tedy translokaci t(1;3). Pomocí sekvenace jsme identifikovali fúzi exonu 4 genu WWTR1 s exonem 9 genu CAMTA1 (obr. 6). Nález jsme uzavřeli jako epiteloidní hemangioendoteliom.

Nádory příčně pruhované svaloviny
Rabdomyosarkom (RMS)
Rabdomyosarkom se nejnověji dělí na čtyři základní typy - embryonální, alveolární, pleomorfní a vřetenobuněčný/sklerozující RMS. Embryonální RMS se vyskytuje především u dětí. Alveolární RMS postihuje spíše starší děti a patří mezi časně metastazující sarkom. Pro tuto variantu rabdomyosarkomu jsou specifické translokace, které nalézáme u více než 80 % pacientů. Nejčastěji jde o t(2;13)(q35;q14) s fúzí genů PAX3/FOXO1, dále t(1;13)(p36;q14) s fúzním genem PAX7/FOXO1. U části nemocných je nutné hledat poměrně vzácné variantní translokace jako je t(X;2)(q13;q35) a fúzní gen PAX3/FOXO4, t(2;2)(q35;p23) a fúzní gen PAX3/NCOA1, či t(2;8)(q35;q13) a fúzní gen PAX3/NCOA2. Právě detekce fúzních genů a popř. sledování exprese transkripčních faktorů zapojených do myogeneze jako jsou MyoD1 a myogenin lze s výhodou využít nejen v diagnostice RMS, ale i ke zhodnocení okrajů excize a ke sledování dynamiky minimální diseminované nemoci (MDN) v kostní dřeni a v periferní krvi (obr. 7).

Kazuistika 6
K histologickému a molekulárnímu vyšetření jsme obdrželi vzorek 31leté pacientky s nádorem nosohltanu a vedlejších dutin nosních. Vyšetřovaná částka 40x25x20 mm byla tvořená prokrváceným a regresivně změněným nádorem. Nádor byl solidně stavěný bez zjevné strukturalizace, či pseudoalveolárního uspořádání. Nádorové buňky byly polygonální, místy vřetenité s výraznými jadernými atypiemi a se světlou eozinofilní cytoplazmou (obr. 8). V cytoplazmě nádorových buněk byl patrný granulární PAS pozitivní materiál. Zastižena byla výrazná mitotická aktivita s četnými atypickými mitotickými vřeteny. Ki-67 pozitivita byla až v 50 % nádorových buněk. Imunohistochemické vyšetření prokázalo pozitivitu v průkazu desminu, myogeninu, MyoD1 a sarkomerického aktinu. Morfologický nález jsme uzavřeli jako pleomorfní rabdomyosarkom. Molekulární vyšetření prokázalo expresi regulačních transkripčních faktorů MyoD1 a myogeninu, které běžně u RMS prokazujeme. U pacientky jsme dále zjistili fúzní gen PAX3/FOXO1 (obr. 8), produkt chromozomální translokace t(2;13). V aspirátu kostní dřeně jsme markery RMS neprokázali (exprese MyoD1, myogeninu ani fúzního genu PAX3/FOXO1). Čtyři měsíce od stanovení diagnózy (po předoperační chemoterapii) pacientka podstoupila resekci nádoru. Pomocí RT-PCR jsme vyšetřili 4 vzorky z okrajů excize, ve 2 vzorcích nebyl přítomen fúzní gen PAX3/FOXO1 a nález koreloval s morfologickým obrazem bez nádorové tkáně, v dalších 2 vzorcích (jeden morfologicky negativní a jeden morfologicky s pozitivním průkazem rabdomyoblastů) byl prokázán fúzní gen PAX3/FOXO1 svědčící pro výskyt nádorových buněk v okraji resekce a tím i pro reziduum rabdomyosarkomu. Definitivně jsme nádor zařadili jako solidní variantu alveolárního RMS a to především s přihlédnutím k výsledku molekulárního vyšetření a k průkazu specifické translokace.

Chondroidní nádory
Mezenchymální chondrosarkom
Je nádorem mladých lidí. Vzniká nejčastěji v orbitě, v oblasti tvrdé pleny mozkové, v retroperitoneu a v končetinové lokalizaci. Jedná se o vysoce maligní, záhy metastazující sarkom. Pro více než 70% mezenchymálních chondrosarkomů je typická inverze chromozómu 8 inv(8)(q13;q21) s fúzí genů HEY1 a NCOA2 (8).
Kazuistika 7
Vyšetřili jsme vzorek od 31letého muže s nádorem baze lební a vedlejších dutin nosních. V histologickém vyšetření byla zastižena atypická nádorová chrupavčitá tkáň s ložiskovými kalcifikacemi, na kterou navazovalo vazivo s dilatovanými cévami a drobný úsek tmavých polygonálních buněk (obr. 9). Chondroidní úseky byly pozitivní s S100 proteinem. Nález jsme uzavřeli jako mezenchymální chondrosarkom. Pacient přichází s opakovanými recidivami nádoru pět a šest let od diagnózy. Vzorky byly vyšetřeny pomocí RT-PCR, pomocí níž byl detekován fúzní gen HEY1/NCOA2 (obr. 9). Dále jsme vyšetřili osm vzorků tkáně po radikální operaci. Ze čtyř vzorků se nepodařilo vyizolovat kvalitní RNA z důvodu fixace materiálu ve 4 % roztoku formaldehydu s následnou degradací nukleových kyselin, ze zbývajících 4 vzorků jsme RNA získali vhodnou pro další molekulární analýzy. V jednom ze čtyř vyšetřovaných vzorků jsme fúzní gen HEY/NCOA2 v korelaci s morfologickým nálezem prokázali. Nález byl tedy v korelaci s morfologicky stanovenou diagnózou mezenchymálního chondrosarkomu. Změnu na molekulární úrovni lze využít k podpoře diagnostiky zejména u nepřehledných nebo zhmožděných vzorků z okrajů, při další recidivě, nebo diseminaci procesu.

Nádory specificky nezařazené
Synoviální sarkom
V 80 % případů se synoviální sarkom nachází v oblasti měkkých tkání kolem kolena a kotníku, ve stěně trupu či břicha a jen výjimečně v tělních dutinách. Postihuje starší děti a mladé dospělé osoby. Z mikroskopického hlediska synoviální sarkom rozlišujeme na monofazický a bifazický. Cytogeneticky jsou synoviální sarkomy dobře definované specifickou translokací t(X;18)(p11.2;q11.2) s fúzními geny SS18-SSX1, SS18-SSX2 a vzácně SS18-SSX4. Synoviální sarkomy patří mezi agresivní sarkomy, zvláště pro jejich značnou chemorezistenci.
Kazuistika 8
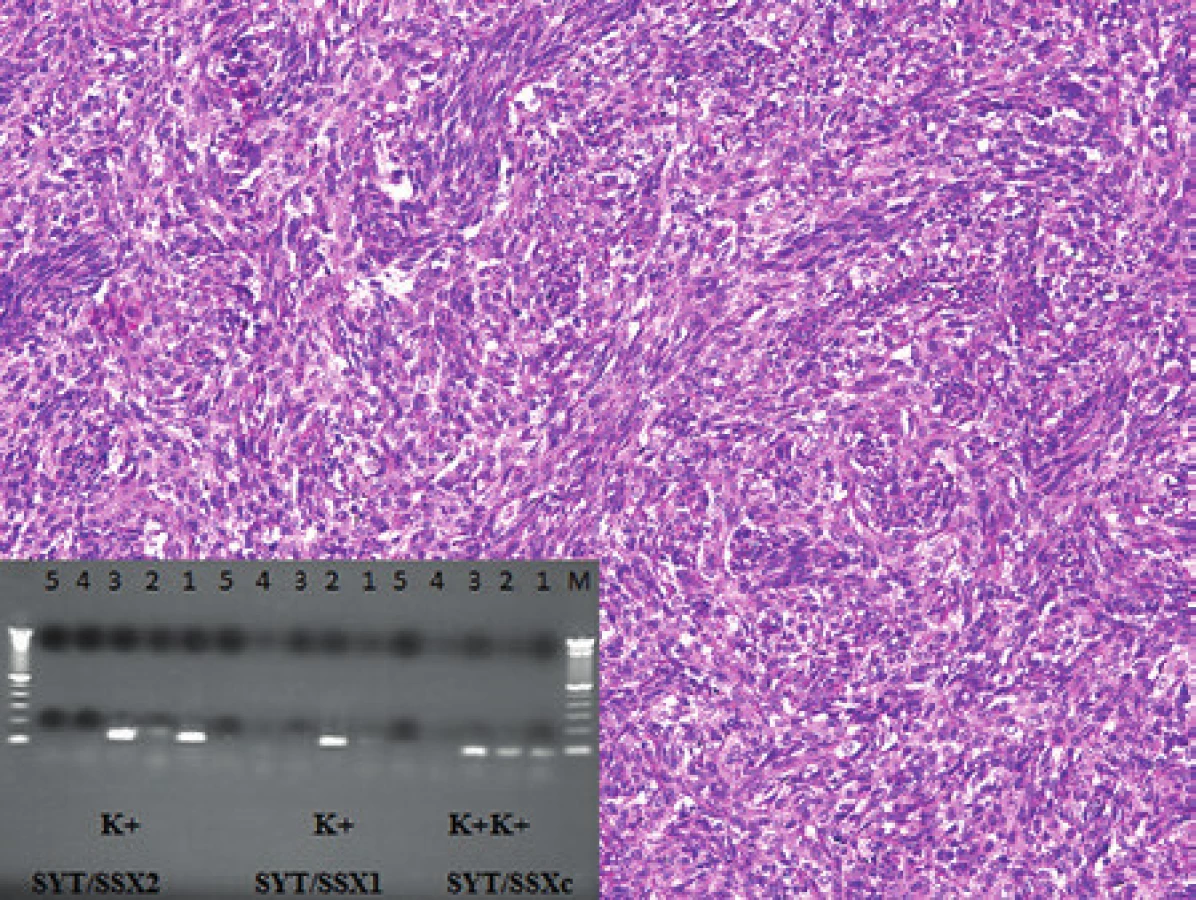
K histologickému a molekulárnímu vyšetření jsme dostali biopsii 49letého pacienta s nádorem v oblasti dolní duté žíly, pravé síně a perikardu. V histologickém vyšetření byl zastižen vřetenobuněčný sarkom s naznačeně epiteloidními ložisky (obr. 10). Pomocí imunohistochemického vyšetření byla prokázána pozitivita nádorových buněk v reakci s vimentinem, epiteliálním membránovým antigenem, proteinem bcl-2 a CD99; negativní byly nádorové buňky v průkazu s cytokeratiny a S100 proteinem. Ki-67 bylo pozitivní až ve 20 % nádorových buněk. Pomocí FISH analýzy jsme prokázali zlom v chromozomální oblasti 18q11.2 (gen SS18) a pomocí RT-PCR fúzní gen SS18/SSX2 (obr. 10). Na základě těchto výsledků jsme nález uzavřeli jako monofázický fibrózní synoviální sarkom. Molekulární vyšetření mělo smysl zejména v podpoře diagnostiky nádoru ve velmi neobvyklé lokalizaci primárního nádoru postihujícího velké cévy a srdce.
Extraskeletální myxoidní chondrosarkom
Extraskeletální myxoidní chondrosarkom je maligní mezenchymální nádor nejisté diferenciace. Jde o nádor dospělých a starších pacientů. Je považován za sarkom s nízkým stupněm malignity. Většinou roste pozvolna, ale často recidivuje. Histologicky je tento nádor zaměňován s nejrůznějšími benigními novotvary. Pro tento typ nádoru je specifická translokace t(9;22)(q22;q12) s fúzním genem EWSR1/NR4A3 (též znám jako TEC či CHN), který bývá detekován u přibližně 75 % pacientů (9). Dále existují variantní translokace t(9;17)(q22;q11) s fúzním genem TAF15/NR4A3 a to u necelých 25 % pacientů. Raritní je translokace t(9;15)(q22;q21) s fúzí genů TCF12/NR4A3.
Angiomatoidní (maligní) fibrózní histiocytom
Je považován za specifickou jednotku. Angiomatoidní maligní fibrózní histiocytom se vyskytuje u dětí. Makroskopicky jde o ohraničený multinodulární nádor měkkých tkání. Nádor může recidivovat a výjimečně metastazovat.
Kazuistika 9
K histologickému a následně i molekulárně-genetickému vyšetření jsme dostali biopsii 4letého chlapce s rezistencí v oblasti levého lokte. Makroskopicky byl nádor uložený v měkkých tkáních a měl uzlovitou strukturu. Mikroskopicky nádor tvořily kompaktní uzly, přičemž v centru některých uzlů byly dutiny vyplněné krví, podobně jako u kavernózních hemangiomů. Kromě těchto dutin byly přítomny nepravidelně distribuované a nikterak hojné štěrbinovité cévy. Nádorové buňky měly polygonální až naznačeně krátce fascikulární uspořádání, okrouhlá až lehce protáhlá vesikulární jádra se zřetelným drobným centrálně postaveným jadérkem (obr. 11). Cytoplazma byla slabě eozinofilní. Nádorové buňky vykazovaly mírný stupeň cytologických nepravidelností. V nádoru a v jeho okolí byl hojný hemosiderin, ve stromatu bohatá lymfocytární a plasmocelulární příměs, místy byly četné makrofágy a obrovské mnohojaderné buňky. Fluorescenční in situ hybridizace (FISH) prokázala zlom v oblasti chromozómu 22q12, kde je lokalizován gen EWSR1 (obr. 11). Následně byl pomocí metody RT-PCR prokázán fúzní gen EWSR1/CREB1, který je produktem translokace t(2;22) a který bývá spolu s další translokací t(12;22) a fúzním genem EWSR1/ATF1 spojován právě s diagnózou angiomatoidního fibrózního histiocytomu. Na základě souboru výše uvedených vyšetření jsme nádor mohli zařadit jako angiomatoidní fibrózní histiocytom.

Alveolární sarkom měkkých tkání
U alveolárního sarkomu měkkých tkání stále není známá výchozí buňka. Jde o vysoce maligní sarkom s nejčastějším výskytem v hlubokých měkkých tkáních stehna a hýždě, ale u dětí také v oblasti hlavy a krku a v některých dalších méně častých lokalizacích. Častěji postiženými bývají mladé ženy. Charakteristickou mikroskopickou strukturou jsou PAS pozitivní krystaly v cytoplazmě nádorových buněk. Ze všech sarkomů nejčastěji metastazuje do mozku. V plicích často tvoří mnohočetné metastázy. Pro tento typ nádoru je specifická nebalancovaná translokace t(X;17)(p11;p25) s fúzním genem ASPL-TFE3. Tím dochází k nadměrné expresi transkripčního faktoru E3 (TFE3) a narušení regulace buněčného cyklu.
Kazuistika 10
K histologickému a molekulárnímu vyšetření jsme dostali biopsii od 18-leté ženy s nádorem v oblasti musculus vastus lateralis. Diagnózu jsme uzavřeli jako alveolární sarkom měkkých tkání. Tři měsíce od diagnózy byly exstirpovány 3 metastázy nádoru v plicích. Další 2 suspektní plicní metastázy jsme měli příležitost vyšetřit o 11 let později, kdy byl nález uzavřen na základě morfologického i molekulárního vyšetření jako metastáza alveolárního sarkomu měkkých tkání. Pomocí RT-PCR jsme prokázali translokaci t(X;17) s fúzním genem ASPL/TFE3 se stejným místem zlomu jak v primárním nádoru, tak v metastázách (obr. 12).

Epiteloidní sarkom
Epiteloidní sarkom vychází především z aponeuróz distálních částí končetin. Pro proximální typ epiteloidního sarkomu je, podobně jako u rabdoidních nádorů, typická delece/mutace v oblasti dlouhého ramene 22q v oblasti genu SMARCB1/INI1.
Desmoplastický nádor z malých kulatých buněk (DSRCT)
DSRCT je vzácné onemocnění zařazené do skupiny sarkomů. Nejčastěji postihuje mladé muže. V typickém případě se manifestuje rozsáhlou infiltrací břišní dutiny. Prognóza onemocnění je nepříznivá, dvouleté přežití se dosahuje pouze ve 20 % případů, a to i při použití kombinovaných protinádorových léčebných postupů. Histopatologicky je typický obraz kulatých až oválných buněk rostoucích v nepravidelných ostrůvcích zavzatých v dezmoplastickém stromatu. Reaktivní novotvorba stromálního vaziva je podmíněna expresí endogenního destičkového růstového faktoru (PDGF), který působí jako mitogen a vede k výrazné stimulaci fibroblastů (10). Původně se mělo za to, že DSRCT vychází z mezotelu, nicméně pravděpodobnější je hypotéza o původu nádoru z progenitorových buněk s multifenotypovou diferenciací (11). Příčina vzniku DSRCT není známa, nicméně patogenetickým podkladem malignity je přestavba genu EWSR1 na 22. chromozómu. V případě DSRCT jde o translokaci t(11;22)(p13;q12) s fúzním genem EWSR1-WT1.
Kazuistika 11
Vyšetřovali jsme nádorovou tkáň 21letého pacienta s rozsáhlým nádorem dutiny břišní (peritoneum včetně omenta a malé pánve) velikosti 170 x 135 x 140 mm a se sekundárním postižením pankreatu, jater a abdominálních lymfatických uzlin. Nádor vycházel nejspíše ze stěny colon transversum, postihoval serózu a svalovinu, ale do submukózy ani do sliznice nezasahoval. Prorůstal do ocasu pankreatu a adheroval pevně ke stěně žaludku, aniž by ji infiltroval. Nádor byl solidně trabekulárně uspořádán, tvořen malými uniformními kulatými buňkami s chudou cytoplazmou a malými hyperchromními jádry (obr. 13). Nádorové elementy byly uloženy v dezmoplastickém stromatu. Nádor byl mitoticky aktivní a fokálně podléhal nekrózám. Imunohistochemické vyšetření prokázalo pozitivitu nádorových buněk v průkazu desminu a v průkazu neuronálně-specifické enolázy. Negativní byly směsi cytokeratinů (OSCAR, KL-1, AE1/3), Wt1, MyoD1, myogenin, chromogranin, synaptofyzin, CD99, CD117, CD34 a hladkosvalový aktin. Ki-67 jaderná pozitivita kolísala, fokálně byla přítomna až ve 30 % nádorových buněk. FISH analýza odhalila zlom v oblasti chromozómu 22q12 (gen EWSR1). Pomocí zavedené RT-PCR nebyl detekován fúzní gen EWSR1/WT1 s typickým místem zlomu jakožto produkt translokace t(11;22). Z důvodu podezření na možnost jiného místa zlomu, jsme použili primery pro RT-PCR, které pokrývají větší oblast výše zmíněných genů. Nově navržená RT-PCR odhalila a následná sekvenace potvrdila fúzní gen EWSR1/WT1 s alternativním dosud nepopsaným místem zlomu (fúze exonu 5 genu EWSR1 s exonem 10 genu WT1 (obr. 13) a dále pak fúze exonu 7 EWSR1 s exonem 8 genu WT1, avšak v minoritním zastoupení). Diagnózu jsme na základě všech výše uvedených vyšetření uzavřeli jako desmoplastický kulatobuněčný nádor. Negativitu proteinu WT1 patrně způsobila alternativní fúze genů.

Sarkom z jasných buněk (CCS)
Sarkom z jasných buněk je maligní nádor typicky postihující hluboké měkké tkáně končetin. Tento nádor byl dříve pokládaný za měkkotkáňovou variantu melanomu. Charakteristický pro tento nádor je histologický, imunohistochemický a elektronmikroskopický obraz těžko odlišitelný od melanomu, k odlišení může posloužit určení specifické translokace t(12;22)(q13;q12) s fúzí genů EWSR1 a ATF1, popř. variantní translokace t(2;22)(q32;q12) s fúzním genem EWSR1-CREB1.
Kazuistika 12
Vyšetřovali jsme nádorovou tkáň 54letého pacienta s rozsáhlým nádorem bérce s metastatickým postižením skeletu. V odebraném materiálu byl zastižen převážně nekrotický a fokálně prokrvácený nádor. Nádorové buňky měly polygonální tvar, byly středně velké, se světlou eozinofilní cytoplasmou. Měly světlá vesikulární jádra s dobře patrnými jadérky, buňky byly uspořádány v solidních formacích (obr. 14). Mitotická aktivita byla zřetelná. Imunohistochemické vyšetření prokázalo pozitivitu s HMB45, S100 proteinem a v průkazu CD57. Negativní byly směsi cytokeratinů, desmin, myogenin, sarkomerický aktin, CD99, epiteliální membránový antigen, LCA, CD138, CD30, PGM1 a HBA71. FISH analýza odhalila zlom v oblasti chromozómu 22q12 (gen EWSR1). Pomocí zavedené RT-PCR jsme detekovali fúzní gen EWSR1/ATF1 jakožto produkt translokace t(12;22) s fúzí exonu 8 genu EWSR1 a exonu 5 genu ATF1. Diagnóza byla na základě všech výše uvedených vyšetření uzavřena jako sarkom z jasných buněk.

Nádory ze skupiny Ewingův sarkom
Ewingův sarkom
Nádory ze skupiny Ewingův sarkom (EWS) tvoří 4,4 % všech nádorů dětského věku a dospívajících. EWS postihuje převážně dlouhé a ploché kosti, ale může se vyskytnout i v měkkých tkáních nebo v orgánech. Jedná se o systémové onemocnění, které se rychle šíří hematogenní cestou. Přibližně 25 % nemocných má již v době stanovení diagnózy metastázy.
Fúzní gen EWSR1/FLI1 je produktem chromozomální aberace t(11;22)(q24;q12) a je označovaný jako molekulární marker Ewingova sarkomu. S onemocněním EWS je tento fúzní gen spojován přibližně v 90 – 95 % všech případů. U nádorových buněk EWS bylo popsáno celkem 18 rozdílných variant fúzního genu EWSR1/FLI1, z nichž nejčastější je varianta, která vzniká fúzí exonu 7 EWSR1 genu a exonu 6 FLI1 genu (v 65 % případů). Vzácně se setkáváme s celou řadou variantních translokací, kde dochází k fúzi genu EWSR1 s geny ETS rodiny jako je například: t(21;22)(q22;q12) a fúze genů EWSR1 a ERG, popřípadě t(7;22)(q22;q12) s fúzí genů EWSR1 a ETV1, a s dalšími, ale podstatně vzácnějšími variantami genových fúzí, jak uvádí Procházka a kol. (12).
Další podskupinou tzv. EWS sarkomů jsou sarkomy, které mají přestavbu v genu FUS, který může být zapojen do translokací se stejnými fúzními partnery ETS rodiny, jak je tomu u genu EWSR1 (13). Popsány jsou v tomto případě translokace t(16;21)(p11;q22) s fúzním genem FUS/ERG a t(2;16)(q36;p11) s fúzí genů FUS a FEV.
Nediferencovaný sarkom
Jde o diagnózu per exclusionem za situace, kdy nelze sarkom zařadit do žádného z výše uvedených typů. Nacházejí se mezi nimi vřetenobuněčné i malobuněčné/kulatobuněčné formy a četnost této popisné diagnózy závisí na specializaci patologa.
V současné době je v rámci této skupiny snaha vyčlenit tzv. „EWS like“ sarkomy, které jsou charakteristické kulatobuněčnou morfologií a přestavbou genu EWSR1 ve fúzi s geny non-ETS rodiny (jako jsou geny PATZ1, POU5F1, SP3, SMARCA5, a další). Další skupinu tvoří „EWS like“ sarkomy s translokací t(4;19)(q35;q13), popř. variantní translokací t(10;19)(q26;q13) s fúzním genem CIC/DUX4 a CIC/DUX10 (14,15). Další podskupinou tzv. „EWS-like“ sarkomů jsou sarkomy, které jsou charakteristické fúzí genů BCOR-CCNB3 na podkladě inverze chromozómu X - inv(X)(p11.2;p11.4). Tato podskupina má odlišný expresní profil oproti EWS a „EWS-like“ sarkomům, což podtrhuje důležitost vyčlenění této skupiny do samostatné jednotky. Tato podskupina však patří spíše do kategorie kostních nádorů.
ZÁVĚR
Optimální léčba pacientů s nádory měkkých tkání úzce souvisí s přesnou diagnostikou. Diferenciální diagnostika, s použitím standardních morfologických vyšetření může být i dnes obtížná, a to jak pro patologa, tak pro klinického lékaře. V průběhu času byly pomocí cytogenetickým analýz nádorů měkkých tkání objeveny nenáhodné a v řadě případů nádorově specifické, a tedy diagnosticky využitelné chromozomální translokace s odpovídající změnou na molekulární úrovni, většinou v podobě fúzního genu. Molekulární techniky PCR umožňují prokázat fúzní gen i v případě nedostupnosti vitální tkáně pro cytogenetické vyšetření, velmi často i z tkáně fixované ve 4% roztoku formaldehydu a zpracované v podobě parafínových bločků (i když tento materiál je pro genetické vyšetření nejméně vhodný z důvodu fragmentace nukleových kyselin v průběhu formaldehydové fixace), proto jsou často využívány pro potvrzení klinické a histopatologické diagnostiky. Je však třeba zdůraznit, že použití samotných molekulárně-genetických metod založených na průkazu specifické genové přestavby není samo o sobě dostatečně spolehlivé k určení nozologické jednotky (16), a to především z důvodu, že některé genetické změny nejsou specifické pouze pro jednu diagnózu. Navíc si musíme být vědomi, že řada molekulárních biologů bez morfologického ověření tkáně patologem nemá potvrzeno, jakou tkáň vlastně vyšetřuje. Z těchto důvodů považujeme korelaci všech dostupných vyšetřovacích metod molekulární patologie s histopatologickým vyšetřením a s klinickým nálezem za nepodkročitelnou podmínku.
Detekce fúzních genů může mít v budoucnu i další význam, a to v terapii fúzně-pozitivních nádorů. Ukazuje se totiž, že nádorově specifické fúzní proteiny mohou být využity v imunoterapii (17). Je prokázáno, že sekvence aminokyselin v podobě fúzních proteinů lze využít jako nádorově specifické antigeny (18).
Klinická diagnóza by se měla opírat o kombinaci histologického nálezu s detekcí genetických změn, které jsou zodpovědné za biologické chování nádorů. Navíc bližší zkoumání nádorově specifických translokací v návaznosti na děje v buňce (vliv na změnu exprese genů, na změny v buněčném cyklu, na schopnosti buněk přežít v prostředí a další) mohou napomoci pochopit patogenezi nádorů měkkých tkání a mohou přispět k identifikaci nových terapeutických cílů v léčbě dosud značně problematicky léčitelných maligních nádorů měkkých tkání.
Poděkování
Podpořeno projektem (Ministerstva zdravotnictví) koncepčního rozvoje výzkumné organizace 00064203 (FN MOTOL) a OPPK CZ.2.16/3.1.00/24022.
Adresa pro korespondenci:
RNDr. Lenka Krsková, Ph.D.
Ústav patologie a molekulární medicíny
2. LF UK a Fakultní nemocnice v Motole
V Úvalu 84, 150 06 Praha 5
tel.: 224435634
email: lenka.krskova@lfmotol.cuni.cz
Zdroje
1. Kodet R, Mrhalová M, Krsková L. Nádory měkkých tkání. Základní orientace v počínání patologa. Referátový výběr Onkologie 2007; 24, speciál 1/07 : 22-30.
2. Veselý K. Histopatologická diagnostika nádorů měkkých tkání. Onkologie 2010; 4 : 293-296.
3. Stejskalová E, Jarošová M, Mališ J, et al. Clinical relevance of chromosomal aberrations in bone and soft tissue tumors in children and young adults. Klinická Onkologie 2009; 22(2): 58-66.
4. Kodet R, Stejskal J, Pilát D, Kocourková M, Šmelhaus V, Eckschlager T. Congenital-infantile fibrosarcoma: A clinicopathological study of five patients entered on the Prague Children´s Tumor Registry. Pathol Res Pract 1996; 192 : 845-853.
5. Malthotra B, Schuetze SM. Dermatofibrosarcoma protuberans treatment with platelet-derived growth factor receptor inhibitor: a review of clinical trial results. Curr Opin Oncol 2012; 24 : 419-424.
6. Zambo I, Veselý K. WHO klasifikace nádorů měkkých tkání a kostí 2013: hlavní změny oproti 3. Vydání. Cesk Patol 2014; 50(2): 64-70.
7. Errani C, Zhang L, Sung YS, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epitheloid hemangioendothelioma of different anatomic sites. Genes, Chromosomes and Cancer 2011; 50 : 644-653.
8. Wang L, Motoi T, Khanin R, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes, Chromosomes and Cancer 2012; 51 : 127-139.
9. Filion C, Labelle Y. Identification of genes regulated by the EWS/NR4A3 fusion protein in extrasceletal myxoid chondrosarcoma. Tumour Biol 2012; 33 : 1599-1605.
10. Lee SB, Kolquist KA, Nichols K, et al. The EWS-WT1 translocation product induces PDGFA in desmoplastic small roundcell tumour. Nature Genetics 1997; 17 : 309–313.
11. Stuart-Buttle CE, Smart CJ, Pritchard S, et al. Desmoplastic small round cell tumour: a review of literature and treatment options. Surgical Oncology 2008; 17(2): 107–112.
12. Procházka P, Vícha A, Kodet R, Kodetová D, Eckschlager T. Nádory ze skupiny Ewingova sarkomu – molekulární biologie a genetika. Klinická onkologie 2007; 20 : 205-208.
13. Shing DC, McMullan DJ, Roberts P, et al. FUS/ERG gene fusion in Ewing´s tumors. Cancer Res 2003; 63 : 4568-4576.
14. Graham C, Chilton-MacNeill S, Zielenska M, Somers GR. The CIC-DUX4 fusion transcript is present in a subgroup of pediatric primitive round cell sarcomas. Hum Pathol 2012; 43 : 180-189.
15. Italioano A, Sung YS, Zhang L, Singer S, Maki RG, Coindre JM, Antonescu CR. High prevalence of CIC fusion with double-homeobox (Dux4) transcription factors in EWSRI – negative undifferentiated small blue round cell sarcomas. Genes, Chromosomes and Cancer 2012; 51 : 207-218.
16. Thorner P, Squire J, Chilton-MacNeil S et al. Is the EWS/FLI-1 fusion transcript specific for Ewing sarcoma and peripheral primitive neuroectodermal tumor? A report of four cases showing this transcript in a wider range of tumor types. Am J Pathol 1996; 148 : 1125–1138.
17. Bennicelli JL, Barr F. Chromosomal translocation and sarcomas. Curr Opin Oncol 2002; 14(4): 412-419.
18. Worley BS, van den Broeke LT, Goletz TJ, et al.: Antigenicity of fusion proteins from sarcoma-associated chromosomal translocations. Cancer Res 2001; 61 : 6868-6875.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2014 Číslo 3
Nejčtenější v tomto čísle
- Intestinální metaplazie žaludku a jícnu: imunohistochemická studie 60 případů včetně porovnání expresí hlenů v normální a zánětlivě změněné sliznici střeva
- Nádory měkkých tkání očima molekulárního patologa
- Komplexní přístup v diagnostice lymfomů v praktických příkladech
- Přehled dosavadních zkušeností s mezinárodní klasifikací tenkojehlové aspirační cytologie štítné žlázy Bethesda 2010
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy