
Molekulárně genetická příčina achromatopsie u dvou pacientů českého původu
Autoři:
L. Hlavatá 1; Ľ. Ďuďáková 1; J. Moravíková 1; A. Zobanová 2; B. Kousal 1,3; P. Lišková 1,3
Působiště autorů:
Klinika dětského a dorostového lékařství, 1. lékařská fakulta, Univerzita Karlova a Všeobecná fakultní nemocnice v Praze, Přednosta: doc. MUDr. Tomáš Honzík, Ph. D.
1; Soukromá oční ordinace MUDr. Anna Zobanová, Poliklinika Barrandov v Praze
2; Oční klinika, 1. lékařská fakulta, Univerzita Karlova a Všeobecná fakultní nemocnice v Praze, Přednostka: prof. MUDr. Jarmila Heissigerová, MBA, Ph. D.
3
Vyšlo v časopise:
Čes. a slov. Oftal., 75, 2019, No. 5, p. 272-276
Kategorie:
Kazuistika
doi:
https://doi.org/10.31348/2019/5/5
Souhrn
Úvod: Achromatopsie je vzácné autozomálně recesivní onemocnění s odhadovanou prevalencí 1 postižený na 30 000 až 50 000 obyvatel. Onemocnění je způsobeno mutacemi v 6 genech. Cílem práce bylo provést molekulárně genetické vyšetření u 11 vzájemně nepříbuzných pacientů českého původu s klinickým nálezem odpovídajícím achromatopsii, u pozitivně testovaných pak detailně popsat jejich oční nález.
Metody: Probandi a jejich asymptomatičtí rodiče podstoupili oftalmologické vyšetření. Po izolaci DNA z venózní krve byl pomocí Sangerova sekvenování proveden cílený screening exonů 6, 7, 9-13 v genu CNGB3, ve kterých se u pacientů evropského původu nejčastěji nacházejí mutace asociované se vznikem achromatopsie.
Výsledky: Analýza sekvenčních dat genu CNGB3 vedla k identifikaci molekulárně genetické příčiny u dvou pacientů. Proband 1 je složeným heterozygotem pro mutace c.819_826del; p.(Arg274Valfs*13) a c.1006G>T; p.(Glu336*) a proband 2 je homozygotem pro mutaci c.1148del; p.(Thr383Ilefs*13). Nejlepší korigovaná zraková ostrost byla u probanda 1 (19 let) oboustranně 0,1 a u probanda 2 (8 let) na pravém oku 0,05 a na levém oku 0,1. Klinicky se achromatopsie projevila u obou postižených jedinců nystagmem, světloplachostí, nízkou zrakovou ostrostí a poruchou barvocitu. Biomikroskopický nález na sítnici byl u obou probandů v normě, nicméně vyšetření pomocí optické koherenční tomografie se spektrální doménou prokázalo mírné bilaterálně symetrické strukturální změny ve fovee.
Závěr: Detekce kauzálních mutací u achromatopsie byla u pacientů českého původu provedena poprvé. Diagnostika na úrovni genu má u tohoto vzácného onemocnění velký přínos, neboť stanovení správné diagnózy není klinicky, zejména v raném dětství, jednoduché. Znalost patogenních mutací dává pacientům naději na zapojení do klinických zkoušek testujících genové terapie a je také základním předpokladem pro prenatální genetickou diagnostiku.
Klíčová slova:
achromatopsie – CNGB3 – česká populace
ÚVOD
Achromatopsie je kongenitální autozomálně recesivní onemocnění sítnice s odhadovanou prevalencí 1 postižený na 30 000 až 50 000 obyvatel. Klinicky se projevuje nystagmem, světloplachostí, poruchou barvocitu, excentrickou fixací a sníženou zrakovou ostrostí, která se ve většině případů pohybuje v pásmu slabozrakosti [23,26].
Biomikroskopický nález na sítnici se jeví u většiny postižených jedinců jako normální, pouze u některých případů lze v makule detekovat jemné přesuny pigmentu nebo atrofické změny [31]. Při elektroretinografickém vyšetření chybí nebo jsou významně redukované odpovědi čípků, zatímco funkce tyčinek je normální nebo pouze mírně snížená [3,25].
Achromatopsie byla do nedávna považována za stacionární onemocnění [26], novější studie se však přiklání k názoru, že se jedná o pomalu progredující chorobu [1,13,30-33]. Analýzou pomocí optické koherenční tomografie se spektrální doménou (SD-OCT) bylo definováno 5 různých stádií [9,27]. Mírné formy jsou charakterizovány porušením kontinuity až absencí vrstvy elipsoidů vnitřních segmentů fotoreceptorů ve fovee, které postupně přecházejí do výraznější atrofie, nejtěžší stadium je doprovázené atrofií pigmentového epitelu [9,27].
Korelace s nálezy na SD-OCT byla demonstrována pomocí další zobrazovací metody autofluorescence fundu [1,9]. V prvních stádiích je signál intenzivnější, pravděpodobně jako známka zvýšeného metabolického obratu provázející přechodové stádium před zánikem buněk. V pokročilejších fázích dochází k poklesu autofluorescence ve fovee v důsledku atrofických změn. Korelace mezi strukturálními a funkčními změnami a věkem nebyla signifikantně prokázána [1,27].
Achromatopsie je způsobena mutacemi celkem v 6 genech: ATF6 [19], CNGA3 [17,34], CNGB3 [14,28], GNAT2 [2,15], PDE6C [29] a PDE6H [16]. Dominuje gen CNGB3 (cyclic nucleotide gated channel beta 3), ve kterém se nacházejí příčinné mutace přibližně u 50 % pacientů evropského původu [18]. CNGB3 kóduje podjednotku modulující funkci CNG (cyclic nukleotid-gated) kanálů v cytoplazmatické membráně čípků, které se podílejí na procesu fototransdukce [18,21].
V české populaci nebyla dosud molekulárně genetická příčina achromatopsie studována. Cílem práce bylo provést u 11 vzájemně nepříbuzných pacientů s klinickým podezřením na achromatopsii screening na přítomnost nejčastějších kauzálních mutací a dále detailně popsat klinické oční nálezy u jedinců s achromatopsií potvrzenou na úrovni DNA.
METODY
Genomová DNA 11 probandů českého původu s klinickým obrazem achromatopsie byla izolována pomocí kitu Gentra Puregene (QIAGEN, Hilden, Německo) ze vzorku venózní krve. Pomocí konvenční metody přímého sekvenování byl proveden screening exonů 6,7,9–13 genu CNGB3 (referenční sekvence NM_019098.4). Tento cílený výběr byl proveden na základě práce Mayera a kol.[22], který otestoval 1074 probandů evropského původu s achromatopsií a zjistil, že u 41,5 % nemocných vzniklo onemocnění na podkladě pouze 12 mutací v genu CNGB3, nacházejících se v námi vybraných oblastech. Použité primery a detailní podmínky reakcí jsou dostupné u autorů na vyžádání. Popis mutací odpovídal doporučením Human Genome Variation Society, kdy první nukleotid kódující sekvence je na pozici 1 [5]. Výzkum byl proveden v souladu s Helsinskou deklarací, probandi nebo jejich zákonní zástupci podepsali písemný informovaný souhlas. Studie byla schválena Etickou komisí Všeobecné fakultní nemocnice v Praze.
U probandů s potvrzenou diagnózou achromatopsie na úrovni DNA jsme provedli podrobné oftalmologické vyšetření. Nejlepší korigovaná zraková ostrost byla zjišťována pomocí optotypů ETDRS (Early Treatment Diabetic Retinopathy Study) nebo Snellenova typu a přepočítaná na decimální hodnoty. Testování barvocitu bylo provedeno pomocí pseudoizochromatických tabulek nebo HRR (Hardy-Rand-Rittler) testu, zorné pole bylo testováno statickým perimetrem (M-700, Medmont International, Nunawading, Austrálie). V rámci biomikroskopického vyšetření sítnice v arteficiální mydriáze bylo fotografováno oční pozadí a autofluorescence fundu u probanda 1 (Visucam 200, Carl Zeiss Meditec AG, Německo), u probanda 2 toto nebylo možné provést pro výrazný nystagmus. Jednotlivé vrstvy makuly byly zobrazeny pomocí SD-OCT (RTVue, Optovue, Inc, Fremont, USA nebo Spectralis, Heidelberg Engineering GmbH, Heidelberg, Německo).
U rodičů bylo provedeno pouze základní oční vyšetření spočívající v měření nejlepší korigované zrakové ostrosti a biomikroskopii fundu. Rodiče probanda 2 podstoupili i vyšetření pomocí SD-OCT.
VÝSLEDKY
Přímým sekvenováním bylo u probanda 1 zjištěno, že je složeným heterozygotem pro mutace v genu CNGB3: v exonu 6 c.819_826del; p.(Arg274Valfs*13) a v exonu 9 c.1006G>T; p.(Glu336*), přičemž c.819_826del byla zděděná od matky, c.1006G>T od otce (Obrázek 1a). U probanda 2 byla detekována v homozygotním stavu mutace c.1148del; p.(Thr383Ilefs*13), každý z rodičů byl tedy heterozygotním nositelem pro tuto variantu (Obrázek 1b).

Proband 1 byl sledován od prvních měsíců života pro nystagmus a výraznou světloplachost. V 19 letech byla nejlepší korigovaná zraková ostrost oboustranně 0,1 s použitím hranových filtrů a korekcí na pravém oku +1,50 D = −1,00 Dcyl ax 170° a na levém oku plan = −0,75 Dcyl ax 180°. Testování barvocitu potvrdilo totální poruchu vnímání barev. Vyšetřením zorného pole byly zjištěny oboustranně centrálně relativní skotomy, ovšem validita provedeného vyšetření byla pro výraznou ztrátu fixace nízká (Obrázek 2c). Biomikroskopicky se zadní pól fundu jevil fyziologický (Obrázek 2a). Intenzita autofluorescence v makule byla také v normě (Obrázek 2b). Kvalita snímků získaných pomocí SD-OCT byla zhoršená pro nystagmus, nicméně jednoznačně byla porušena kontinuita vrstvy fotoreceptorů (Obrázek 2d), tedy nález odpovídající stadiu 1 dle Greenberga a kol. [9]. Vyšetření obou rodičů neprokázalo žádné oční abnormality.
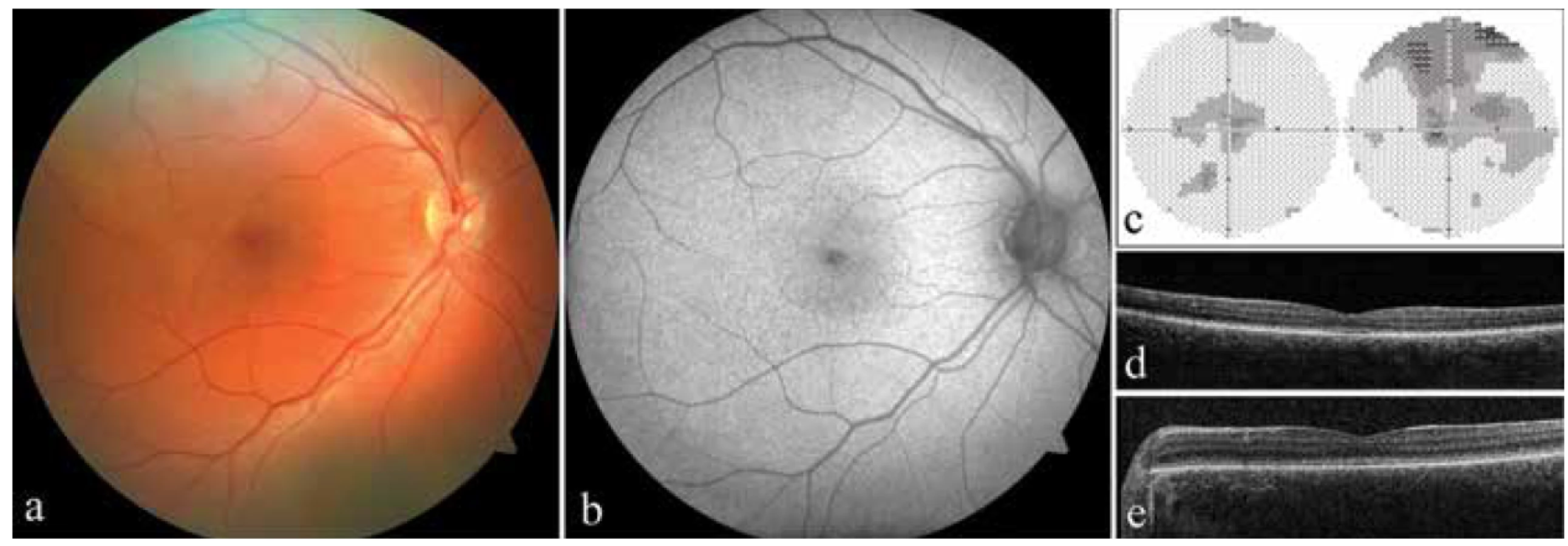
U probanda 2 se také onemocnění projevilo světloplachostí a nystagmem od prvních měsíců života. Diagnóza achromatopsie byla stanovena ve 4 letech. Zraková ostrost činila v té době 0,1 na obou očích bez korekce a cykloplegická refrakce byla +3,25 D = −3,00 Dcyl ax 6° na pravém oku a +3,00 D = −1,50 Dcyl ax 13° na levém oku, ta však zrakovou ostrost nelepšila. Ve věku 8 let byla zraková ostrost bez korekce 0,05 na pravém oku a 0,1 na levém oku. K jejímu zlepšení nedocházelo ani při použití hranových filtrů, nicméně tyto alespoň významně redukovaly světloplachost. Vyšetřením barvocitu byla potvrzena totální barvoslepost oboustranně. Biomikropický nález na sítnici byl bez patrných strukturálních změn, pro nystagmus a světloplachost však nebylo možno pořídit kvalitní fotografii očního pozadí a rovněž nebylo možné zdokumentovat autofluorescenci fundu. Kvalita SD-OCT snímků makuly byla snížená (Obrázek 2e), nicméně vrstva elipsoidů byla přítomna, stádium onemocnění jsme tedy vyhodnotili jako 1 [9]. Oční nález rodičů byl v normě.
DISKUSE
Poprvé jsme u pacientů českého původu s klinickou diagnózou či podezřením na achromatopsii provedli detekci kauzálních mutací. Stanovení diagnózy na úrovni genu má u achromatopsie velký přínos, neboť zejména v raném dětství nelze validně provést řadu nezbytných objektivních a subjektivních vyšetření, jako jsou SD-OCT, autofluorescence fundu, vyšetření barvocitu nebo elektroretinografické vyšetření.
Signifikantní korelace mezi fenotypem a genotypem nebyla u achromatopsie prokázaná [8,31], a proto nelze dle klinického nálezu určit gen, ve kterém by se mohly nacházet patogenní varianty. Podle dostupné literatury se na vzniku onemocnění nejvíce podílejí kauzální mutace v genu CNGB3 [18]. Z 10 nejčastějších patogenních variant v tomto genu vyskytujících se u jedinců s achromatopsií původem převážně ze západní Evropy [22] jsme u 11 pacientů českého původu nalezli pouze tři: c.1148del, c.819_826del, c.1006G>T. Mutace c.1148del, která byla zjištěna u probanda 2, je vůbec nejčastější patogenní alelou, v homozygotním stavu byla přítomna v rozsáhlé studii čítající 1074 probandů u více než pětiny rodin [22].
To, že jsme prokázali obě patogenní varianty pouze u dvou probandů z 11 (18 %), oproti očekávaným 42 % naznačuje, že u českých pacientů je spektrum a frekvence kauzálních mutací jiné než v dosud studovaných populacích z jiných evropských zemí. Toto zjištění však může být zkresleno malým souborem pacientů.
Klinický oční nález byl u našich probandů typický. Achromatopsie se manifestovala již v prvních měsících života nystagmem a světloplachostí, rovněž zraková ostrost byla u obou v pásmu těžké zrakové vady, které je definováno rozmezím zrakové ostrosti od 0,05 do 0,1 [11]. Biomikroskopicky se sice nález na sítnici jevil v normě, nicméně pomocí SD-OCT bylo možno v makule prokázat u obou probandů diskrétní strukturální změny.
Účinná léčba achromatopsie zatím neexistuje, pacienti jsou odkázáni pouze na zrakové korekční pomůcky. Světloplachost lze částečně eliminovat nošením speciálních absorpčních čoček, tzv. hranových filtrů. Pokud jsou známé kauzální mutace, mají rodiče postiženého dítěte v případě další gravidity možnost preimplantační genetické diagnostiky [10,35]. V současnosti také probíhají klinické zkoušky genové terapie u pacientů s mutacemi v genech CNGA3 a CNGB3 [6,7,12]. Jejich možný efekt byl podpořen v preklinických studiích na zvířecích modelech, které prokázaly zlepšení elektroretinografických nálezů i zrakové ostrosti [4,20,24].
ZÁVĚR
Achromatopsie je sice vzácné onemocnění, ale lze se s ním v klinické praxi potkat. Diagnostické rozpaky, které u malých dětí achromatopsii často provázejí, představují pro rodinu často značnou psychickou zátěž. Při podezření na tuto klinickou jednotku doporučujeme genetické testování, které může diagnostický proces výrazně zkrátit.
Děkujeme Ing. Martinu Meliškovi za provedení vyšetření pomocí SD-OCT a zpracování výsledků. Práce byla podpořena granty Univerzity Karlovy PROGRES Q26, UNCE 204064 a SVV 260367/2017.
Autoři práce prohlašují, že vznik i téma odborného sdělení a jeho zveřejnění není ve střetu zájmů a není podpořeno žádnou farmaceutickou firmou.
Do redakce doručeno dne: 28. 8. 2019
Do tisku přijato dne: 21. 10. 2019

MUDr. Lucia Hlavatá
Klinika dětského a dorostového lékařství
1. lékařská fakulta, Univerzita Karlova a Všeobecná fakultní nemocnice v Praze
Ke Karlovu 2
128 00 Praha 2
Korespondující autor:
doc. MUDr. Petra Lišková, M.D., Ph.D.
Zdroje
1. Aboshiha, J., Dubis, AM., Cowing, J., et al.: A prospective longitudinal study of retinal structure and function in achromatopsia. Invest Ophthalmol Vis Sci, 55 (9); 2014 : 5733-5743.
2. Aligianis, IA., Forshew, T., Johnson, S., et al.: Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2). J Med Genet, 39 (9); 2002 : 656-660.
3. Andreasson, S., Tornqvist, K.: Electroretinograms in patients with achromatopsia. Acta Ophthalmol (Copenh), 69 (6); 1991 : 711-716.
4. Carvalho, LS., Xu, J., Pearson, RA., et al.: Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet, 20 (16); 2011 : 3161-3175.
5. Den Dunnen, JT., Dalgleish, R., Maglott, DR., et al.: HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat, 37 (6); 2016 : 564-569.
6. Dudakova L., Kousal B., Kolarova H., et al.: Gene Therapy for inherited retinal and optic nerve disorders: Current Knowledge. Cesk Slov Oftalmol. 72(4); 2016 : 128-136.
7. Fu, X., Huu, VAN., Duan, Y., et al.: Clinical applications of retinal gene therapies. Precis Clin Med, 1 (1); 2018 : 5-20.
8. Genead, MA., Fishman, GA., Rha, J., et al.: Photoreceptor structure and function in patients with congenital achromatopsia. Invest Ophthalmol Vis Sci, 52 (10); 2011 : 7298-7308.
9. Greenberg, JP., Sherman, J., Zweifel, SA., et al.: Spectral-domain optical coherence tomography staging and autofluorescence imaging in achromatopsia. JAMA Ophthalmol, 132 (4); 2014 : 437-445.
10. Hlavata L., Dudakova L., Trkova M., et al.: Preimplantation genetic diagnosis and monogenic inherited eye diseases. Cesk Slov Oftalmol.72(5); 2016 : 167-171.
11. ICD-11 for Mortality and Morbidity Statistics [online]. [cit. 2019-07-04]. Dostupné z: https://icd.who.int/browse11/l-m/en#/http%3a%2f%2fid.who.int%2ficd%2fentity%2f1103667651.
12. Kahle, NA., Peters T., Zobor D., et al.: Development of Methodology and Study Protocol: Safety and Efficacy of a Single Subretinal Injection of rAAV.hCNGA3 in Patients with CNGA3-Linked Achromatopsia Investigated in an Exploratory Dose-Escalation Trial. Hum Gene Ther Clin Dev, 29 (3); 2018 : 121-131.
13. Khan, NW., Wissinger, B., Kohl, S., et al.: CNGB3 achromatopsia with progressive loss of residual cone function and impaired rod-mediated function. Invest Ophthalmol Vis Sci, 48 (8); 2007 : 3864-3871.
14. Kohl, S., Baumann, B., Broghammer, M., et al.: Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet, 9 (14); 2000 : 2107-2116.
15. Kohl, S., Baumann, B., Rosenberg, T., et al.: Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet, 71 (2); 2002 : 422-425.
16. Kohl, S., Coppieters, F., Meire, F., et al.: A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am J Hum Genet, 91 (3); 2012 : 527-532.
17. Kohl, S., Marx, T., Giddings, I., et al.: Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet, 19 (3); 1998 : 257-259.
18. Kohl, S., Varsanyi, B., Antunes, GA., et al.: CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur J Hum Genet, 13 (3); 2005 : 302-308.
19. Kohl, S., D. Zobor, W.C. Chiang, et al.: Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat Genet, 47 (7); 2015 : 757-65.
20. Komaromy, AM., Alexander, JJ., Rowlan, JS., et al.: Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet, 19 (13); 2010 : 2581-2593.
21. Lamb, TD., Pugh, EN. Jr.: Phototransduction, dark adaptation, and rhodopsin regeneration the proctor lecture. Invest Ophthalmol Vis Sci, 47 (12); 2006 : 5137-5152.
22. Mayer, AK., Van Cauwenbergh, C., Rother, C., et al.: CNGB3 mutation spectrum including copy number variations in 552 achromatopsia patients. Hum Mutat, 38 (11); 2017 : 1579-1591.
23. Michaelides, M., Hunt, DM., Moore, AT.: The cone dysfunction syndromes. Br J Ophthalmol, 88 (2); 2004 : 291-297.
24. Michalakis, S., Muhlfriedel, R., Tanimoto, N., et al.: Restoration of cone vision in the CNGA3-/ - mouse model of congenital complete lack of cone photoreceptor function. Mol Ther, 18 (12); 2010 : 2057-2063.
25. Moskowitz, A., Hansen, RM., Akula, JD., et al.: Rod and rod-driven function in achromatopsia and blue cone monochromatism. Invest Ophthalmol Vis Sci, 50 (2); 2009 : 950-958.
26. Simunovic, MP., Moore, AT.: The cone dystrophies. Eye (Lond), 12 ( Pt 3b) 1998 : 553-6.
27. Sundaram, V., Wilde, C., Aboshiha, J., et al.: Retinal structure and function in achromatopsia: implications for gene therapy. Ophthalmology, 121 (1); 2014 : 234-245.
28. Sundin, OH., Yang, JM., Li, Y., et al.: Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet, 25 (3); 2000 : 289-293.
29. Thiadens, AA., den Hollander, AI., Roosing, S., et al.: Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am J Hum Genet, 85 (2); 2009 : 240-247.
30. Thiadens, AA., Slingerland, NW., Roosing, S., et al.: Genetic etiology and clinical consequences of complete and incomplete achromatopsia. Ophthalmology, 116 (10); 2009 : 1984-1989.e1.
31. Thiadens, AA., Somervuo, V., van den Born, LI., et al.: Progressive loss of cones in achromatopsia: an imaging study using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci, 51 (11); 2010 : 5952-5957.
32. Thomas, MG., Kumar, A., Kohl, S., et al.: High-resolution in vivo imaging in achromatopsia. Ophthalmology, 118 (5); 2011 : 882-887.
33. Thomas, MG., McLean, RJ., Kohl, S., et al.: Early signs of longitudinal progressive cone photoreceptor degeneration in achromatopsia. Br J Ophthalmol, 96 (9); 2012 : 1232-1236.
34. Wissinger, B., Jagle, H., Kohl, S., et al.: Human rod monochromacy: linkage analysis and mapping of a cone photoreceptor expressed candidate gene on chromosome 2q11. Genomics, 51 (3); 1998 : 325-331.
35. Yahalom, C., Macarov, M., Lazer-Derbeko, G., et al.: Preimplantation genetic diagnosis as a strategy to prevent having a child born with an heritable eye disease. Ophthalmic Genet, 39 (4); 2018 : 450-456.
Štítky
OftalmologieČlánek vyšel v časopise
Česká a slovenská oftalmologie

2019 Číslo 5
- Selektivní laserová trabekuloplastika nesnižuje nitroční tlak více než argonová laserová trabekuloplastika
- Progresi glaukomu je třeba hodnotit strukturálními i funkčními parametry
- Ztráta centrálního vidění po filtrujících operacích glaukomu
- Od PGF-2 alfa-isopropyl esteru k latanoprostu: přehled vývoje Xalatanu
- Compliance u pacientů s glaukomem
Nejčtenější v tomto čísle
- Molekulárně genetická příčina achromatopsie u dvou pacientů českého původu
- Intravitreálna liečba endogénnej endoftalmitídy pri urosepse – Kazuistika
- Senzitivita a specificita spektrálního OCT u pacientů s počínajícím glaukomovým onemocněním.
- Dlouhodobé sledování pacientů se sympatickou oftalmií z diagnosticko léčebného pohledu. Literární přehled.
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy