
První zkušenosti s biosimilárním infliximabem CT-P13 u nemocných se zánětlivými revmatickými onemocněními v České republice v národním registru ATTRA
First experience with biosimilar infliximab CT-P13 in patients with inflammatory rheumatic diseases in the Czech Republic in the ATTRA national registry
Biosimilar infliximab CT-P13, developed by Korean company Celltrion, is the first EMA approved biosimilar agent. In double-blind studies in rheumatoid arthritis and ankylosing spondylitis biosimilar infliximab was equally effective and had the same safety profile as the original infliximab.
CT-P13 was introduced into clinical practice in the Czech Republic at the end of 2013. All the patients have been treated according to the indications of the Czech Society for Rheumatology at the centers of biological treatment and enrolled in the ATTRA registry. The aim of this study was to evaluate the first year of experience with the CT-P13 agent in the ATTRA national registry.
Results:
A total of 100 patients with rheumatoid arthritis (RA) were enrolled in the registry. Analysis was then performed on 93 adult patients. The mean age of these patients was 54 years and the disease duration was 10 years. Disease activity was high, the average baseline DAS 28 was 5.7 ± 0.8, number of swollen joints was 8.8 ± 4.3 and CRP levels were 28.1 ± 22.5 mg / l. After nine months of treatment there was a significant decrease in all monitored parameters. At the start of treatment 73.3% of patients had high disease activity according to EULAR and 26.7% of patients had moderate activity. After nine months of treatment 87.5% of patients were in remission, 93.7% had low activity, 6.3% had moderate activity, and none of the patients had high disease activity. The treatment survival after nine months was 78.0%.
Seventy-two patients were enrolled in the registry of ankylosing spondylitis (AS) and 68 adult patients thereof were further analyzed. Their average age was 44 years and disease duration of 9 years. Disease activity at the start of treatment was high, and the average BASDAI was 6.2 ± 1.7 and CRP levels were 31.1 ± 26.7 mg / l. After nine months of treatment BASDAI decreased to 0.9 ± 1.4, and quality of life improved (HAQ decreased from 1.1 ± 0.6 to 0.4 ± 0.5). There were improvements in all dimensions of quality of life according to SF-36.
In the group of RA patients 21 adverse events and 7 serious adverse events were reported. In patients with AS there were 7 adverse events and no serious adverse event. No new signals of toxicity were reported.
Conclusion:
First experience with biosimilar CT-P13 in the ATTRA national registry is positive. No new signals of toxicity were reported.
Key words:
Biosimilar infliximab CT-P13, inflammatory rheumatic diseases
Autoři:
K. Pavelka 1; K. Jarošová 1; D. Suchý 2; M. Uher 3; K. Hejduk 3
![]()
Působiště autorů:
Revmatologický ústav Praha
1; Oddělení klinické farmakologie Fakultní nemocnice Plzeň
2; Institut biostatistiky a analýz, Lékařská fakulta, Masarykova univerzita Brno
3
Vyšlo v časopise:
Čes. Revmatol., 24, 2016, No. 1, p. 15-30.
Kategorie:
Originální práce
Souhrn
Prvním biosimilárním lékem schváleným EMA je biosimilární infliximab CT-P13 korejské firmy Celltrion. Ve dvojitě slepých studiích u revmatoidní artritidy a ankylozující spondylitidy byl stejně účinný a měl stejný bezpečnostní profil jako originální infliximab.
Do klinické praxe v České republice byl CT-P13 uveden na konci roku 2013. Všichni pacienti jsou léčeni dle indikací České revmatologické společnosti v centrech biologické léčby a zavedeni do registru ATTRA. Cílem studie bylo vyhodnocení prvního roku zkušeností s preparátem CT-P13 v národním registru ATTRA.
Výsledky.
Do registru bylo zařazeno celkem 100 pacientů s revmatoidní artritidou (RA). Analýza je dále provedena na 93 dospělých pacientech. Průměrný věk těchto pacientů byl 54 let a trvání onemocnění 10 let. Aktivita onemocnění byla vysoká, průměrný vstupní DAS 28 byl 5,7 ± 0,8, počet oteklých kloubů 8,8 ± 4,3 a CRP 28,1 ± 22,5 mg/l. Po devíti měsících léčby došlo k signifikantnímu poklesu všech sledovaných parametrů. Na začátku léčby mělo 73,3 % pacientů vysokou aktivitu onemocnění dle EULAR a 26,7 % pacientů aktivitu střední. Po devíti měsících léčby mělo 87,5 % pacientů remisi, 93,7 % nízkou aktivitu a 6,3 % střední aktivitu, vysokou aktivitu onemocnění neměl žádný pacient. Přežívání na léčbě bylo po devíti měsících 78,0 %.
Do registru ankylozující spondylitidy (AS) bylo zařazeno 72 pacientů, z nichž je dále analyzováno 68 dospělých pacientů. Jejich průměrný věk byl 44 let a trvání nemoci 9 let. Aktivita onemocnění při zahájení léčby byla vysoká, průměrný BASDAI byl 6,2 ± 1,7 a CRP 31,1 ± 26,7 mg/l. Po devíti měsících léčby došlo k poklesu BASDAI na 0,9 ± 1,4 a ke zlepšení kvality života (pokles HAQ z 1,1 ± 0,6 na 0,4 ± 0,5). Došlo ke zlepšení všech dimenzí kvality života dle dotazníku SF-36.
Ve skupině s RA bylo zaznamenáno 21 nežádoucích účinků, z toho 7 závažných, ve skupině s AS 7 nežádoucích účinků, žádný závažný. Nebyly zaznamenány žádné nové signály toxicity.
Závěr.
První zkušenosti s biosimilárním CT-P13 v národním registru ATTRA jsou pozitivní. Nebyly zaznamenány žádné nové signály toxicity.
Klíčová slova:
Biosimilární infliximab CT-P13, zánětlivá revmatická onemocnění
Úvod
Biologické léky přinesly výraznou změnu do léčby zánětlivých revmatických onemocnění, především pak revmatoidní artritidy (RA) a spondyloartritid (SpA). Jsou výrazně účinnější než konvenční syntetické, chorobu modifikující léky (DMARDs), jako je např. methotrexát (MTX), a to jak při potlačení zánětlivé aktivity onemocnění, tak při zpomalení strukturální progrese (1, 2). Potlačení aktivity RA se manifestuje z pohledu pacienta jako snížení bolesti, zkrácení ztuhlosti, zmenšení únavnosti a dalších systémových projevů, zpomalení rentgenové strukturální progrese pak znamená zpomalení destrukcí a deformit kloubů, snížení disability a zachování funkce. Výsledkem je zlepšení kvality života nemocných s těmito závažnými, celoživotními onemocněními (3). Výrazným problémem bránícím širšímu využití biologických léků je jejich cena. Dostupnost biologických léků je výrazně nižší v chudších státech Evropské unie. V Česku je dle odhadu léčeno kolem 5 % pacientů s RA, zatímco např. ve Švédsku, Německu a Rakousku kolem 20 % a v Norsku dokonce 30 % pacientů. Výrazným pokrokem může být snížení ceny těchto léků, čemuž mohou napomoci tzv. léky biosimilární.
Biosimilární léky jsou definovány jako bioterapeutické léky, které jsou prokazatelně podobné biologickému léku co se týče kvality, účinnosti a bezpečnosti a které jsou v dané indikaci a dávce registrovány regulační agenturou (4). Existuje však velký rozdíl mezi klasickým lékem s malou molekulou a mezi biologickým lékem, který se skládá z velké, komplexní proteinové molekuly. Zatímco při registraci generického léku k malé molekule postačuje doložení identické biologické dostupnosti, při registraci biosimilárního léku je situace složitější, protože biosimilární lék nemůže být nikdy zcela identický s originálem biologického léku. Proto regulační agentury, jako jsou např. EMA a FDA, stanovily složitý a rigorózní proces, který se skládá z předklinické a klinické části a který má dokumentovat podobnost s referenčním biologickým lékem (5).
Biosimilární ekvivalent infliximabu CT-P13 byl vyvinut korejskou firmou Celltrion a je prvním EMA schváleným biosimilárním lékem pro léčbu autoimunitních, zánětlivých onemocnění. Referenčním biologickým originálem byl infliximab (INF) Remicade. Biosimilární infliximab CT-P13 je distribuován pod obchodními názvy Remsima a Inflectra. CT-P13 byl schválen pro všechny indikace jako referenční INF, tzn. revmatoidní artritidu, ankylozující spondylitidu, psoriatickou artritidu, Crohnovu nemoc, ulcerózní kolitidu a psoriázu.
CT-P13 je IgG1 chimerická monoklonální protilátka, která obsahuje humánní a myší část. CT-P13 je produkován stejnou buněčnou linií jako INF a má identickou sekvenci aminokyselin. V řadě studií prokázal CT-P13 stejné farmakodynamické (PD) a farmakokinetické (PK) vlastnosti jako INF (6). CT-P13 a INF prokázaly především stejnou účinnost ve vazbě na monomerickou i trimerickou formu lidského TNF alfa na několika experimentálních modelech (6). Srovnatelné byly i komplement dependentní cytotoxické a apoptotické účinky CT-P13 a INF.
Po ukončení preklinické fáze, která kromě řady testů účinnosti a PK obsahovala i celou rozsáhlou baterii testů toxicity a bezpečnosti CT-P13, bylo přistoupeno ke klinickému zkoušení CT-P13. Po konzultaci s regulační agenturou EMA byl navržen program klinického zkoušení, který obsahoval dvě studie. První byla studie fáze 1, jejímž primárním cílem bylo prokázat identické farmakokinetické vlastnosti (PK) a která srovnávala CT-P13 a INF u nemocných s ankylozující spondylitidou (AS) a byla publikována pod názvem PLANETAS (7). Druhou pak byla studie fáze 3, která srovnávala primárně účinnost CT-P13 oproti INF u nemocných s revmatoidní artritidou (RA) a byla publikována pod názvem PLANETRA (8). Do studie PLANETAS byli zařazeni pacienti s ankylozující spondylitidou s vysokým stupněm aktivity (BASDAI skóre > 4). Primárním cílem bylo hodnocení plochy pod křivkou (AUC) plazmatické koncentrace a maximální sérové koncentrace (Cmax) CT-P13 a INF po nejméně 5 dávkách léku. Mezi oběma preparáty nebyl zjištěn žádný signifikantní rozdíl. Žádný rozdíl nebyl zjištěn ani v klinické účinnosti. Randomizovaná, dvojslepá fáze trvala 24 týdnů a po ní následovala otevřená extenze, ve které byli pacienti původně na INF převedeni na CT-P13 a pacienti původně na CT-P13 pokračovali v léčbě. Při hodnocení účinnosti léčby po 48 týdnech nebyly zjištěny žádné rozdíly mezi skupinou léčenou kontinuálně CT-P13 a skupinou převedenou z INF na CT-P13 (9). Ve skupině po převedení léčby byl mírně vyšší počet nežádoucích účinků, což se ale týkalo pouze nezávažných forem a žádný rozdíl nebyl v závažných nežádoucích účincích.
Do studie PLANETRA byli zařazeni pacienti s aktivní RA, kteří měli nedostatečný efekt methotrexátu. Primárním cílem byla odpověď na léčbu (ACR 20) v týdnu 30. V primárním kritériu nebyl žádný rozdíl mezi CT-P13 a INF a stejné platí o řadě sekundárních kritérií. V druhé, otevřené části pacienti na CT-P13 pokračovali v léčbě, zatímco pacienti původně na INF byli switchováni na CTP 13. Při hodnocení účinnosti po 52 týdnech nebyl mezi skupinami zjištěn žádný signifikantní rozdíl. V této studii nebyl zjištěn žádný rozdíl ani ve výskytu nežádoucích účinků mezi skupinou léčenou kontinuálně CT-P13 a skupinou switchující z INF na CT-P13 (10). Žádné rozdíly nebyly zjištěny ani ve výskytu protilátek proti léku, ani v jiném testu na potenciální imunogenitu.
Biosimilární infliximab CT-P13 byl po registraci SÚKL zaveden do běžné klinické praxe v České republice v roce 2014, a to pod názvem Remsima a Inflectra. Cílem naší práce bylo ověřit bezpečnost a účinnost léku v běžné klinické praxi.
Statistické metody
Spojité parametry jsou popsány průměrem a směrodatnou odchylkou (SD) a případně mediánem s 5% a 95% kvantilem, u kategoriálních parametrů jsou použity absolutní a relativní četnosti. Testování významnosti změny v jednotlivých časových bodech bylo provedeno pomocí Wilcoxonova párového testu (u spojitých proměnných), respektive pomocí McNemarova testu (u kategoriálních proměnných). Hodnocení času setrvání pacientů na léčbě bylo provedeno pomocí Kaplan-Meierovy metodiky. Výsledky byly zpracovány programem IBM SPSS Statistics, verze 22.
Studie u revmatoidní artritidy
Všichni pacienti léčení biologickými léky v České republice jsou zařazeni do národního registru ATTRA, který byl založen již v roce 2001 (11). Biologická léčba je prováděna v centrech biologické léčby, které mají zvláštní smlouvu s pojišťovnami. K biologické léčbě je dle kritérií České revmatologické společnosti indikován pacient, který má RA a který:
- a) má aktivitu onemocnění dle DAS28 vyšší než 5,1;
- b) selhává na léčbě methotrexátem (alternativně při nesnášenlivosti MTX jiným sDMARD) v plné dávce po dobu nejméně 3 měsíců (12).
V průběhu studie byly sledovány následující parametry: DAS 28, FW, CRP, HAQ, EuroQol, SF-36, globální hodnocení pacientem, dosažení remise či stavu nízké aktivity, setrvání na léčbě, důvody přerušení léčby, nežádoucí účinky. Pacient byl hodnocen v průběhu prvního roku léčby ve tříměsíčních intervalech a dále v intervalech 6 měsíců.
Jako stav remise byl hodnocen DAS28 nižší než 2,6, stav nízké aktivity jako DAS28 nižší než 3,2, střední aktivita pak jako DAS28 v rozmezí 3,2 a 5,1 a vysoká aktivita jako DAS28 vyšší než 5,1.
Biosimilární infliximab CT-P13 byl podáván v obvyklé dávce 3 mg/kg hmotnosti v intervalech 0, 2, 6 a dále 8 týdnů v intravenózních infuzích identickým způsobem jako referenční originální infliximab. Byl používán biosimilární CT-P13 výrobce Celltrion, který je u nás dostupný pod obchodními názvy Remsima a Inflectra.
Registr je veden jako fáze IV klinického zkoušení v SÚKL. Všichni pacienti podepisují před vstupem do registru informovaný souhlas.
Výsledky
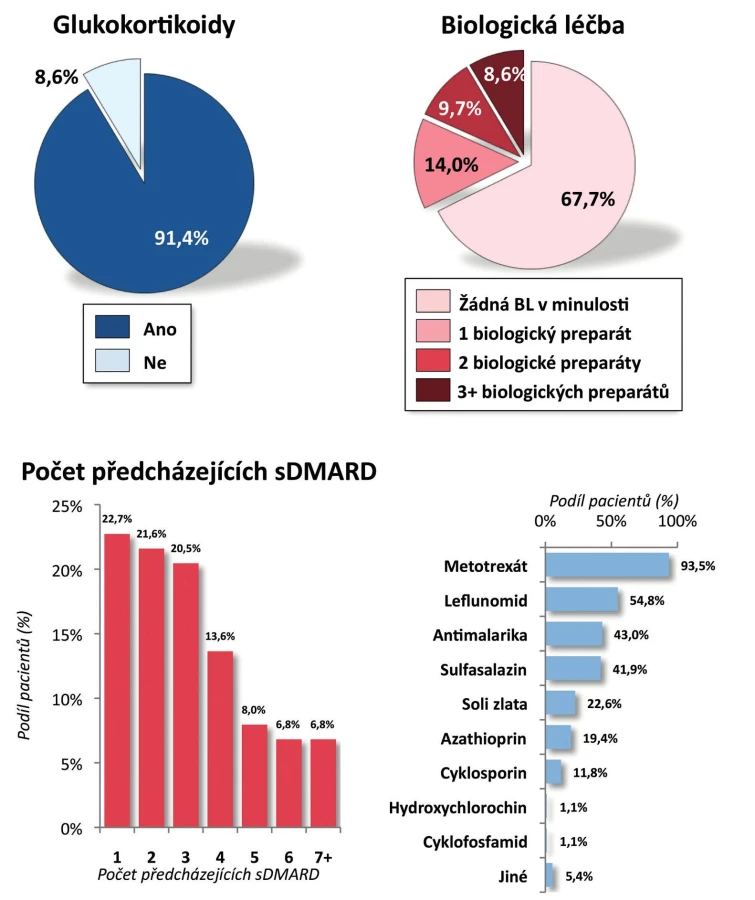
Pacienti. Do registru bylo zařazeno celkem 100 pacientů s RA. Pro další zpracování byli vyřazeni dětští pacienti (s věkem při diagnóze menším než 16 let), analýza tedy byla provedena na 93 dospělých pacientech. Jejich průměrný věk byl 54 ± 13 let, trvání nemoci 10 ± 9 let; 28,0 % pacientů tvořili muži a 72,0 % ženy. Revmatoidní faktor byl pozitivní u 80,6 % pacientů a anti-CCP protilátky u 72,0 % pacientů (tab. 1). Aktivita onemocnění při zahájení léčby byla vysoká, průměrný vstupní DAS28 byl 5,7 ± 0,8, počet oteklých kloubů 8,8 ± 4,3, počet citlivých kloubů 12,6 ± 5,7. Rovněž průměrné reaktanty akutní fáze byly vysoké, sedimentace 41,1 ± 24,0 mm/h a CRP 28,1 ± 22,5 mg/l (tab. 1). Celkem 86,0 % pacientů užívalo v době zahájení léčby glukokortikoidy a 98,9 % chorobu modifikující lék (sDMARD). Nejčastěji užívaným chorobu modifikujícím lékem byl MTX (84,9 %), dále leflunomid (18,3 %), sulfasalazin (8,6 %) a v menší frekvenci další sDMARD. V minulosti užívali pacienti řadu sDMARD, nejčastěji jeden (22,7 %) a dva (21,6 %) a jako určitý unikát lze uvést, že 6,8 % pacientů udávalo v minulosti dokonce léčbu sedmi sDMARD (obr. 1). Celkem 67,7 % pacientů mělo CT-P13 jako první biologický lék, 14,0 % jako druhý, 9,7 % jako třetí a 8,6 % jako čtvrtý nebo další. Pacienti byli před zahájením léčby CT-P13 nejčastěji léčeni certolizumabem (31,3 %), golimumabem (18,8 %) a adalimumabem (15,6 %) (obr. 2). Důvodem přechodu z předcházejícího bDMARD na CT-P13 bylo ve 21,9 % primární selhání a v 43,8 % sekundární selhání.


Všichni pacienti prošli před zahájením léčby screeningem na vyloučení latentní tuberkulózy, dle doporučení ČRS (13).
Výsledky. Po 9 měsících léčby došlo k poklesu průměrného DAS28 z 5,7 ± 0,8 na 2,0 ± 0,7 (p < 0,001, obr. 3). Na začátku léčby mělo 73,3 % pacientů vysokou aktivitu onemocnění dle EULAR a 26,7 % aktivitu střední, po 9 měsících léčby docílilo 87,5 % pacientů remise, 93,7 % nízké aktivity onemocnění, 6,3 % střední aktivity onemocnění a žádný pacient již neměl vysokou aktivitu onemocnění (p < 0,001). Průměrný HAQ na začátku léčby byl 1,5 ± 0,5 a po 9 měsících léčby poklesl na 0,4 ± 0,5 (p = 0,002, obr. 4), rovněž došlo k signifikantnímu zlepšení EuroQolu (p = 0,002). Při hodnocení kvality života došlo po 3 a 6 měsících sledování k signifikantnímu zlepšení všech dimenzí hodnocených v SF-36 (u všech p < 0,001) kromě dimenze SF-36 – Emoční stav (p = 0,316 po 3 měsících, p = 0,750 po 6 měsících). V řadě aspektů se hodnoty kvality života vyrovnaly průměrným hodnotám české populace (obr. 5).



Přežívání na léčbě CT-P13 bylo po uplynutí intervalu 9 měsíců 78,0 % (67,8; 88,1).
Při hodnocení nežádoucích účinků léčby je nutné konstatovat, že expozice pacientů je zatím relativně malá. Bylo hodnoceno 93 pacientů, medián sledování byl 5,7 měsíce, celkem bylo vyhodnoceno 50,9 paciento-roků. Bylo zaznamenáno celkem 21 nežádoucích příhod, z toho 7 bylo hodnoceno jako závažné. Ve dvou případech šlo o alergickou infuzní reakci, po které musela být léčba ukončena, ve dvou případech o závažnou infekci. Dále se vyskytl jeden případ symptomů z oblasti CNS, jeden případ kardiovaskulárních NÚ a jeden případ neurologických příznaků, které však nebyly hodnoceny lékařem jako mající vztah k léčbě anti-TNF. Zatím se dá konstatovat, že nebyly zaznamenány žádné nové signály, které by nebyly známy ze studií s biologickými léky (tab. 2).

Studie u ankylozující spondylitidy
Pacienti jsou v České republice léčeni dle indikačních kritérií České revmatologické společnosti pro ankylozující spondylitidu (AS) (14). Pacienti musí mí vysokou aktivitu onemocnění definovanou jako BASDAI > 4 dvakrát v odstupu 2 měsíců, dále selháváním léčby nejméně u dvou NSA, selháním léčby sulfasalazinem u periferních forem a lokálním opichem u forem s monoartritidou a CRP vyšším než 10 mg/l.
Pacienti. Do registru bylo zařazeno celkem 72 pacientů. Z analýzy bylo vyřazeno 9 dětských pacientů (s věkem při diagnóze menším než 16 let), analyzováno tedy bylo 63 dospělých pacientů. Jejich průměrný věk byl 44 ± 13 let, průměrné trvání nemoci 9 ± 9 let, 67,6 % tvořili muži a 32,4 % ženy (tab. 3). Periferní artritidu mělo 32,8 % pacientů a pozitivitu HLA-B27 83,8 % pacientů. Aktivita nemoci byla vysoká, průměrná sedimentace při zahájení léčby byla 37,8 ± 25,4 mm/h, průměrný CRP 31,1 ± 26,7 mg/l a průměrný BASDAI 6,2 ± 1,7. Glukokortikoidy při zahájení léčby užívalo 17,6 % pacientů, sDMARD 52,9 % pacientů, a to nejčastěji MTX (39,7 %) a sulfasalazin (30,9 %). Celkem 70,6 % pacientů mělo CT-P13 jako první biologický lék, 22,1 % jako druhý a 7,4 % jako třetí. Nejčastěji byl jako předcházející biologický lék před CT-P13 uváděn golimumab (30,4 %), dále etanercept (17,4 %) a infliximab (Remicade, 17,4 %) (obr. 6).


Účinnost CT-P13 byla velmi dobrá. Došlo k poklesu BASDAI z iniciálních 6,2 ± 1,7 na 0,9 ± 1,4 po 9 měsících (p = 0,005, obr. 7). Pokud bychom považovali hodnotu BASDAI = 4 za hranici mezi nízkou a vysokou aktivitou onemocnění, pak na začátku léčby mělo vysokou aktivitu 88,3 % pacientů a po 9 měsících léčby měli všichni pacienti aktivitu nízkou (p = 0,003). Došlo rovněž ke zlepšení kvality života, HAQ skóre se v průběhu 9 měsíců snížilo ze 1,1 ± 0,6 na 0,4 ± 0,5 (p = 0,007, obr. 8). Naopak hodnoty EuroQolu, kde zvýšená hodnota znamená pozitivní efekt, se signifikantně zvýšily (p = 0,004, obr. 8). Rovněž došlo k signifikantnímu zlepšení kvality života dle dotazníku SF-36 (u všech dimenzí po 3 měsících p < 0,001) kromě dimenze SF-36 – Emoční stav (po 3 měsících p = 0,053, obr. 9). Přežívání na léčbě preparátem bylo po 9 měsících 91,0 % (82,3; 99,8).



Bylo zaznamenáno celkem 7 nežádoucích příhod, přičemž žádná nebyla závažná.
Diskuse
Biosimilární léky mohou přinášet podstatné snížení nákladů na biologickou léčbu. Např. Kim demonstroval, že při 30% redukci ceny může být během 5 let ve velkých státech EU (Německo, Francie, Itálie, Británie) ušetřeno až 433 miliónů EUR (14). Snížení nákladů je také hlavním racionálním důvodem zavádění biosimilárních léků.
Samozřejmou obavou při zavádění biosimilárních léků je deklarace o jejich bezpečnosti. V tomto kontextu je také poukazováno na fakt, že v době registrace je počet pacientů v klinickém hodnocení menší než při registraci originálního biologického léku. Je ale nutné zdůraznit, že při testování biosimilárních léků je kladen obrovský důraz na analytickou a předklinickou část hodnocení biosimilárních léků, které musí jednoznačně prokázat, že existující odchylky ve struktuře léku v žádném případě nemění žádný důležitý farmakodynamický či farmakokinetický parametr (5). Za biosimilární lék lze také považovat pouze ten, který byl rigorózně zkoušen dle doporučení EMA a FDA a byl těmito agenturami také schválen. Po ukončení předklinického zkoušení následuje klinická studie, která musí být provedena proti referenčnímu biologickému léku v jeho uznané indikaci, v přesně stejném dávkování a na dostatečném počtu pacientů po dobu nejméně 6 měsíců. Kromě parametrů účinnosti a bezpečnosti je také kladen velký důraz na testování imunogenicity. Infliximab je chimérická monoklonální protilátka, která vyvolává vznik protilátek proti léku (tzv. anti drug antibodies – ADA) až ve 40 % případů. Tyto protilátky mohou být příčinou nižší nebo klesající účinnosti infliximabu. Proto byly ADA protilátky pečlivě sledovány ve studiích PLANETRA a PLANETAS (7, 9), přičemž nebyl mezi oběma preparáty zjištěn žádný signifikantní rozdíl. Otázky imunogenicity zvláště vystupují do popředí, pokud je originální infliximab zaměněn za biosimilární infliximab v procesu, pro který se vžívá anglický výraz switching. Obě studie daly i na tuto otázku částečnou odpověď, když po 24týdenní dvojslepé fázi byli všichni pacienti původně na originálním infliximabu převedeni na CT-P13 a dále sledováni do týdne 52. Při tomto procesu nebyl zjištěn zvýšený výskyt ADA protilátek. Ve studii PLANETRA byl výskyt nežádoucích účinků stejný ve skupině léčené kontinuálně CT-P13 i ve skupině switchované (10). Ve studii PLANETAS byl ve skupině switchované mírně zvýšený výskyt nezávažných nežádoucích účinků (nikoliv závažných), což může být také následkem malého počtu pacientů. Globálně lze ale říci, že otázky bezpečnosti switchingu z referenčního na biosimilární lék nejsou zcela vyřešeny a odpověď mohou dát až analýzy velkého počtu pacientů z registrů pacientů, jako např. z velké studie probíhající v Norsku. EMA ve svém stanovisku také otázku zaměnitelnosti (interchangibility) neřeší a přenechává toto rozhodnutí na národních registračních agenturách, ale v tuto chvíli je nutné zdůraznit, že největší odpovědnost spočívá na ošetřujícím lékaři. Ten jediný může po zvážení všech medicínských aspektů individuálního pacienta rozhodnout. V každém případě je však nutné v tento moment odmítnout tzv. administrativní povinnou interchangibilitu, protože získaná bezpečnostní data nejsou zatím dostatečná. S ještě větším důrazem je nutné odmítnout tzv. substituci, kdy výběr biologického léku provádí lékárník. Bezpečná léčba biologickými léky vyžaduje péči erudovaného specialisty (revmatologa) a substituci v lékárnách je nutné z principiálních důvodů odmítnout i do budoucna především z hlediska bezpečnosti pacientů.
Česká revmatologická společnost zatím doporučuje biosimilární lék především u pacientů, kteří zatím nebyli léčeni biologickými léky. Těchto pacientů bylo ve skupině s RA 67,7 % a ve skupině s AS 70,6 %. Zbývající pacienti již byli v minulosti biologickými léky léčeni.
U těchto pacientů je méně zkušeností s potenciálními nežádoucími účinky, ale zatím nic nesvědčí při aplikaci CT-P13 o zvýšeném riziku. Opatrnost je doporučována při switchi z originálního infliximabu na biosimilar CT-P13. V našem souboru byli tři takoví pacienti s ankylozující spondylitidou. Žádné nežádoucí účinky u nich nebyly zaznamenány.
Závěr
První zkušenosti s biosimilárním infliximabem CT-P13 v národním registru ATTRA jsou pozitivní, ale je potřeba zařadit do sledování větší počet pacientů a mít delší dobu sledování. EMA žádá výrobce o poskytování dat z běžné klinické praxe, tzn. observačních studií a registrů, přičemž výsledky českého registru ATTRA jsou jejich součástí a jsou také jedny z prvních v Evropě. Prozatímní zkušenosti s CT-P13 jsou pozitivní, účinnost je v očekávané míře jako u dalších anti TNF preparátů v registru, přičemž zatím dostupná bezpečnostní data nepřinášejí žádné nové signály.
Podpořeno projektem (Ministerstva zdravotnictví) koncepčního rozvoje výzkumné organizace 023728 (Revmatologický ústav).
Adresa pro korespondenci:
Prof. MUDr. Karel Pavelka, DrSc.
Revmatologický ústav a Revmatologická klinika 1. LF UK
Na Slupi 4
128 50 Praha 2
tel.: 234 075 244
e-mail: pavelka@revma.cz
Zdroje
1. Braun J, Brandt J, Listing J, et al. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet 2002; 359 : 1187–93.
2. Nam JL, Winthrop KL, van Vollenhoven R, et al. Current evidence for management of rheumatoid arthritis with biological disease – modifying antirheumatic drugs: a systematic literature review inform in the EULAR recommendations for the management of RA. Ann Rheum Dis 2010; 69 : 976–986.
3. Závada J, Uher M, Jarkovský J, et al. Hodnocení skóre užitku EW-5D a odhad nákladové užitečnosti prvního roku léčby inhibitory TNF u pacientů s revmatoidní artritidou – výsledky analýzy z národního registru biologické léčby ATTRA. Čes Revmatol 2014; 22 : 10–16.
4. World Health Organization, Guidelines on evaluation of similar biotherapeutic products (SBPs), 2009. http://wwwwho.int/biologicals/areas/biological - therapeutics/BIOTHERAPEUTICS-FOR WEB.22APRIL 2010.pdf, Accessed 19 Dec 2013
5. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on similar biological medicinal products containing biotechnology – derived proteins as active substance: non clinical and clinical issues. Draft: 2013.http//www.ema.europa.eu/docs/en-GB/document-library ESPAR-Public assessment - report/human/ 002778/WC50 0151490.pdf Accessed 17 Febr 2014
6. McKeage K: A review of CT-P13: An infliximab biosimilar. BioDrugs. 2014 DOI 10.1007/s 40259-014-0094-1
7. Park W, Hrycaj P, Jeka BS, et al. A randomised, double blind, multicentre, parallel-group, prospective study comparing the pharmacocinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: The Planetas study. Ann Rheum Dis 2013; 72 : 1605–12.
8. Park W, Jaworski J, Brzeziecki J, et al. A randomised, double blind, paralel – group, phase 1 study comparing the pharmacokinetics safety and efficacy of CT-P13 and infliximab in patients with active ankylosing spondylitis: 54 weeks results from the PLANETAS study. Arthritis Rheum 2013; 72 (Suppl.3): 516.
9. Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double blind, parallel - group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when co administered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis 2013; 72 : 1613–1620.
10. Yoo DH, Racewicz A, Brzeziecki J, et al. A phase 3 randomised controlled trial to compare CT-P13 with infliximab in patients with active rheumatoid arthritis: 54 week results from the PLANETRA study. Ann Rheum Dis 2013; 65(Suppl.3): 73.
11. Pavelka K, Gatterová J, Tegzová D, Jarošová K, et al. Radiographic progression in RA patients treated with infliximab, data from the Czech national registry (ATTRA). Clin Exp Rheum 2007; 25 : 540–545.
12. Pavelka K, Vencovský J. Doporučení České revmatologické společnosti pro léčbu revmatoidní artritídy. Čes Revmatol 2010; 18 : 182–191.
13. Vencovský J., et al. Bezpečnost biologické léčby – doporučení České revmatologické společnosti. Čes Revmatol 2009; 3 : 146–160.
14. Kim JS, Hong JA, Kudrin A. 5 year budget impact analysis of biosimilar infliximab for the treatment of rheumatoid arthritis in UK, Italy, France and Germany. Arthritis Rheum 2014; 66 (Suppl): 512.
Štítky
Dermatologie Dětská revmatologie RevmatologieČlánek vyšel v časopise
Česká revmatologie

2016 Číslo 1
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Ingenol-mebutát rozšiřuje možnosti léčby aktinické keratózy
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
Nejčtenější v tomto čísle
- Hematologické manifestace u pacientů se systémovým lupus erythematodes
- První zkušenosti s biosimilárním infliximabem CT-P13 u nemocných se zánětlivými revmatickými onemocněními v České republice v národním registru ATTRA
- Novinky a poznatky z kongresu ACR 2015
- Zprávy z kongresu EULAR 2015
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy