
Plicní postižení u systémových onemocnění pojiva
Lung involvement in connective tissue diseases
Lung involvement in connective tissue diseases (CTDs) is relatively frequent and may present as diverse clinical-pathological manifestations. Using sensitive detection methods such as HRCT of lungs we may find lung involvement in as many as 70% with systemic sclerosis (SSc). In some CTDs the lung involvement is the leading cause of death and thus it should be actively looked for. Prognosis of different types of lung involvement in CTDs varies and depends on the proportion of inflammatory and fibrotic interstitial changes. Interstitial lung processes with prominent inflammation have usually better prognosis than those with extensive fibrotic changes. Treatment depends on the type of the autoimmune disease and on the phenotype of interstitial lung involvement and is usually based on steroids, immunosuppressive agents and targeted immunological, so-called biological, treatment.
Key words:
connective tissue diseases, lung involvement, prognosis, treatment
Autoři:
M. Vašáková
Působiště autorů:
Pneumologická klinika 1. LF UK, Fakultní Thomayerova nemocnice s poliklinikou
Vyšlo v časopise:
Čes. Revmatol., 18, 2010, No. 4, p. 192-200.
Kategorie:
Přehledné referáty
Souhrn
Plicní postižení se vyskytuje u všech systémových onemocnění pojiva v relativně vysoké prevalenci a může probíhat pod různými klinicko-patologickými obrazy. Pokud použijeme citlivé metody detekce, jako je HRCT hrudníku, tak můžeme najít například u systémové sklerodermie (SSc) plicní manifestaci až u 70 % pacientů. U některých sytémových onemocnění pojiva je plicní postižení nejčastější příčinou smrti, proto je třeba na ně myslet a cíleně je vyhledávat. Prognóza jednotlivých typů plicních projevů se liší v závislosti na zastoupení zánětlivých a fibrotických změn v plicní tkáni. Intersticiální plicní procesy (IPP) s výraznou buněčností mají obvykle lepší prognózu než ty, kde převažují změny fibrotické. Léčba je dána jednak povahou autoimunitního procesu a jednak fenotypem plicní manifestace a obvykle využívá kortikoidy, imunosupresiva nebo imunologickou cílenou, tzv. biologickou, léčbu.
Klíčová slova:
systémová onemocnění pojiva, plicní postižení, prognóza, léčba
Úvod
Systémová onemocnění pojiva (SOP), tzv. kolagenózy, jsou nemoci charakterizované autoimunitním procesem s tvorbou autoprotilátek zaměřených proti řadě buněčných epitopů. Řada z těchto nemocí ve svém průběhu postihuje i plicní tkáň (tab. 1). Fenotypy postižení plicního intersticia jsou nespecifické pro dané onemocnění a obvykle kopírují fenotypy idiopatických intersticiálních pneumonií (IIP) (tab. 2). Léčba a prognóza jednotlivých intersticiálních plicních procesů (IPP) se liší dle toho, do jaké míry je zastoupena reverzibilní složka difuzního procesu, tj. zánět, a do jaké míry je přítomna složka ireverzibilní, tj. fibróza a remodelace plicní tkáně. Procesy charakterizované větší buněčností, tzn. větším podílem zánětlivých změn, lépe reagují na léčbu než procesy s rozsáhlou fibrózou. V rámci jedné SOP se může vyskytnout několik různých fenotypů plicního postižení. Jednoznačné rozklíčování fenotypu je nutné pro určení závažnosti plicního postižení a pro jeho léčbu. Někdy je obtížné diferenciálně diagnosticky odlišit, zda se jedná o primární IPP v rámci systémového onemocnění, nebo zda je obraz intersticiálního postižení dán polékovým postižením při léčbě SOP nebo difuzní pneumonií v imunokompromitovaném terénu u nemocného léčeného imunosupresivy. V některých případech může být postižena buď izolovaně, nebo spolu s IPP i pleura a klouby a svaly hrudního koše (1, 2).


EPIDEMIOLOGIE
Nejvyšší výskyt IPP u SOP (až u 70 % nemocných) je u systémové sklerodermie (SSc) a plicní manifestace je u SSc také nejčastější příčinou smrti. U Sjögrenova syndromu (SS) se plicní postižení vyskytuje u 30 % pacientů z toho u 10 % jde o plicní manifestaci klinicky významnou. U revmatoidní artritidy (RA) se vyskytuje IPP u cca 20 % pacientů a nebývá klinicky závažné. U polymyozitidy/dermatomyozitidy (PM/DM) se vyskytuje plicní manifestace až u 30 % pacientů. U systémového lupusu erythematodu (SLE) je klinicky významné postižení popisováno u 5 % a subklinické formy IPP jsou nalezeny u 30 % pacientů. U ankylozující spondylitidy (AS) bývají intersticiální změny minimální a nespecifické, klinicky významné onemocnění ve formě bulózního emfyzému v horních lalocích je zřídkavé.
KLINICKÝ OBRAZ
Klinicky se většina IPP u SOP projevuje progredující námahovou a posléze klidovou dušností, snadnou unavitelností, kašlem a v pozdějších fázích při nastupující hypoxémii i cyanózou. U některých IPP (fenotyp intersticiální plicní fibrózy obvyklého typu - IPF-UIP) se také vyskytují fenotypové projevy, jako jsou paličkovité prsty s nehty tvaru hodinového sklíčka a poslechový fenomen krepitu slyšitelný nad plicními bazemi. Hemoptýza obvykle nebývá u IPP v rámci SOP dominantní, může se zřídka vyskytovat v rámci kapilaritidy u SLE (1, 2, 3, 4).
DIAGNOSTIKA
Laboratorní nálezy u IPP většinou nejsou patognomické a ve většině případů nenapomohou diagnóze. Výjimkou jsou některé autoprotilátky, které predikují vyšší pravděpodobnost IPP u SOP (protilátky proti topoizomeráze u SSc). V případě IPP spojených s alveolární hemorhagií (kapilaritida u SLE, Behćetův syndrom) bývá přítomna anémie, většinou sideropenická. Naopak v případě pokročilých IPP s hypoxémií je často přítomna nápadná polyglobulie.
Funkční vyšetření plic
IPP u SOP jsou většinou charakterizovány restriktivní ventilační poruchou, snížením plicní difuze (transfer faktoru) a sníženou plicní poddajností.
Mechanismy redukované plicní poddajnosti u IPP mají několik příčin:
- ztráta plicního objemu
- redukovaná distensibilita alveolů
- změny v elastických vlastnostech plíce
- zvýšené povrchové napětí v alveolech
Statické plicní objemy. Statické plicní objemy jsou u IPP v typických případech redukovány, ale v počátečních stadiích nemoci může být hodnota vitální kapacity (VC) v normě. Redukovaná VC je odrazem redukovaného počtu funkčních alveolárních jednotek, daného obliterací nebo alveolární výplní. Totální plicní kapacita (TLC) může být při bodypletysmografickém měření i zcela v normě, zvláště v případě konkomitantního postižení malých dýchacích cest s obrazem air-trapping. Reziduální objem je u IPP obvykle zachován a poměr RV/TLC je často zvýšen. Je to dáno přítomností špatně ventilovaných cystických prostor (honey-combing), které přispívají ke zvětšení plicního objemu měřeného bodypletysmograficky a koexistencí postižení drobných dýchacích cest jako součásti nemoci nebo následkem kouření.
Funkce dýchacích cest. Funkce dýchacích cest je u IPP obvykle dobře zachována. Spirometrické hodnoty včetně usilovné vitální kapacity (FVC), usilovné vitální kapacity za 1 vteřinu (FEV1) a jejich poměr jsou typicky normální, nebo je zachován jejich normální poměr i při absolutně redukovaných hodnotách. V případě některých IPP bývají histologicky patrné změny i v oblasti dýchacích cest, funkčně by se tedy mohlo postižení drobných dýchacích cest projevit ponejprve změnami FEF 25–75.
Porucha výměny plynů. Charakteristickým nálezem u IPP je klidová hypoxémie a zvýšený alveoloarteriální kyslíkový gradient. V počátečních fázích onemocnění ale může být klidový parciální tlak kyslíku v tepenné krvi (paO2) v normě. Parciální tlak oxidu uhličitého (paCO2) v arteriální krvi je typicky normální nebo snížen s výjimkou terminálních fází onemocnění a pH je také normální.
Transfer faktor (TLCO) je významně snížen, a to ve větší míře než by odpovídalo redukci TLC. Porucha difuze spolu s hypoxémií je dána ztrátou kapilárního řečiště, abnormální tloušťkou bazální membrány a nepoměrem ventilace a perfuze, který je dán nepravidelnou distribucí patologických změn v plicním parenchymu. Pravolevý zkrat se na poruše výměny plynů u IPP prakticky nepodílí.
Patofyziologie zátěže. U IPP je typicky výrazné zhoršení dušnosti již při submaximální zátěži a je snížená vrcholová spotřeba kyslíku a ventilační rezerva. Vysoký je fyziologický mrtvý prostor s výraznou arteriální desaturací při zátěži. Pacienti s IPP mají patologický dechový vzorec s vysokou dechovou frekvencí a nízkým dechovým objemem. Navíc mají povětšinou zvýšen již klidový tlak v plicnici, který se při zátěži ještě výrazně zvyšuje.
Zobrazovací metody u IPP
Zadopřední skiagram hrudníku. Vyhodnocení obrazu IPP je závislé na použití vysoce kvalitní radiologické techniky. Základem zobrazení je samozřejmě prostý zadopřední skiagram hrudníku, ale podstatně více informací o postižení intersticia nám podává výpočetní tomografie hrudníku s vysokou rozlišovací schopností (HRCT). V iniciálních fázích IPP dokonce prostý skiagram hrudníku nezachytí patologické změny vůbec a 10% pacientů s histologicky prokázaným IPP má normální skiagram hrudníku. Mezi změny, které můžeme pozorovat u IPP na skiagramu hrudníku patří: změny velikosti plicních polí, obraz nodulace, obraz retikulace a obraz nepravidelných neostře ohraničených stínů. Velmi obtížně detekovatelné jsou takové změny, jako opacity mléčného skla (GGO), které se zvláště u obézních pacientů mohou zaměnit se sumací měkkých tkání (tab. 3).

HRCT hrudníku. Pro dobré zobrazení intersticia je nutné užití techniky tenkých řezů, většinou o tloušťce 1 mm a rekonstrukce obrazu tzv. high-resolution algoritmem CT hrudníku. Scany se většinou provádějí v poloze na zádech, ale při diskrétních změnách v oblasti dorzobazálních partií plic je dobré provést ještě scany na břiše, aby se odlišila zvýšená cévní náplň v poloze vleže. Vyšetření se provádí v inspiriu, ale jsou určité indikace pro vyšetření v exspiriu, a to hlavně při podezření na postižení periferních dýchacích cest, tzv. fenomén air-trapping při chronické obstrukční plicní nemoci nebo obliterující bronchiolitidě. Obraz CT je hodnocen podle rozdílů hustot zobrazovaných tkání v tzv. Hounsfieldových jednotkách, kdy vzduch má denzitu –1 000 HU, voda 0 HU, měkké tkáně okolo 40-80 HU. Střední denzita normální plíce je obvykle v rozsahu -820 až -860 HU, ale značně závisí na množství inspirovaného vduchu, čili na aktuální dechové fázi. Při hodnocení HRCT obrazu je důležité určit i vztah patologických lézí k bronchovaskulárním strukturám, pohrudnici a sekundárnímu plicnímu lobulu. Jednou z nejčastějších intersticiálních patologických struktur je nodulus, při jeho hodnocení si kromě výše uvedeného uložení (náhodné, perilymfatické, centrilobulární, vázané na dýchací cesty) všímáme i velikosti, ohraničení a denzity (GGO, denzita měkkých tkání, denzita kalcifikační). Dále hodnotíme lineární denzity, mezi které patří ztluštělá septa, retikulární linie typické hlavně pro IPF - UIP (obr. 1). Zvláštním případem drobných cyst je obraz voštiny (honey-combing), který je typickým radiologickým a patologickým obrazem konečného stadia některých IPP. V případě rozsáhlých fibrotických změn mohou vznikat sekundárně trakční bronchiektasie. Pleurální změny u IPP většinou přímo souvisí se subpleurálními fibrotickými změnami a přímo na ně navazují. Změny denzity parenchymu se u IPP většinou vyskytují ve smyslu jejího zvýšení. Typickým příkladem zvýšení denzity, které však ještě není schopno překrýt obraz cév, jsou opacity mléčného skla (GGO), pokud změna denzity zakryje i cévní struktury, hovoříme o konzolidaci. (obr. 2) O GGO se dříve soudilo, že reprezentuje vždy tzv. aktivní alveolitidu, zdá se však, že v případě IPF-UIP může být obrazem jemné počínající fibrózy. Sníženou plicní denzitu vídáme nejčastěji v případě emfyzému, u IPP se s ní můžeme setkat většinou v případě hyperinflace při obrazu air-trapping se sekundární oligémií při postižení malých dýchacích cest.


Mediastinální adenopatie se vyskytuje u řady IPP, kromě sarkoidózy (84 %) i u IPF-UIP (67 %), azbestózy, SOP, hypersenzitivní pneumonitidy (53 %), OP (36 %) a je reaktivní (5, 6, 7).
Bronchoalveolární laváž (BAL).
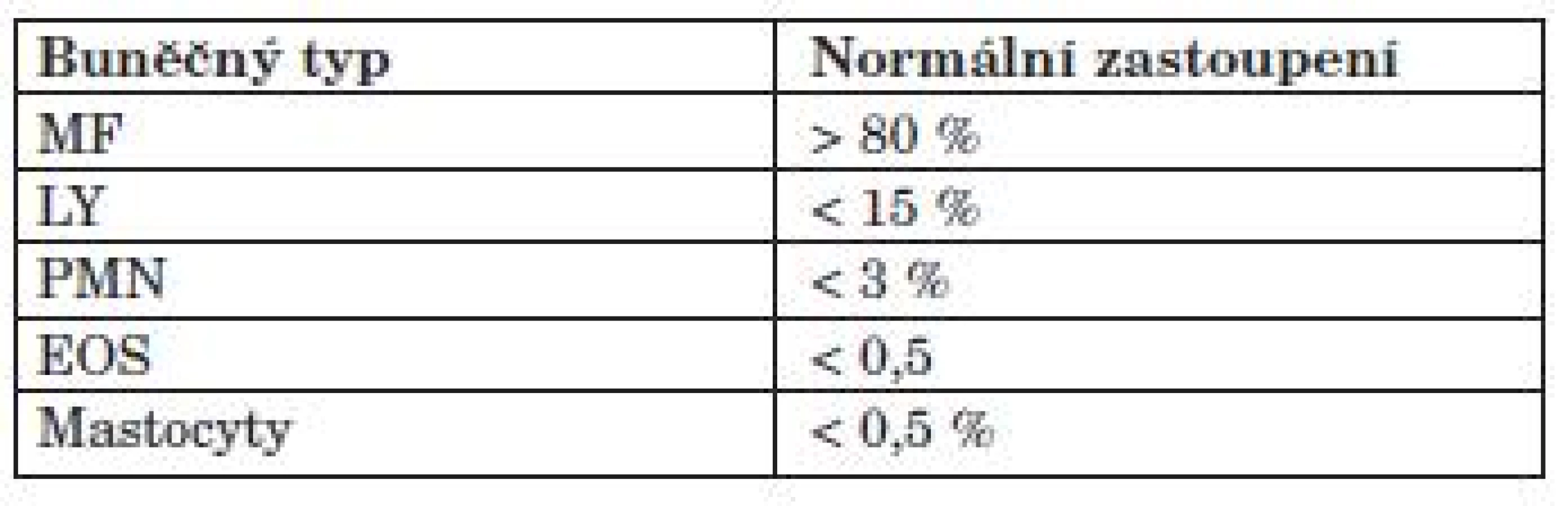
Dle definice se jedná o bronchoskopickou techniku používanou ke sběru buněk, inhalovaných částic, infekčních organismů a roztoků z distálních partií dýchacího traktu. Při její technické realizaci je nutné k získání dostatečného množství materiálu instilovat do příslušného bronchu dostatečné množství tekutiny, u dospělých minimálně 100 ml. Technické provedení je dáno mezinárodními standardy, obvykle nevelké odchylky od standardu jsou dané zvyky na různých pracovištích: množství instilované tekutiny se liší obvykle v rozmezí 100–300 ml a dle toho se liší počet porcí tekutiny (3–5 porcí). Nové práce ukazují, že BAL z různých laloků se může lišit, a proto je výhodné materiál získávat z té oblasti plic, kde jsou změny maximální. Návratnost tekutiny při BAL je obvykle 40–70 % původního objemu a je významně snížena u obstrukčních chorob (např. i IPF s emfyzémem), u kuřáků a starých osob. Lavážní tekutinu (BALT) vyšetřujeme cytologicky buď necentrifugovanou, nebo po centrifugaci a obarvení. Při cytologickém hodnocení BAL zaznamenáváme počet buněk, jejich typ a morfologii (tab. 4). Dle zastoupení jednotlivých populací buněk v BALT lze s jistou pravděpodobností soudit na typ IPP, avšak vždy je nutná korelace s klinickým obrazem, typem kolagenózy, HRCT a eventuálně histologickým obrazem (tab. 5). Zásadní roli má BAL v případě difuzní alveolární hemorhagie (DAH), kdy je lavážní tekutina zbarvena buď čestvou krví nebo hnědavě, v závislosti na časovém odstupu od krvácení a cytologicky jsou alveolární makrofágy naplněné hemosiderinem a někdy v sobě mají čerstvě pohlcené erytrocyty. Důležité je, že se při dalších frakcích výplachů tekutina barví stále více hemorhagicky na rozdíl od krvácení z velkých dýchacích cest, což je jasná známka alveolárního zdroje krvácení.
V rámci vyšetření BALT průtokovou cytometrií můžeme spočítat celkový počet buněk a typizovat jednotlivé populace buněk pomocí velikosti, granularity a pozitivity zvolených imunocytochemických barvení. Obvykle vyšetřujeme základní diferenciační a aktivační povrchové markery lymfocytů – CD45, CD3, CD4, CD8, HLA DR+. CD4+ T lymfocyty bývají zvýšené v BALT u sarkoidózy (CD4+/CD8 + T lymfocyty > 3,5-diagnostické), beryliózy a azbestózy. Naopak CD8+ T lymfocyty jsou zvýšené u exogenní alergické alveolitidy, silikózy, blastomykózy, SOP, při rejekci štěpu, polékové reakci intersticia, při reakci štěpu proti hostiteli a HTLV-1 a HIV infekci.
Přínos BAL v diagnostice a sledování IPP. BAL by měla být provedena u každého podezření na IPP. Může být diagnostická, nebo alespoň zužuje diferenciální diagnostiku IPP. Nicméně její role při určení dynamiky, pravděpodobné léčebné odpovědi a prognózy IPP je prozatím sporná (1, 2, 3, 4).
Histopatologický obraz u IPP u SOP
Ve většině případů IPP u SOP není chirurgická plicní biopsie nutná, pokud je fenotyp postižení dobře definován dle klinického obrazu, funkčního vyšetření, HRCT, BAL a eventuálně transbronchiální biopsie (TBB). Pokud však je typ IPP nejistý a na jeho přesné definici závisí další léčba (kupříkladu IPP v rámci SOP versus polékové poškození či chronická infekce) je chirurgická plicní biopsie vyžadována. Je třeba si ale uvědomit, že plíce má pouze omezený repertoár reakcí na poškození, což znamená, že v případě některých IPP je histopatologický obraz společný pro různé klinické jednotky, a proto je nutné korelovat histopatologický obraz s klinickým obrazem. Navzdory těmto omezením, histologický nález poskytuje informace o etiologii, aktivitě, stáří a reverzibilitě procesu. Například některé histologické obrazy mohou být reverzibilní a svědčí pro předpokládanou dobrou léčebnou odpověď a dobrou prognózu, naopak jiné svědčí pro špatnou prognózu a rezistenci na léčbu.
IPP u SOP je reakcí plicní tkáně na poškození, což po histopatologické stránce znamená obvykle směs konsolidace parenchymu a intersticiálních infiltrátů. V závislosti na stáří reakce na poškození můžeme diferencovat časnou fázi s obrazem akutního difuzního alveolárního poškození (DAD) s edémem a hyalinními blankami, subakutní fázi s obrazem organizující (se) pneumonie s nálezem organizace exsudátu a proliferace pneumocytů II. typu (OP) a chronickou fázi s obrazem voštinovité plíce, intersticiální fibrózy a proliferací vláken hladkého svalu. Granulomy se mohou vyskytovat u všech stadií reakce na poškození (tab. 6).

Fenotypy IPP u systémového onemocnění pojiva
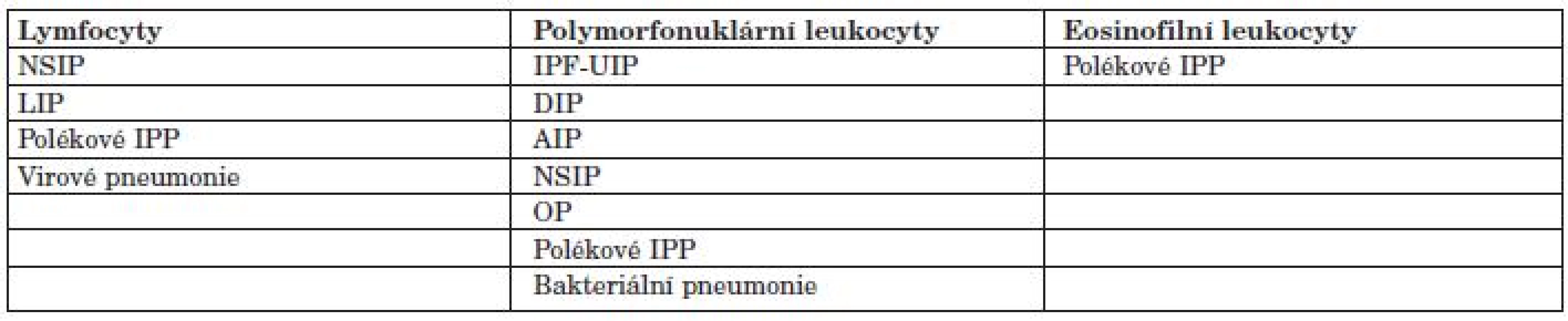
Plicní postižení u SOP může mít různé fenotypy, většinou kopírují jednotlivé podtypy idiopatických intersticiálních pneumonií. Obvykle se u kolagenóz vyskytují fenotypy: intersticiální plicní fibróza histologicky obvyklého typu (IPF-UIP), nespecifická intersticiální pneumonie (NSIP), lymfocytární intersticiální pneumonie (LIP) a a organizující (se) pneumonie (OP). Méně často se vyskytuje obraz DAH nebo granulomatózního plicního postižení (tab. 2) (1, 2, 9).
Intersticiální plicní fibróza typu UIP
IPF je specifickou formu chronického fibrotizujícícho intersticiálního procesu s obrazem obvyklé intersticiální pneumonie (UIP). Je prognosticky nejzávažnějším podtypem IIP. Má typické fenotypové projevy: poslechový nález krepitu nad bazemi plicními a paličkovité prsty s nehty tvaru hodinových sklíček. Typický radiologický nález popisuje zmnoženou plicní kresbu až retikulaci na prostém zadopředním skiagramu hrudníku a obraz plicní fibrózy s obrazem voštinovité plíce v bazích plicních a minimální okrsky tzv. aktivních změn při vyšetření HRCT. V BALT je typické zmnožení granulocytů, obvykle s malou příměsí eozinofilů, lymfocyty bývají zvýšeny minimálně. UIP se vyskytuje jako idiopatická, nebo v rámci SOP (RA, SSc, SS)(10).
Nespecifická intersticiální pneumonie
NSIP je prognosticky méně závažnou formou IPP než IPF-UIP. V rámci NSIP rozeznáváme 3 podtypy, a to podtyp buněčný, fibrotický a smíšený. Podtyp fibrotický má prognózu nejhorší, obvykle se blíží prognóze IPF-UIP, podtyp buněčný a smíšený mají prognózu lepší, což je dáno tím, že určitá porce změn v plicní tkáni odpovídá aktivním zánětlivým změnám, které obvykle lze ovlivnit protizánětlivou, většinou kortikoidní, léčbou. NSIP se může vyskytovat izolovaně jako idiopatická, ale velmi často ji vídáme jako plicní postižení u SOP (RA, SSc, SS), polékového postižení, imunopatologických stavů, ale i u exogenní alergické alveolitidy.
Deskvamativní intersticiální pneumonie
Deskvamativní intersticiální pneumonie (DIP) byla poprvé popsána v roce 1965. Onemocnění je histopatologicky charakterizováno masivní proliferací alveolárních makrofágů. Kromě kouření se DIP může vyskytovat i jako IPP dětského typu a v souvislosti s expozicí jiným škodlivinám a s jinými patologickými stavy ( SOP, leukémie, virové infekce).
ORGANIZUJÍCÍ (SE) PNEUMONIE (OP)
představuje klinicko - patologický syndrom, který se může vyskytovat v důsledku různých nemocí nebo expozic škodlivinám (OP sekundární), pokud nenajdeme příčinu, pak stav označíme jako kryptogenní organizující se pneumonie (COP). Histopatologicky je OP charakterizována obliterací periferních dýchacích cest granulační tkání. OP se může manifestovat po plicních zánětech, může být sekundární při reakci štěpu proti hostiteli (graft-versus-host-disease) nejčastěji u transplantací plic a kostní dřeně, po radioterapii hrudníku, při SOP (RA) a při maligních onemocněních. Diagnóza se opírá o klinický obraz, HRCT nález popisující peribronchiálně a periferně vázané okrsky konzolidace a obraz mléčného skla, převahu CD8 pozitivních lymfocytů v BALT a chirurgickou plicní biopsii s obrazem granulační tkáně v alveolech a terminálních bronchiolech (11).
AKUTNÍ INTERSTICIÁLNÍ PNEUMONIE
Akutní intersticiální pneumonie (AIP) byla první popsanou IIP, a to v roce 1944 Hammanem a Richem. Její průběh je na rozdíl od ostatních IPP akutní. Histologický obraz difuzního alveolárního poškození (DAD) je neodlišitelný od postižení při ARDS (adult respiratory distress syndrome) a je někdy proto označován jako idiopatický ARDS. Pod obrazem AIP také probíhá akutní exacerbace IPF. Někdy AIP doprovází SOP, může být viděna i jako následek infekcí, aspirace, drogového abusu. Diagnóza se opírá o klinický průběh a HRCT nález mlhovitých opacit, v BALT je převaha neutrofilních granulocytů a můžeme zachytit amorfní materiál zastupující zbytky hyalinních blanek, chirurgická biopsie s obrazem DAD pak diagnózu AIP podpoří (9,12).
LYMFOCYTÁRNÍ INTERSTICIÁLNÍ PNEUMONIE
Lymfocytární intersticiální pneumonie (LIP) je charakterizována infiltrací plicní tkáně lymfocyty a stále se spekuluje o jejím vztahu k plicnímu lymfomu. Často se vyskytuje jako součást SOP, typicky Sjögrenova syndromu. Diagnóza se opírá o klinický obraz, HRCT obraz mlhovitých opacit a centrilobulárních nodulů se ztluštěním bronchovaskulárních svazků a interlobulárních sept a s obrazem drobných cyst a o chirurgickou plicní biopsii s nálezem infiltrace plicní tkáně lymfocyty, plasmatickými buňkami a histiocyty se zárodečnými centry. LIP většinou uspokojivě regreduje po léčbě systémovými kortikoidy.
SYNDROM KOMBINOVANÉ FIBRÓZY A EMFYZÉMU (CPFE)
CPFE je kombinací emfyzému a fibrotizujícího plicního procesu. Fibrotizující složka je obvykle tvořena některým z obrazů idiopatických intersticiálních pneumonií (IIP), nejčastěji IPF-UIP nebo NSIP. Prognóza závisí na typu fibrotizujícího procesu, rozsahu změn a přítomnosti plicní hypertenze. V případě SOP CPFE bývá nejčastěji u RA, SSs a smíšeném onemocnění pojiva (MCTD). Klinicky se projevuje CPFE většinou progredující námahovou dušností, někdy bývají přítomny známky velmi podobné klinickému obrazu chronické obstrukční plicní nemoci. Na HRCT jsou známky emfyzému i fibrózy (13).
DIFUZNÍ ALVEOLÁRNÍ HEMORHAGIE (DAH)
Difuzní alveolární hemorhagie (DAH) je obecným označením pro klinický syndrom charakterizovaný difuzním poškozením plicních kapilár, arteriol a venul, vedoucí k výronu krve do oblasti alveolů a periferních dýchacích cest. Je definován klinickou triádou hemoptýzy, anémie a progresivní hypoxémie. V radiologickém obraze dominují difuzní nebo skvrnité oboustranné infiltráty. V bronchoskopickém obraze nemusí být patrny stopy hemoptýzy, ale důležité je vyšetření BAL, neboť pro DAH je charakteristické progresivní hemorhagické zbarvení v následujících porcích BALT. DAH je vzácný syndrom, jehož nejčastější příčinou je Wegenerova granulomatóza a za ním dle četnosti následují Goodpastureův syndrom, idiopatická plicní hemosideróza a plicní postižení u SOP (14).
AKUTNÍ EXACERBACE INTERSTICIÁLNÍCH PLICNÍCH PROCESŮ
Akutní exacerbace (AE) IPP je charakterizována klinicky souborem kritérií: zhoršením dušnosti a kašle v posledním měsíci, novými opacitami mléčného skla nebo konsolidacemi v radiologickém obraze, zhoršením stávající hypoxémie a rychlým rozvojem respiračního selhání v nepřítomnosti infekce, plicní embolie, městnavého srdečního selhání, pneumothoraxu a jiných chorobných stavů, které by mohly být příčinou zhoršení. Plicní biopsie, pokud jsou provedeny v tomto stadiu nemoci, prokáží většinou obraz difuzního alveolárního poškození (DAD). V některých případech AE IPP byl popsán histopatologický vzorec organizující se pneumonie (OP), či progrese fibroblastických fokusů, tito pacienti pak mají podstatně lepší prognózu, než ti s obrazem DAD. I když nejčastěji se AE IPP objevuje v průběhu IPF, byly popsány AE i u pacientů s IPP jinými než IPF, a to u idiopatické nonspecifické pneumonie (NSIP), u NSIP při SOP a u exogenní alergické alveolitidy.
Stran léčebných postupů jsou ve většině případů aplikovány parenterálně kortikosteroidy v pulzním režimu, někdy spolu s dalšími imunosupresivy a antibiotiky. Kromě těchto léčebných postupů byly zkoušeny také metody méně obvyklé, kupříkladu hemoperfuze polymyxinovou kolonou, cyklosporin A v kombinaci s kortikoidy, inhibitory neutrofilní elastázy (sivelestat) a lecitinizovná superoxid dismutáza.
AE představuje velmi závažnou příhodu v průběhu nemoci u pacientů s IPP, která ve většině případů končí fatálně navzdory jakékoli léčbě. Pacienti, kteří mají v rámci AE IPP klinický obraz AIP a tomu odpovídající histopatologický obraz DAD, umírají prakticky všichni. Jistou šanci na přežití mají ti, kteří mají jiný vzorec postižení (OP) a kteří nepotřebují neinvazivní či invazivní plicní ventilaci.(8)
SPECIFIKA IPP U JEDNOTLIVÝCH SOP
Systémová sklerodermie (SSc). Plicní postižení u SSc se vyskytuje relativně často, a to hlavně u pacientů s pozitivními autoprotilátkami proti topoizomeráze (ATA), anticentromerové autoprotilátky (ACA) jsou spojené hlavně s plicním cévním postižením. Nejčastějším histopatologickým typem plicního postižení u SSc je NSIP, pouze minimum pacientů má obraz UIP, někdy se objevuje obraz tzv. centrilobulární fibrózy, která může mít souvislost s opakovanými aspiracemi. Vzácně se může objevit v rámci SSc OP nebo DAH. Nutné je myslet i na případnou plicní arteriálni hypertenzi (PAH) u SSc, která modifikuje obraz plicního postižení, nebo se vyskytuje izolovaně a významně zhoršuje prognózu pacientů (9, 15, 16).
Revmatoidní artritida (RA). U RA je plicní postižení geneticky svázáno s HLA genotypem HLA-B8 a HLA-Dw3. Plicní fibróza u RA je také častější u pacientů s vysokým titrem revmatoidního faktoru (RF) a s revmatickými uzly v plicích. V rámci RA můžeme zachytit postižení plic typu UIP, NSIP, OP nebo LIP a také nekavitující či kavitující revmatické uzly, obliterující bronchiolitidu (OB) a folikulární bronchitidu (FB). Někdy může onemocnění akutně exacerbovat pod obrazem DAD (17, 18, 19).
Polymyositis/Dermatomyositis (PM/DM). Kombinace plicního postižení a PM/DM bývá u antisyntetázového syndromu, který je spojen s protilátkami proti aminoacyl tRNA syntetázám, nejčastěji anti-histidyl (Jo-1). Nejčastějším typem postižení plic je NSIP, někdy OP. Obraz může být modifikován svalovým postižením dechových svalů a svalů hrtanu (opakované aspirace) (20, 21).
Systémový lupus erythematodes (SLE). U SLE je plicní postižení málo časté. Je popsána vazba plicního postižení s pozitivitou anti Ro/SSa. Nejčastějším typem postižení je akutní lupoidní pneumonitida a alveolární hemorhagie, může se vyskytnout vzácně i fibróza. U SLE je časté postižení pleury a může se objevit plicní vaskulitida, plicní hypertenze a tromboembolické komplikace (14, 22).
Sjögrenův syndrom (SS). SS může být buď primární, nebo sekundární v rámci jiných kolagenóz. V případě IPP při SS se setkáme s obdobnými podtypy IPP jako u výše popsaných kolagenóz, navíc u SS může být vyjádřena lymfoproliferace, a to od benigní až po maligní, od LIP až po pseudolymfom a lymfom (23, 24).
Ankylozující spondylitida (AS). Plicní postižení u AS je většinou sekundární, dané limitací ventilace při postižení skeletu hrudníku, občas se setkáme s fibrobulózním postižením horních laloků. Při tomto postižení dochází k plicní distorzi a retrakci plicních hilů s tvorbou kavit, které jsou nezřídka sekundárně kolonizované či infikované patogeny (mykobakteria, houby) (25).
LÉČBA
Rozhodnutí o léčbě IPP při SOP záleží na řadě faktorů. Obecně léčbu indikujeme, pokud je doba trvání SOP do manifestace IPP méně než 5 let, dále pak při rozsáhlém (HRCT obraz) a funkčně závažném (transfer faktor) postižení a při progresivním zhoršování plicního nálezu. V případě plicního postižení u SSc, neexistuje prokazatelně účinná léčba u pacientů se SSc s plicní fibrózou. Zánětlivý fenotyp IPP s obrazem GGO na HRCT odpovídá na protizánětlivou léčbu výrazně lépe. Základem léčby je cyklofosfamid, dle posledních doporučení podávaný pulzním režimu 500–1000 mg i.v./m2/měsíc, s kortikosteroidy v dávce do 15 mg denně p.o. Tato léčba dokáže ve většině případů zmírnit kožní ztluštění a stabilizovat plicní funkce. Alternativou léčby cyklofosfamidem může být mykofenolát mofetil, eventuálně azathioprin. U agresivních forem SSc jsou popisovány remise po vysokých dávkách CPH s lymfoablací a autologní transplantací kostní dřeně. V případě léčby plicního postižení u RA jsou základem kortikoidy (prednison 0,5 mg/kg/den), někdy v kombinaci s imunosupresivy (azathioprin 1–2 mg/kg/den). U některých pacientů je indikována léčba metotrexátem (MTX) nebo tzv. biologická ( anti-TNF) léčba (etanercept). Plicní postižení u PM/DM léčíme kortikoidy v dávce 0,75–1 mg/kg, u akutních forem volíme pulzní podání v dávce 1000 mg Solu Medrolu denně v úvodu léčby, většinou přidáváme i imunosupresiva: cyklofosfamid v pulzním režimu 500–700 mg měsíčně, alternativou může být azathioprin 1–2 mg/kg/den nebo cyklosporin 2–3 mg/kg/den. U akutních fulminantních průběhů můžeme zkusit intravenózní podání gamaglobulinů. Základem léčby IPP při SLE jsou kortikoidy v dávce 1 mg/kg, někdy v kombinaci s imunosupresivy (cyklofosfamid v úvodu, s přechodem na mykofenolát mofetil nebo azathioprin v udržovací fázi). V případě refrakterní nemoci nebo LIP je užitečný rituximab. V případě DAH je nutné zahájit léčbu vysokými dávkami i.v. kortikoidů (1 g methylprednisolonu 3 dny po sobě) s následným podáváním 60 mg prednisonu denně. Současně je podáván cyklofosfamid 500–1000 mg/m2 i.v každé 4 týdny. Někdy v případě refrakterní DAH indikujeme plasmaferézu. Základem léčby IPP u SS jsou orálně podávané kortikoidy, a to 1 mg/kg Prednisonu úvodem. Pokud se stav pacienta nelepší, přidáváme hydroxychlorochin, azathioprin, nebo cyklofosfamid.
Plicní hypertenze u SOP (SSc, SLE) v řadě případů odpovídá na imunosupresivní léčbu, pokud nikoli, je na místě specifická léčba (epoprostenol, bosentan, sitaxsentan, sildenafil).
V případě pokročilého IPP u SOP s hypoxemií je indikována dlouhodobá domácí oxygenoterapie (DDOT), případně transplantace plic, pokud není prokazatelné jiné orgánové postižení.
PROGNÓZA
Prognóza jednotlivých podtypů plicních postižení u SOP se značně liší a je závislá na histologickém podtypu, funkčním postižení v době diagnózy (transfer faktor) a také na komplikujících faktorech (postižení cév, plicní infekce). Fenotyp IPF-UIP má u systémového onemocnění obvykle lepší prognózu než v případě izolovaného výskytu (4).
Prognóza IPP u SSc je významně lepší než u idiopatických intersticiálních pneumonií, to platí pro oba základní fenotypy UIP a NSIP. Deset let přežívá 70 % pacientů, pokud je však transfer faktor snížen pod 40 %, pak pětileté přežití klesá pod 10 %. Při plicním postižení typu UIP 5 let přežívá 82 % a při NSIP 5 let přežívá 91 % pacientů. Špatným prognostickým faktorem je pokles transfer faktoru v posledních 3 letech a hypereozinofilie v BALT. Dalším významným faktorem zhoršujícím přežití je plicní hypertenze, zvláště je-li nepoznaná a tudíž neléčená.
5leté přežití pacientů se SS s plicním postižením je 85 %, špatnou prognózu mají pacienti s maligním lymfomem nebo s rozsáhlým fibrotickým postižením plic.
10leté přežití u SLE je udáváno přibližně 90 %. Příčinou úmrtí je nejčastěji aktivita SLE, trombotické komplikace a infekce. Velmi špatná je prognóza DAH, 50 % umírá již za hospitalizace pro tuto příhodu.
Akutní IPP u PM/DM může mít i fulminantní průběh navzdory léčbě, až polovina těchto pacientů zemře na respirační selhání do 1–2 měsíců (5leté přežití 35 %). Chronické IPP mají prognózu jednoznačně lepší, 5 let přežívá 100 % pacientů.Vzhledem k závažnosti akutních a subakutních forem plicního postižení při PM/DM musíme po IPP aktivním screeningem pátrat.
Prognóza IPP u RA závisí na fenotypu plicního postižení, na rozdíl od SSc je významně horší u těch, kteří mají fenotyp UIP oproti pacientům s NSIP. Při porovnání prognózy pacientů s IPF a s plicním postižením při RA typu UIP, nebyl na rozdíl od SSc pozorován významný rozdíl v délce přežívání.
Závěr
Plicní postižení u SOP je velmi častou manifestací, nejčastěji se s ním potkáme v případě SSc. Závažnost IPP a léčba je dána klinicko-patologickým fenotypem postižení. IPP u SOP je třeba aktivně vyhledávat, aby pacienti dostali včas adekvátní léčbu.
Doc. MUDr. Martina Vašáková, Ph.D
Pneumologická klinika 1. LF UK
Fakultní Thomayerova nemocnice s poliklinikou
Vídeňská 800
140 59 Praha 4
Zdroje
1. Costabel U, duBois RM, Egan JJ. Diffuse parenchymal lung disease. Progress in Respiratory Research, editor Bolliger CT, vol. 36. Basel Switzerland, Karger, 2007.
2. Vašáková M. Intersticiální plicní onemocnění. In Zatloukal P., Fiala P., Votruba J. et al. Pneumologie, Vnitřní lékařství, díl IIIa. Praha 2001. Galén, s.145-169.
3. Vašáková M, Šterclová M, Anton J. Intersticiální plicní procesy – přehled, diferenciální diagnostika, vyšetřovací metody. Praktický lékař 2007; (87) 8 : 461-468
4. Antoniou KM, Margaritopoulos G, Economidou F, et al. Pivotal clinical dilemmas in collagen vascular diseases associated with interstitial lung involvement. Eur Respir J 2009; 33(4): 882-96.
5. Bellia M, Cannizzaro F, Scichilone N, et al. HRCT and scleroderma: semiquantitative evaluation of lung damage and functional abnormalities. Radiol Med; 114(2): 190-203.
6. Kartikheyan D. High-resolution computed tomography of the lungs. A pattern approach. London, Hodder Arnold, 2005.
7. Afeltra A, Zennaro D, Garzia P, et al. Prevalence of interstitial lung involvement in patients with connective tissue diseases assessed with high-resolution computed tomography. Scand J Rheumatol 2006; 35(5): 388-94.
8. Ambrosini V, Cancellieri A, Chilosi M, et al. Acute exacerbation of idiopathic pulmonary fibrosis: report of series. Eur Respir J 2003; 22 : 821-826.
9. Boin F, De Fanis U, Bartlett SJ, et al. T cell polarization identifies distinct clinical phenotypes in scleroderma lung disease. Arthritis Rheum 2008; 58(4): 1165-74.
10. Vašáková M. Idiopatická plicní fibróza – Novinky v diagnostice a léčbě. Interní Med 2007; 5; 233-236.
11. Cavallasca JA, Caubet M, Helling CA, et al. Cryptogenic organizing pneumonia (COP), as presentation of rheumatoid arthritis. Rheumatol Int 2008; 29(1): 99-101.
12. Parambil JG, Myers JL, Ryu JH. Diffuse alveolar damage: uncommon manifestation of pulmonary involvement in patients with connective tissue diseases. Chest 2006; 130(2): 553-8.
13. Vašáková M, Žáčková P, Matěj R. Syndrom kombinované fibrózy a emfyzému - CPFE syndrom. Praktický lékař 2009; 89(6): 287-289.
14. Swigris JJ, Fischer A, Gillis J, et al. Pulmonary and thrombotic manifestations of systemic lupus erythematosus. Chest 2008;133(1):271-80.
15. Gilson M, Zerkak D, Wipff J, et al. Prognostic factors for lung function in systemic sclerosis: prospective study of 105 cases. Eur Respir J. 2010;35(1):112-7.
16. Renzoni EA. Interstitial lung disease in systemic sclerosis. Monaldi Arch Chest Dis 2007;67(4):217-28.
17. Metafratzi ZM, Georgiadis AN, Ioannidou CV, et al. Pulmonary involvement in patients with early rheumatoid arthritis. Scand J Rheumatol. 2007;36(5):338-44.
18. Gauhar UA, Gaffo AL, Alarcón GS. Pulmonary manifestations of rheumatoid arthritis. Semin Respir Crit Care Med. 2007;28(4):430-40.
19. Brown KK. Rheumatoid lung disease. Proc Am Thorac Soc. 2007;15;4(5):443-8.
20. Fathi M, Lundberg IE, Tornling G. Pulmonary complications of polymyositis and dermatomyositis. Semin Respir Crit Care Med 2007;28(4):451-8.
21. Ideura G, Hanaoka M, Koizumi T, et al. Interstitial lung disease associated with amyopathic dermatomyositis: review of 18 cases. Respir Med. 2007;101(7):1406-11.
22. Al-Mutairi S, Al-Awadhi A, Raghupathy R, et al. Lupus patients with pulmonary involvement have a pro-inflammatory cytokines profile. Rheumatol Int. 2007;27(7):621-30.
23. Lazor R. Lung Involvement in Sjogren’s Syndrome: Interstitium, Airways, or Both? Respiration. 2009;78(4):375-376.
24. Fagundes MN, Caleiro MT, Navarro-Rodriguez T, et al. Esophageal involvement and interstitial lung disease in mixed connective tissue disease. Respir Med. 2009;103 (6):854-60.
25. Quismorio FP Jr. Pulmonary involvement in ankylosing spondylitis. Curr Opin Pulm Med 2006; 12(5): 342-5.
Štítky
Dermatologie Dětská revmatologie RevmatologieČlánek vyšel v časopise
Česká revmatologie

2010 Číslo 4
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Riziko roztroušené sklerózy u pacientů s psoriázou
- Diagnostika osteoporózy v kontextu současných doporučení
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
Nejčtenější v tomto čísle
- Plicní postižení u systémových onemocnění pojiva
- Ruptura šlachy m. tibialis posterior u pacientů s revmatoidní artritidou
- Doporučení České revmatologické společnosti pro léčbu revmatoidní artritidy
- OBSAH ROČNÍKU ČASOPISU ČESKÁ REVMATOLOGIE 18/2010
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy