Buněčné imunodeficience patří k těm vzácnějším, ale bohužel též závažnějším poruchám imunity, při nichž zejména imunitní buňky, ale sekundárně též i tvorba protilátek selhávají, a pro něž mezinárodní klasifikace vyčleňuje samostatnou skupinu „imunodeficiencí postihujících buněčnou a protilátkovou imunitu“ (1). Ve většině případů se jedná zejména o postižení T či B lymfocytů, ač v některých případech mohou být postiženy i natural killer (NK) buňky či být snížen počet granulocytů.
Nejzávažnější skupinu tvoří takzvané těžké kombinované imunodeficience (severe combined immunodeficiency, SCID). Jedná se o vzácná onemocnění s incidencí cca 1–2 : 100 000 nově narozených dětí (2), která – nejsou-li včas odhalena – mohou mít extrémně špatnou prognózu.
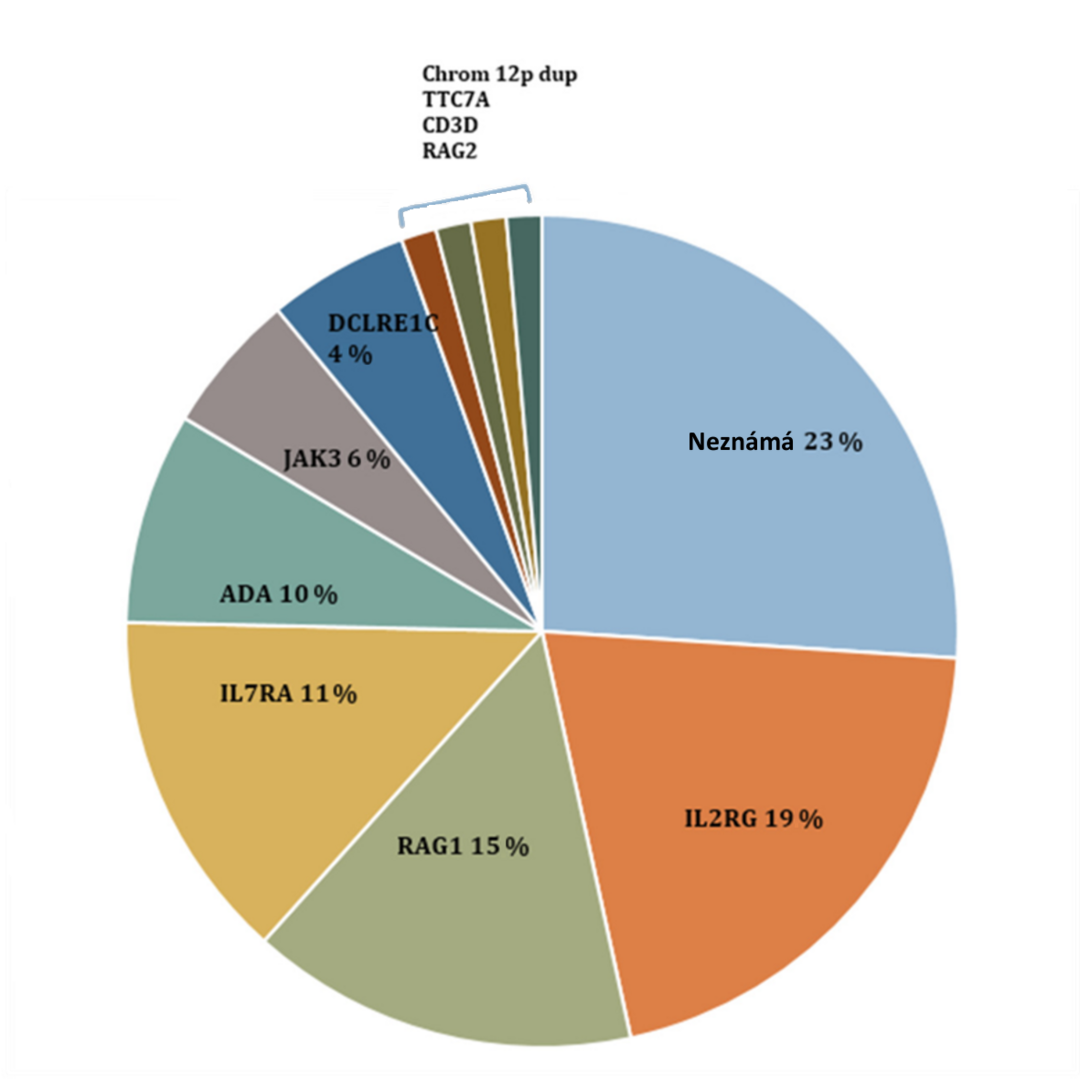
Mechanismus vzniku SCID onemocnění ve většině případů zasahuje zásadní procesy vývoje lymfocytů. Nejčastější formou je tzv. X-vázaný SCID, způsobený mutací v genu IL2RG na X chromozomu, která vede k chybění gamma-řetězce přítomného v celé řadě receptorů pro cytokiny, jež jsou kritické pro vývoj T lymfocytů, jako jsou např. IL-2, IL-7 a další. Výsledkem je T-B+ SCID s úplným chyběním T lymfocytů a nízkými NK buňkami, avšak s přítomnými B lymfocyty. Jiným příkladem příčiny mohou být mutace v genech RAG1 či RAG2, které kódují enzymy potřebné pro vznik specifických T a B buněčných receptorů, či mutace v genu ADA pro adenosindeaminázu, enzym odbourávající toxické produkty buněčného metabolismu. V těchto případech pak chybí jak T, tak i B lymfocyty. V posledních letech se též díky lepší dostupnosti genomických technologií, zejména celoexomového sekvenování (whole exome sequencing, WES), objevila celá řada tzv. „hypomorfních“ mutací v genech, dříve spojovaných s onemocněním SCID a velmi těžkým klinickým obrazem, které však byly identifikovány u pacientů s méně závažnou formou imunodeficience až ve starším dětství či dospělosti (3).
Obr 1: Prevalence mutací způsobujících SCID, zachycených v rámci screeningu více než 3 milionů novorozenců v USA, který odhalil 52 případů SCID. Adaptováno z Dorsey et al., 2017 (2).
Zásadní problém vyplývající z chybění či špatné funkce lymfocytů je zvýšená náchylnost k infekcím, která v případě dětí se SCID nabývá život ohrožujících rozměrů. Pacienti mohou trpět všemi druhy bakteriálních, virových i mykotických infekcí, včetně infekcí oportunními patogeny. Ač první cca 3 měsíce života přetrvává částečný protektivní vliv mateřských imunoglobulinů, které plod obdrží od matky transplacentárním přenosem in utero, infekce se mohou rozvinout již v tomto období a bývají často doprovázeny poruchou prospívání a perzistentním průjmem. V některých případech může též dojít k nekontrolované proliferaci malého množství lymfocytů, které potom v těle způsobí de facto reakci štěpu proti hostiteli, projevující se obrazem těžké erytrodermie, lymfadenopatie a hepatosplenomegalie. V takové situaci mluvíme o tzv. Omennově syndromu, kdy hladiny lymfocytů v krvi mohou být normální, či dokonce zvýšené –, jedná se však o oligoklonální populaci, která není schopna zajistit dobrou obranyschopnost organismu a způsobuje více škody než užitku.
Vzhledem k závažnosti těchto onemocnění a nutnosti jejich rychlé identifikace a následných izolačních a terapeutických kroků byl např. ve Spojených státech, Kanadě a pilotně též v řadě zemí EU zaveden screeningový program novorozenců testující správnou tvorbu T a B lymfocytů pomocí tzv. TREC/KREC metody, která je schopna v suché kapce krve detekovat excizní kroužky DNA vznikající při vývoji lymfocytů, a odhalit tak řadu forem SCID (4, 5). Toto vyšetření je dostupné i v České republice, momentálně ve 2 laboratořích v Praze a v Brně (6), a je připravován i pilotní projekt novorozeneckého screeningu.
V případě podezření na závažnou buněčnou imunodeficienci, ať již na základě klinického obrazu, rodinné anamnézy, či laboratorních nálezů, je nejdůležitějším prvním krokem izolace pacienta od všech potenciálních zdrojů infekce. Pro riziko přenosu CMV infekce od matky se též doporučuje přerušit kojení a samozřejmě neočkovat živými vakcínami, jako např. proti rotavirům. Dalším krokem je detailní vyšetření imunologem, který se soustředí na osobní i rodinnou anamnézu včetně možné konsangvinity, přítomnost vyrážky a syndromických rysů a stav dýchacích cest. Následovat musí podrobnější laboratorní vyšetření včetně subpopulací lymfocytů, naivních T lymfocytů včetně jejich nejmladších forem, tzv. recent thymic emmigrants (RTE), a jejich schopnosti proliferace. Další přístup se bude lišit dle podezření na konkrétní formy SCID, avšak prakticky ve všech případech bude zahrnovat genetické vyšetření k upřesnění příčiny. Prvním stadiem terapie je zpravidla substituce imunoglobulinů k podpoře obranyschopnosti organismu a profylaktické podávání antibiotik a antimykotik. Pacienti s Omennovým syndromem mohou vyžadovat imunosupresivní léčbu.
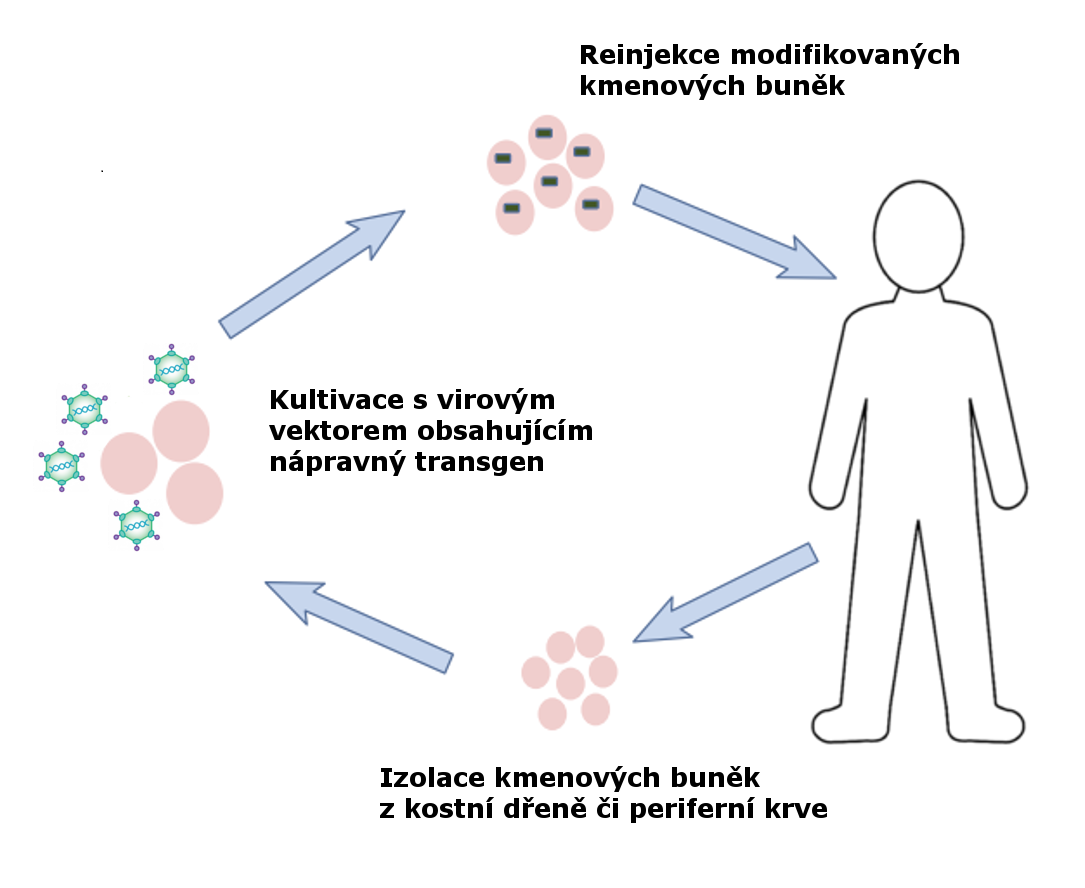
Obr 2: Schéma genové terapie, adaptováno z Booth et al., 2016 (7).
U většiny forem SCID je jediným kurativním řešením transplantace kostní dřeně, která umožní pacientovi tvorbu nových, zdravých lymfocytů. Průměrná doba přežití se liší dle centra, věku a množství komplikací v okamžiku záchytu a dle genetické příčiny, avšak pohybuje se nad hranicí 70 % po 5 letech (8).
U některých forem SCID se v posledních letech objevily vzrušující možnosti specifické léčby metodami genové terapie, kdy jsou pacientovi odebrány buňky kostní dřeně, in vitro pomocí virových vektorů opraven genetický defekt a následně jsou buňky navráceny zpět do těla pacienta. Tato metoda je nyní dostupná pro mutace v genech ADA a IL2RG (9, 10) a dle dostupných údajů se zdá, že do budoucna bude nabízet lepší prognózu a menší množství komplikací než tradiční transplantace kostní dřeně.
Závažné buněčné imunodeficience patří mezi nejzávažnější a nejvíce potenciálně život ohrožující situace, na něž je v pediatrické péči nutno pomýšlet. Komplexita jejich diagnostiky, terapie, následného sledování a péče si vyžaduje týmový přístup řady specialistů a dobře vybavená zdravotnická zařízení. V České republice je o tyto pacienty skvěle postaráno a díky mezinárodní spolupráci našich imunologů a hematologů je jim dostupná péče na světové úrovni.
MUDr. Adam Klocperk
Ústav imunologie 2. LF UK a FN Motol, V Úvalu 84, Praha 5, 150 00
Literatura:
Další informace naleznete na webových stránkách o primární imunodeficienci, které můžete doporučit svým pacientům.