
Investigativní genetická genealogie – nový přístup k určování příbuznosti osob
Investigative genetic genealogy – new approach to detect relatedness of individuals
The development of molecular genetic techniques, coupled with the development of bioinformatic approaches and the expansion of genealogy as a hobby, also brings new possibilities for forensic genetics. The review article compares today’s classical methods of genotyping and determination of kinship based on the analysis of polymorphisms such as short tandem repeats, with the possibilities of investigative genetic genealogy, which uses genotyping based on analysis of hundreds of thousands of single nucleotide polymorphisms, bioinformatics and sharing individual profiles within public databases. We describe the principles of these laboratory techniques and the interpretation of their results. We compare these new approaches with the current practice in forensic laboratories. We stress the importance of the introduction of whole genome sequencing into forensic genetics. Whole genome sequencing allows due its ability to work with degraded DNA samples the preparation of proper material for investigative genetic genealogy which has already proven its usefulness in successfully solving serial murder cases as well as in identifying unknown victims of violent crimes. In addition to methodological approaches and achievements, the limits and ethical aspects of this new field of forensic genetics are also mentioned.
Keywords:
DNA analysis – kinship determination – STR genotyping – investigative genetic genealogy
Autoři:
Korabečná Marie 1; Šimková Halina 1; Stenzl Vlastimil 2
Působiště autorů:
Ústav biologie a lékařské genetiky 1. lékařské fakulty UK a Všeobecné fakultní nemocnice Praha
1; Kriminalistický ústav Policie České republiky, Praha
2
Vyšlo v časopise:
Soud Lék., 66, 2021, No. 4, p. 58-65
Kategorie:
Přehledový článek
Souhrn
Rozvoj molekulárně genetických technik, provázený vývojem bioinformatických přístupů a rozšířením genealogie jako hobby, přináší nové možnosti také forenzní genetice. V přehledném článku jsou porovnány dnes již klasické metody genotypizace a určování příbuznosti, založené na analýze polymorfismů typu krátkých tandemových repetic, s možnostmi investigativní genetické genealogie, která využívá genotypizaci založenou na analýze stovek tisíců jednonukleotidových polymorfimů, bioinformatiky a sdílení profilů jednotlivců v rámci veřejných databází.
Klíčová slova:
DNA analýza – určení příbuznosti – STR genotypizace – investigativní genetická genealogie
Rozvoj metod molekulární genetiky v posledních desetiletích vedl k rutinnímu zavedení analýzy DNA jako metody pro individuální identifikaci osob v kriminalistice i při řešení paternitních sporů v občanskoprávních řízeních. Tato analýza byla umožněna poznáním rozsahu variability sekvencí lidské DNA a vývojem technologických možností jejího zkoumání. Genomy kterýchkoliv dvou osob se navzájem liší ve více než 1% sekvence genomické DNA (1). Současné metody rutinně používané v praxi forenzních laboratoří jsou založeny zejména na analýze polymorfismů lidské DNA označovaných jako krátké tandemové repetice (Short Tandem Repeats – STR), které představují více než 3% lidského genomu (2). Jejich výhodou je existence velkého počtu variant (alel) v každém z lokusů ve všech populacích (viz schéma na Obr.1A). Tento tzv. multialelismus spolu se znalostí četnosti jednotlivých alel ve zkoumané populaci tak představuje vhodný nástroj pro individuální identifikaci. Na základě mezinárodní spolupráce byly vybrány konkrétní STR polymorfismy jako markery vhodné coby součást tzv. STR profilů, určených k přímé identifikaci osob a posuzování blízké biologické příbuznosti, a současně vhodných pro uchovávání ve forenzních databázích (4). Technologický vývoj sledoval tuto linii, na trh byly uvedeny kity dovolující vyšetření panelu forenzně významných STR polymorfismů pomocí jediné multiplexové PCR (Polymerase Chain Reaction) s minimálním nutným množstvím vstupní analyzované DNA a následnou separací amplifikovaných fragmentů pomocí kapilární elektroforézy (3). Začaly vznikat průběžně doplňované databáze pro práci s polymorfismy typu STR (4), obsahující vlastní polymorfní sekvence i sekvence přilehlých oblastí DNA, frekvence jednotlivých alel v různých populacích i popisy metodických aspektů vyšetřování těchto polymorfismů.
STR polymorfismy jsou běžně používány i pro řešení paternitních sporů. Tento typ lokusů je lokalizován rovněž na pohlavních chromozomech, takže jejich sledování může být využito i pro určení paternální či maternální linie. Se vzrůstající příbuzenskou vzdáleností však tento způsob vyšetřování naráží na své limity.
STR polymorfismy plní důležité funkce ve forenzní genetice, avšak nejsou nejčastějšími polymorfismy, se kterými se můžeme v sekvencích lidské DNA setkat. Mnohem frekventovanější jsou tzv. jednonukleotidové polymorfismy (Single Nucleotide Polymorphisms – SNP), které se v lidském genomu vyskytují v průměru jednou na tisíc párů bazí (5). Na rozdíl od multialelických STR lokusů jsou SNP polymorfismy většinou pouze bialelické (v populaci existují jen dvě varianty daného místa se záměnou jediného nukleotidu), proto je k dosažení diskriminační síly porovnatelné s vyšetřením založeném na STR markerech třeba vyšetřit mnohem větší počet těchto lokusů (6). Obdobná omezení se týkají polymorfismů typu INDEL (7), kde alela buďto obsahuje dotyčnou sekvenci (inserční varianta) nebo tuto sekvenci neobsahuje (deleční varianta). Individuální lidský genom nese více než půl milionu takovýchto polymorfních míst (8).
K rutinnímu vyšetřování stupně příbuznosti osob tyto polymorfismy (SNP a INDEL) nejsou zatím v praxi příliš využívány, přesto si ale jejich analýza cestu do forenzní genetiky našla, a to především v oblasti tzv. DNA fenotypizace a určování biogeografického původu (BGA). Byly nalezeny geny, v nichž jsou lokalizovány polymorfismy typu SNP nebo INDEL ,které zásadním způsobem ovlivňují lidskou pigmentaci: barvu duhovky, kůže a vlasů (9). Některé z těchto polymorfismů mají zároveň vztah k biogeografického původu dotyčné osoby (10). Vyšetřování panelů jednonukleotidových polymorfismů za účelem zjištění biogeografického původu neznámé osoby, která je zdrojem zkoumaného biologického materiálu, se stalo reálným zejména s nástupem metod sekvenování nové generace do forenzních laboratoří. Tyto polymorfismy jsou právě pro svou schopnost přinést informaci o biogeografickém původu osoby souhrnně označovány jako AIMs -Ancestry Informative Markers) (11).
Nástup nových technologických platforem umožnil současné testování řádově stotisíců SNP v genomu každého jednotlivce a vedl ke vzniku tzv. genetické investigativní genealogie jako nástroje forenzní genetiky, jenž slouží k vyhledávání vzdálenějších příbuzných pro účely objasňování závažných trestných činů či při pátrání po zmizelých osobách (12).
V následujícím textu se proto zaměřujeme na porovnání dnes již klasických metodických přístupů založených na polymorfismech STR ve vztahu k určování příbuznosti osob s možnostmi investigativní genetické genealogie (IGG), včetně jejích technických i etických aspektů.
PŘEHLED METOD DNA ANALÝZY UMOŽŇUJÍCÍCH URČENÍ PŘÍBUZNOSTI OSOB
Určení příbuzenských vztahů na základě STR analýzy
Metodika
Klasický metodický přístup je založen na enzymatické amplifikaci všech polymorfních lokusů v jediné reakci. Pro běžné případy se zpravidla využívá vyšetření 15 STR lokusů rozmístěných na nepohlavních chromozomech a amplifikace části genu pro amelogenin lokalizovaného v pseudoautozomální oblasti pohlavních chromozomů. (Obrázek 1B). Amplifikace vybrané sekvence genu pro amelogenin umožnuje detekovat přítomné pohlavní chromozomy, a určit tak genetické pohlaví původce DNA vzorku. Zbylé primerové páry hybridizují k DNA sekvencím sousedícím s oblastmi jednotlivých vyšetřovaných tandemových repetic. Design těchto reakcí vede ke tvorbě různě dlouhých amplifikovaných fragmentů, přičemž citlivost reakce zaručuje úspěšný výsledek i u malého výchozího množství DNA ve vzorku - běžně i hluboko pod 1 ng na analýzu (3). Podle typu fluorescenčního značení použitých PCR primerů a délkového rozmezí získaných fragmentů je po jejich separaci kapilární elektroforézou možné přesně identifikovat jednotlivé alely polymorfních lokusů, a sestavit tak DNA profil vyšetřované osoby. Každá alela je označována počtem opakování tandemových repetic, které obsahuje (viz Obrázek 1C). V nekomplikovaném případě paternitního sporu, kdy je možné získat profily od celého tria (matka, dítě, pravděpodobný otec), pak porovnáním STR profilů matky a dítěte (černě značený profil v Obr.1C) získáme informaci, které alely v profilu dítěte jsou otcovského původu, a můžeme porovnat, zda se tento set alel vyskytuje v STR profilu nařčeného muže (šedě označená část profilu v Obr.1C). V případě nálezu shody následuje výpočet paternitního indexu, který vychází ze znalosti populačních frekvencí jednotlivých alel obsažených v tomto profilu, jak bude vysvětleno dále. K vyloučení otcovství musí být nalezena neshoda v nejméně třech polymorfních lokusech, protože u polymorfismů typu STR dochází v důsledku tzv. „prokluzu polymerázy“ poměrně často k mutacím při replikaci DNA, kdy výsledkem je změna počtu opakování základního motivu v repetici, a tedy změna alely (13). Frekvence těchto mutací je známa pro každý vyšetřovaný polymorfismus a lze ji zahrnout do výpočtu paternitního indexu (14).
Fig. 1. Basic principles of STR analysis. (A) Example of the structure of a polymorphic STR locus, alleles are indicated by the
number of repeats they contain. (B) Distribution of polymorphic STR loci on chromosomes on the example of a panel included
in a widely used kit AmpF/STR Identifiler. (C) Complete STR profiles of the father-child-mother trio with maternal (black) and
paternal (gray) profile, these profiles together with the frequencies of individual alleles are used to calculate the paternity index.

Porovnání STR polymorfismů na Y chromozomu slouží k potvrzení paternální linie v rodokmenu, nicméně nález shodného STR haplotypu (tj. setu STR polymorfismů tvořících vazebnou skupinu nepodléhající rekombinaci) na Y chromosomech syna a domnělého otce nepotvrzuje přímo otcovství tohoto muže, protože jeho bratři, strýcové i otec nesou tentýž haplotyp. Nález shodného Y chromozomálního haplotypu ve forenzních databázích může ale vést k nalezení mužských příbuzných mužského původce DNA vzorku (15). Pro sledování maternální linie lze rovněž využít STR polymorfismů lokalizovaných na X chromozomech (16) či vazebných skupin INDEL polymorfismů vyšetřovaných obdobnou metodikou (7, 17). Sledování maternální linie umožňuje také sekvenování mitochondriální DNA (18).
Aplikace metod sekvenování nové generace vede k získání sekvence všech vybraných polymorfních STR lokusů, a dovoluje tak odlišení tzv. mikrosekvenčních variant, u nichž došlo zpravidla k bodové mutaci v jedné či více tandemových repeticích, eventuálně k vložení krátké nerepetetivní sekvence mezi repetice. Tímto způsobem došlo ke zvýšení počtu polymorfních alel jednotlivých STR lokusů a dalšímu zvýšení jejich diskriminační síly (19).
Interpretační možnosti
Pro každý typ biologického příbuzenského vztahu zároveň existuje očekávaná míra genetické shody, daná koeficientem příbuznosti, který charakterizuje míru sdílení genetického materiálu od společného předka či předků (20). Znalost populační frekvence jednotlivých alel STR markerů spolu s aplikací Hardy-Weinbergova zákona a principů bayesovské statistiky, pak umožňuje posoudit věrohodnost skutečně pozorované shody při platnosti jednotlivých zvažovaných hypotéz (21). Pro každou dvojici zvažovaných tvrzení je pak možné tyto věrohodnosti poměřit pomocí věrohodnostního poměru (likelihood ratio – LR), pro který se v případě posouzení biologického otcovství vžil termín paternitní index (22). Způsobu interpretace výsledků STR genotypizace pro určování příbuzenských vztahů je dlouhodobě věnována pozornost a k dispozici jsou osvědčené softwarové nástroje (23) – k nejrozšířenějším patří například program Familias (24).
Potřeba nepřímé genetické identifikace tělesných ostatků je evidentní například v případech identifikací obětí hromadných neštěstí (Disaster Victim Identification – DVI), forenzně genetická identifikace je využitelná i v souvislosti s migračními krizemi. Společnou vlastností těchto případů je to, že biologický materiál identifikované osoby není již často intaktní, ale dosáhl určitého – někdy i velmi pokročilého – stupně degradace. To v praxi znamená mnohdy omezenější možnosti správně a úplně genotypovat všechny vybrané markery. Typickým důsledkem pak je situace, kdy ze standardně testované sady STR markerů je úspěšně genotypováno jen několik málo, což vede k velmi nízké hodnotě paternitního indexu, a tedy k nemožnosti identifikaci uzavřít jako nespornou. Další komplikací je, pokud v rámci daného testování není z jakéhokoli důvodu možné využít klasické trio a je nutné identifikovat osobu prostřednictvím jiných, vzdálenějších příbuzenských vztahů. Zatímco identifikace pomocí sourozeneckých vzorků může být díky standardní dostupnosti dat z 24 STR markerů poměrně průkazná, už i v případě polovičních sourozenců a omezeného množství STR markerů je interpretace výsledků obtížná, v případě vzdálenějších příbuzenských vztahů (např. prvostupňoví bratranci) prakticky nemožná. Stejná omezení platí i v případech, kdy je identifikace založena na využití forenzních genetických databází, které sice mohou obsahovat stovky tisíc až miliony profilů DNA, ovšem založených na limitovaném počtu 20 (USA), 23 (Čína) nebo 16 (Evropa) STR autozomálních markerů (25). Tento typ vyhledávání potenciálních příbuzných v databázi, tzv. příbuzenské prohledávání neboli familiar searching, má proto v kriminalistické praxi značná omezení (26), jakkoli se z laického hlediska jeví jako potenciálně velmi mocný nástroj. Investigativní genetická genealogie (IGG), jejíž principy jsou popsány v následujících odstavcích, pomáhá limity STR genotypizace v této oblasti eliminovat.
Určení příbuzenských vztahů na základě IGG
Privátní společnosti poskytující komerční geneticko-genealogické testování věnovaly v posledních cca 10 letech značné úsilí hledání a rozvoji nových metod uspokojujících požadavky zájemců z hobby komunity genealogů. Tyto společnosti, typově označované jako DTC (direct to customer), nabídly již desítkám milionů svých zákazníků testování založené na polymorfismech typu SNP. Hnacím motorem pokroku v této oblasti zůstává raketový rozvoj technologií umožňujících získání až několika set tisíc SNP během jediné analýzy (především v rámci tzv. high-density microarrays genotypování – viz dále), což sebou přináší výrazné snížení ceny analýzy, a tím zásadní zlepšení jejich dostupnosti. Takovéto počty genotypovaných markerů již umožňují identifikovat jedince i prostřednictvím srovnávání shody v mnohem vzdálenějších příbuzenských vztazích, jako jsou například druzí a třetí bratranci (12, 27).
Díky dostupnosti tohoto nového typu genetických dat se především v USA začal rozvíjet nový obor, takzvaná investigativní genetická genealogie (IGG), která kombinuje výstupy z prohledávání geneticko-genealogických databází s genealogickým výzkumem širších rodinných rodokmenů. IGG nabízí poskytovatelům forenzních služeb pomoc především ve dvou oblastech:
1) Poskytnutí operativních informací orgánům činným v trestním řízení, týkajících se vzdáleně příbuzenských vztahů mezi neznámým zůstavitelem biologického materiálu na místě činu a jeho třeba i vzdálenými biologickými příbuznými, kteří zanesli svoje profily DNA do veřejně přístupných geneticko-genealogických databází a vyjádřili souhlas s jejich využitím pro tento účel (12, 25, 27).
2) Identifikace neznámé osoby (kupříkladu mrtvoly neznámé totožnosti) analýzou jejího biologického (většinou degradovaného) materiálu a porovnáním s profily DNA možných vzdálených příbuzných v geneticko-genealogických databázích (28).
Způsob aplikace IGG je znázorněn na Obr.2. Zde je třeba zdůraznit, že k využití IGG dochází až poté, co nebyly nalezeny žádné k porovnávání vhodné profily ve forenzních STR databázích (25). Shoda studovaného vzorku DNA s DNA osoby nalezené pomocí IGG musí být rovněž v závěrečné fázi vyšetřování prokázána shodou v STR profilu. Průlom ve využívání IGG znamenal případ označovaný jako „Golden State Killer“, kdy byl vrah po desetiletích neúspěšného pátrání v roce 2018 nalezen metodou IGG (27). Přístup IGG byl rovněž úspěšně aplikován v evropském prostoru k identifikaci neznámého zavražděného muže nalezeného ve Švédsku, kdy IGG vedla k dohledání jeho blízkých příbuzných v Chorvatsku (28).
Fig. 2. Incorporation of the method of investigative forensic genealogy into the scheme of forensic analysis of an unknown
DNA sample. The extent of shared parts of the genome between individuals of a given degree of relatedness (r = coefficient of relatedness)
is indicated on the chromosomes. The extent of shared areas decreases with the decrease of the coefficient of relatedness.

Metodika IGG
Zatímco STR profilování je ve svém základním provedení spojené s polymerázovou řetězovou reakcí (PCR) v multiplex formátu a následnou separací amplifikovaných fragmentů pomocí kapilární elektroforézy, metodické postupy, na nichž je založena IGG, jsou o poznání komplexnější. Metody genetické analýzy dovolující vyšetření vysokého počtu polymorfismů typu SNP u každého jedince jsou buď sekvenační nebo založené na tzv. „high density microarrays“(12). Principy obou technologií jsou schematicky znázorněn na Obr.3. Ze schématu je patrná nutnost amplifikace genetického materiálu, jednotlivé amplifikační kroky jsou označeny šedými ovály. Zatímco celogenomové sekvenování umožňuje za podmínky pečlivé bioinformatické analýzy (především filtrování získaných dat dle jejich kvality) analyzovat všechny polymorfismy v daném individuálním genomu, ostatní metody předem definují analyzovaný set SNP polymorfismů aplikací PCR na tyto vybrané úseky a následným sekvenováním získaných amplikonů nebo zachycením požadovaných úseků genomické DNA pomocí hybridizace na předem připravené sondy. Problémem zůstává požadavek na relativně vysoké množství vstupního vzorku DNA a jeho kvalitu. Rutinní metody forenzních laboratoří založené na STR profilování bohatě vystačují s 1ng DNA, v případě aplikace SNP microarray byly publikovány uspokojivé výsledky s množstvím DNA ve vzorku o něco málo nižším než 25 ng (29).
Fig. 3. Basic laboratory methodological principles used in the process of investigative forensic genealogy.

Z hlediska získání potřebného SNP profilu se tak jeví být vhodnou metodou volby pro forenzní laboratoře celogenomové sekvenování, které je aplikovatelné na fragmentovanou DNA, a dovoluje tudíž i práci s degradovanou DNA, běžně se vyskytující ve forenzních vzorcích. Jeho hlavní nevýhodou zůstává vyšší cena a náročné bioinformatické zpracování. Cesta celogenomového sekvenování vzorku na akademickém pracovišti byla zvolena i v prvním evropském úspěšně ukončeném případu aplikace IGG. V tomto případě byly použity 3 ng DNA izolované s kosti a celogenomové sekvenování bylo provedeno s průměrným pokrytím 32,2x (28). Tato hodnota označuje průměrný počet přečtení každého analyzovaného fragmentu DNA při aplikaci metody masívního paralelního sekvenování.
Interpretační možnosti IGG
Přístup využívající věrohodnostní poměr (likelihood ratio - LR) je založen na posouzení, jak věrohodná (tj. očekávatelná) jsou pozorovaná genetická data (genotypy) za předpokladu platnosti předem formulovaných tvrzení týkajících se vzájemných příbuzenských vztahů mezi zkoumanými jedinci (22). Především tento přístup je široce rozšířen při formulování závěrů forenzně genetických expertíz, a to z toho důvodu, že umožňuje vyjádřit váhu důkazu formou číselné hodnoty. Jeho nevýhodou je ovšem nutnost vytvoření rozšířené frekvenční databáze jednotlivých alelických variant, čítajících stovky až tisíce markerů. Znalost populačních frekvencí jednotlivých haplotypů lze pak využít při interpretaci sledu jednotlivých detekovaných SNP na chromozomech vyšetřovaného jedince (30). Dalším z problémů, které je nutné brát v úvahu, je zvýšené riziko vazebné nerovnováhy mezi blízkými lokusy, které je v případě jejich většího počtu mnohem pravděpodobnější (31), přičemž tato nerovnováha má vliv na výpočet jednotlivých věrohodností.
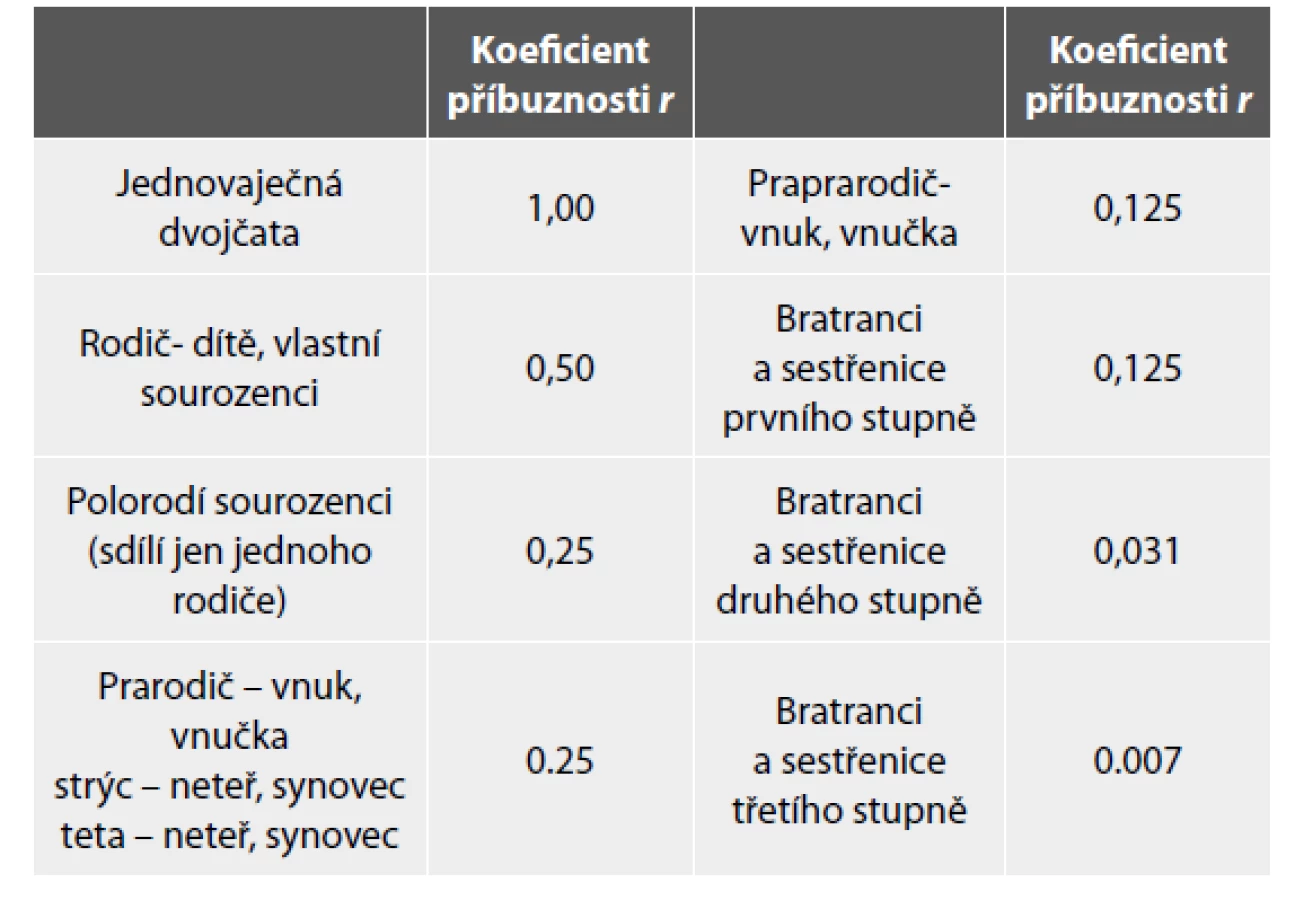
Exploratorní přístup nepotřebuje východisko ve formě znalosti frekvencí jednotlivých variant sekvencí, vychází z prostého posouzení délky a počtu segmentů DNA nesoucích shodné varianty sledovaných SNP v genomech porovnávaných jedinců (32). V zásadě je nutné posoudit, nakolik jsou oblasti DNA mezi těmito jedinci sdílené, tzn. zděděné od společných předků (identical by descent - IBD). O síle této příbuznosti potom může vypovídat koeficient příbuzenství (kinship coefficient) (33), který říká, jaká je pravděpodobnost, že shodná alela u dvou jedinců náhodně vybraných z populace je identická původem. Po vynásobení tohoto koeficientu dvěma získáváme obecně více známý v genetice běžně používaný koeficient příbuznosti (coefficient of relatedness) (34). Příklady koeficientů příbuznosti pro nejběžnější příbuzenské vztahy jsou uvedeny v Tabulce 1. Alternativně lze v rámci exploratorního přístupu posuzovat délku segmentů DNA, v kterých jsou zkoumaní jedinci shodně homozygotní pro sledované alely SNP. Čím delší jsou takto detekované segmenty, tím je pravděpodobnější, že tyto segmenty pocházejí od společného předka. Tento model je především využíván pro posouzení genetické příbuznosti v rámci komerčních geneticko-genealogických testů. Aplikací exploratorního přístupu na set 20 tisíc profilů se podařilo správně detekovat 90% třetích bratranců a 46% čtvrtých bratranců (35). Oba typy exploratorních modelů neudávají přesný stupeň příbuznosti, ale spíše možný rozsah příbuzenských vztahů, které musí být dále ověřeny genealogickým výzkumem (12).
Úspěch IGG je předurčen jak metodologickým zázemím v podobě pokročilých molekulárně genetických a bioinformatických metod schopných produkovat a následně zpracovávat obrovské objemy dat, tak charakterem dostupných databází. Charakteristiky hlavních databází jsou stručně shrnuty v Tabulce 2. Pro forenzní účely je porovnáním s databázemi vyhledán soubor vzorků s nejbližším detekovatelným potenciálním příbuzenským vztahem ke zkoumanému vzorku, poté následuje objasňování rodokmenových vztahů včetně využití volně dostupných dat ze sociálních sítí (12) a sběr kontrolních vzorků společně s vyhodnocováním ostatních důkazů daného případu. Vyšetřování je úspěšně uzavřeno nalezením osoby, jejíž STR profil se plně shoduje se zkoumaným vzorkem DNA klíčovým pro řešení případu (25).

SOUČASNÉ PROBLÉMY A MOŽNOSTI DALŠÍHO VÝVOJE
Forenzní implementace IGG je redukovaná především metodickými možnostmi standardních forenzně genetických laboratoří. Nové technologie vyžadují zavedení finančně nákladného přístrojového vybavení a sofistikovaných bioinformatických přístupů k interpretaci dat. Další omezení vyplývá ze samé podstaty forenzních vzorků, které nejsou díky svému množství a kvalitě v případě identifikace degradovaných biologických materiálů nejvhodnějším vstupním materiálem pro standardní SNP genotypování na čipech („high density microarrays“). Tento nedostatek je možné kompenzovat metodikou zaměřenou na tzv. celogenomové sekvenování. Historicky byly tyto postupy zkoumány v souvislosti s výzkumem starobylé DNA (ancient DNA) (36). V dnešní době jsou využitelné právě kvůli citlivosti analýzy vzhledem k forenzním vzorkům (28). Z tohoto důvodu probíhá v ČR výzkumný projekt VI20172020102 Ministerstva vnitra ČR o názvu „Využití DNA polymorfismů typu CNV pro určení stupně příbuznosti osob“ zaměřený na hledání vzácných genetických variant pro využití právě při zjišťování vzdálenejší příbuznosti osob a na implementaci metody celogenomového sekvenování do praxe forenzních labotratoří . Tato metoda v kombinaci se specializovanými bioinformatickými přístupy je univerzálně využitelná pro detekci jakýchkoliv polymorfismů v lidské DNA. Nalezení shodné populačně extrémně vzácné varianty (například krátké sekvence, která se na dotyčném místě v genomu může vyskytovat v různém počtu kopií, tzv. polymorfismus typu CNV - Copy Number Variation) v genomu dvou jedinců poskytuje potenciální důkaz jejich příbuznosti, při čemž každá takováto varianta může obsahovat různý počet SNP polymorfismů.
Protože data založená na SNP genotypování nejsou kompatibilní s forenzními DNA databázemi naplněnými daty získanými STR profilováním, je nutné v případě porovnávání SNP profilů uvažovat o jiných typech databází, které splňují nároky především s ohledem na jejich velikost, spolehlivost generování hitů a nároků na zabezpečení dat. Privátní společnosti nabízející tento typ analýz využívají svoje vlastní databáze, které jsou však z komerčních důvodů vzájemně nekompatibilní (alespoň ne plně) viz Tabulka 2. SNP profil vhodný pro nahrání do databáze musí mít potřebnou velikost a kvalitu, musí obsahovat kolem 700 000 SNP genotypů jedince, kterého chceme porovnávat s obsahem databáze. Celogenomové sekvenování je schopno poskytnout profily potřebné kvality ke vložení do těchto databází (12, 28), dosažení podobné velikosti databází tohoto typu (Tabulka 2) je v současné době pro forenzní laboratoře nereálným úkolem.
Z popudu geneticko-genealogické komunity vznikla v roce 2010 otevřená databáze GEDMatch, která umožnila zájemcům databázovat svoje SNP profily bez ohledu na to, u které společnosti si nechali provést geneticko-genealogický test. I do dnešních dnů je to prakticky jediná veřejná geneticko-genealogická databáze, která umožňuje nahrávat genetické profily z mnoha různých zdrojů včetně orgánů činných v trestním řízení za předpokladu získání souhlasu majitelů databáze, který odráží postoj uživatelů k danému případu. (12, 25). Každá osoba mající svůj profil v databázi tak může rozhodnout, zda jej dá k dispozici pro právě probíhající vyšetřování. Není překvapením, že využitelnost této databáze je vázána především na severoamerický kontinent, protože právě z tohoto prostoru pochází většina zájemců o geneticko - genealogické testování. Nicméně je doložena její využitelnost i v evropském prostoru, kdy vlastní SNP profil forenzního vzorku byl generován akademickým pracovištěm (28), z tohoto důvodu je zatím v západních právních systémech tento přístup vnímán jako operativní informace. Rozšířené používání SNP dat by mělo přinést v budoucnu nové biostatistické metody, které by zefektivnily využití exploratorního přístupu jako spolehlivého nástroje k vyhledávání skutečných biologických příbuzných v dostupných databázích.
Případy úspěšně vyřešené pomocí IGG se v současné době již pohybují v řádu stovek (27). Další rozvoj a úspěšné aplikace IGG přístupu stejnou měrou jako na vývoji technologie závisí na paralelním vývoji v oblasti legislativy, který musí zohledňovat také řadu etických aspektů.
PODĚKOVÁNÍ
Práce byla podpořena projektem VI20172020102 Ministerstva vnitra ČR.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikaci tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádným podnikatelským subjektem. Toto prohlášení se týká i všech spoluautorů.
Received: April 19, 2021
Accepted: August 4, 2021
Adresa pre korespondenci:
Prof. RNDr. Marie Korabečná, PhD
Ústav biologie a lékařské genetiky 1. LF UK a VFN Praha
Albertov 4, 12800 Praha 2
e-mail: marie.korabecna@lf1.cuni.cz
tel.: 224968047
Zdroje
1. Pang AW, MacDonald JR, Pinto D et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol 2010; 11: R52.
2. Tang H, Nzabarushimana E. STRScan: targeted profiling of short tandem repeats in whole-genome sequencing data. BMC Bioinformatics 2017; 18(Suppl 11): 398.
3. Butler JM. Short tandem repeat typing technologies used in human identity testing. Biotechniques 2007; 43(4):2–5.
4. Ruitberg CM, Reeder DJ, Butler JM. STRBase: a short tandem repeat dna database for the human identity testing community. Nucleic Acids Res 2001; 29(1): 320–322.
5. Santurro A, Vullo AM, Borro M et al. Personalized Medicine Applied to Forensic Sciences: New Advances and Perspectives for a Tailored Forensic Approach. Curr Pharm Biotechnol 2017; 18(3): 263-273.
6. Butler JM, Coble MD, Vallone PM. STRs vs. SNPs: thoughts on the future of forensic DNA testing. Forensic Sci Med Pathol 2007; 3(3): 200-205.
7. Zidkova A, Horinek A, Kebrdlova V, Korabecna M. Application of the new insertion - deletion polymorphism kit for forensic identification and parentage testing on the Czech population. Int J Legal Med 2013 ; 127(1): 7-10.
8. Chen J, Guo Jt. Comparative assessments of indel annotations in healthy and cancer genomes with next-generation sequencing data. BMC Med Genomics 2020; 13 : 170-181.
9. Breslin K, Wills B, Ralf A et al. HIrisPlex-S system for eye, hair, and skin color prediction from DNA: Massively parallel sequencing solutions for two common forensically used platforms. Forensic Sci Int Genet 2019;43 : 102152.
10. Palencia-Madrid L, Xavier C, de la Puente M et al. Evaluation of the VISAGE basic tool for appearance and ancestry pediction using PowerSeq chemistry on the MiSeq FGx system. Genes (Basel) 2020; 11(6): 708-718.
11. Bulbul O, Filoglu G. Development of a SNP panel for predicting biogeographical ancestry and phenotype using massively parallel sequencing. Electrophoresis 2018; 39(21): 2743-2751.
12. Kling D, Phillips C, Kennett D, Tillmar A. Investigative genetic genealogy: Current methods, knowledge and practice. Forensic Sci Int Genet 2021;52 : 102474.
13. Abecasis GR, Altshuler D, Auton A et al. A map of human genome variation from population - scale sequencing. Nature 2010; 467 : 1061–1073.
14. Droździok K, Kabiesz J, Tomsia M, Skowronek R, Rębała K. Mutation analysis of short tandem repeats in a population sample from Upper Silesia (southern Poland). Leg Med (Tokyo) 2018; 33 : 1-4.
15. Gusmão L, Butler JM, Carracedo A, et al. DNA Commission of the International Society of Forensic Genetics (ISFG): an update of the recommendations on the use of Y-STRs in forensic analysis. Int J Legal Med 2006; 120(4): 191-200.
16. Tillmar AO, Kling D, Butler JM et al. DNA Commission of the International Society for Forensic Genetics (ISFG): Guidelines on the use of X-STRs in kinship analysis. Forensic Sci Int Genet 2017; 29 : 269-275.
17. Pereira R, Pereira V, Gomes I et al. A method for the analysis of 32 X chromosome insertion deletion polymorphisms in a single PCR. Int J Legal Med 2012; 126(1): 97-105.
18. Parson W, Gusmão L, Hares DR et al., DNA DNA Commission of the International Society for Forensic Genetics: revised and extended guidelines for mitochondrial DNA typing. Forensic Sci Int Genet 2014; 13 : 134-142.
19. Parson W, Ballard D, Budowle B et al. Massively parallel sequencing of forensic STRs: Considerations of the DNA commission of the International Society for Forensic Genetics (ISFG) on minimal nomenclature requirements. Forensic Sci Int Genet 2016; 22 : 54-63.
20. Rousset, F. Inbreeding and relatedness coefficients: what do they measure? Heredity 2002; 88 : 371–380.
21. Egeland T, Mostad PF, Mevâg B, Stenersen M. Beyond traditional paternity and identification cases. Selecting the most probable pedigree. Forensic Sci Int 2000; 110(1): 47-59.
22. Gjertson DW, Brenner CH, Baur MP et al., ISFG: Recommendations on biostatistics in paternity testing. Forensic Sci Int Genet 2007; 1(3-4): 223-231.
23. Drábek J. Validation of software for calculating the likelihood ratio for parentage and kinship. Forensic Sci Int Genet 2009; 3(2): 112-118.
24. Kling D, Tillmar AO, Egeland T. Familias 3 - Extensions and new functionality. Forensic Sci Int Genet 2014; 13 : 121-127.
25. Ge J, Budowle B. Forensic investigation approaches of searching relatives in DNA databases. J Forensic Sci 2021; 66(2): 430-443.
26. Karantzali E, Rosmaraki P, Kotsakis A, Le Roux-Le Pajolec MG, Fitsialos G. The effect of FBI CODIS Core STR Loci expansion on familial DNA database searching. Forensic Sci Int Genet 2019; 43 : 102129.
27. Greytak EM, Moore C, Armentrout SL. Genetic genealogy for cold case and active investigations. Forensic Sci Int 2019; 299 : 103 - 113.
28. Tillmar A, Sjölund P, Lundqvist B, Klippmark T, Älgenäs C, Green H. Whole-genome sequencing of human remains to enable genealogy DNA database searches - A case report. Forensic Sci Int Genet 2020;46 : 102233.
29. Wendt FR, Rahikainen AL, King JL, Sajantila A, Budowle B. A genome-wide association study of tramadol metabolism from post-mortem samples. Pharmacogenomics J 2020; 20 : 94–103.
30. Tillmar AO, Phillips C. Evaluation of the impact of genetic linkage in forensic identity and relationship testing for expanded DNA marker sets. Forensic Sci Int Genet 2017; 26 : 58–65.
31. Kling D. On the use of dense sets of SNP markers and their potential in relationship inference, Forensic Sci Int Genet 2019; 39 : 19–31.
32. Browning BL, Browning SR. Improving the accuracy and efficiency of identity-by-descent detection in population data, Genetics 2013; 194 (2): 459–471.
33. Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies, Bioinformatics 2010; 26(22): 2867 – 2873.
34. Wright S. Coefficients of inbreeding and relationship. Am Nat 1922; 56(645): 330–338.
35. Henn M, Hon L, Macpherson JM, et al. Cryptic distant relatives are common in both isolated and cosmopolitan genetic samples, PLoS One 2012; 7(4): e34267.
36. Burbano HA, Hodges E, Green RE, et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science 2010; 328(5979): 723-725.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Soudní lékařství

2021 Číslo 4
Nejčtenější v tomto čísle
- Samovražedné úmrtia elektrinou: tri neobvyklé kazuistiky
- Investigativní genetická genealogie – nový přístup k určování příbuznosti osob
- Generalizovaná trombóza u pacienta s prebiehajúcim bezpríznakovým ochorením Covid-19 – kazuistika
- František Novomeský, Akin Savas Toklu: Fundamentals of Diving Medicine.
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy