
Using Routine Surveillance Data to Estimate the Epidemic Potential of Emerging Zoonoses: Application to the Emergence of US Swine Origin Influenza A H3N2v Virus
Background:
Prior to emergence in human populations, zoonoses such as SARS cause occasional infections in human populations exposed to reservoir species. The risk of widespread epidemics in humans can be assessed by monitoring the reproduction number R (average number of persons infected by a human case). However, until now, estimating R required detailed outbreak investigations of human clusters, for which resources and expertise are not always available. Additionally, existing methods do not correct for important selection and under-ascertainment biases. Here, we present simple estimation methods that overcome many of these limitations.
Methods and Findings:
Our approach is based on a parsimonious mathematical model of disease transmission and only requires data collected through routine surveillance and standard case investigations. We apply it to assess the transmissibility of swine-origin influenza A H3N2v-M virus in the US, Nipah virus in Malaysia and Bangladesh, and also present a non-zoonotic example (cholera in the Dominican Republic). Estimation is based on two simple summary statistics, the proportion infected by the natural reservoir among detected cases (G) and among the subset of the first detected cases in each cluster (F). If detection of a case does not affect detection of other cases from the same cluster, we find that R can be estimated by 1−G; otherwise R can be estimated by 1−F when the case detection rate is low. In more general cases, bounds on R can still be derived.
Conclusions:
We have developed a simple approach with limited data requirements that enables robust assessment of the risks posed by emerging zoonoses. We illustrate this by deriving transmissibility estimates for the H3N2v-M virus, an important step in evaluating the possible pandemic threat posed by this virus.
Please see later in the article for the Editors' Summary
Published in the journal:
. PLoS Med 10(3): e32767. doi:10.1371/journal.pmed.1001399
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1001399
Summary
Background:
Prior to emergence in human populations, zoonoses such as SARS cause occasional infections in human populations exposed to reservoir species. The risk of widespread epidemics in humans can be assessed by monitoring the reproduction number R (average number of persons infected by a human case). However, until now, estimating R required detailed outbreak investigations of human clusters, for which resources and expertise are not always available. Additionally, existing methods do not correct for important selection and under-ascertainment biases. Here, we present simple estimation methods that overcome many of these limitations.
Methods and Findings:
Our approach is based on a parsimonious mathematical model of disease transmission and only requires data collected through routine surveillance and standard case investigations. We apply it to assess the transmissibility of swine-origin influenza A H3N2v-M virus in the US, Nipah virus in Malaysia and Bangladesh, and also present a non-zoonotic example (cholera in the Dominican Republic). Estimation is based on two simple summary statistics, the proportion infected by the natural reservoir among detected cases (G) and among the subset of the first detected cases in each cluster (F). If detection of a case does not affect detection of other cases from the same cluster, we find that R can be estimated by 1−G; otherwise R can be estimated by 1−F when the case detection rate is low. In more general cases, bounds on R can still be derived.
Conclusions:
We have developed a simple approach with limited data requirements that enables robust assessment of the risks posed by emerging zoonoses. We illustrate this by deriving transmissibility estimates for the H3N2v-M virus, an important step in evaluating the possible pandemic threat posed by this virus.
Please see later in the article for the Editors' Summary
Introduction
The 2009 A(H1N1)pdm09 influenza pandemic [1], the SARS epidemic in 2003 [2], and the recent emergence of a novel coronavirus [3] are recent reminders of the global health threat posed by zoonotic viruses. Prior to widespread emergence in human populations, such pathogens can cause occasional infections in sub-populations that have been exposed to reservoir species (common reservoir species include for example bats, birds, swine, non-human primates). Whilst viruses causing such “spill-over” infections are usually poorly adapted for sustained human-to-human transmission, they are under strong selection pressure to increase transmissibility once in humans [4]. If the reproduction number R (i.e., the average number of persons infected by a case) evolves to exceed 1, a large scale epidemic in humans may result. Over the last decade, particular concerns were raised regarding highly pathogenic H5N1 avian influenza, due to the high mortality rate seen in humans and the virus's rapid spread in avian populations. However, as the A(H1N1)pdm09 influenza pandemic demonstrated, H5N1 is not the only influenza virus that may pose a pandemic risk. Recently, a swine-origin triple reassortant influenza A(H3N2) variant virus has emerged in the United States, carrying the matrix gene (M) from the H1N1pdm09 virus (H3N2v-M) [2]–[4]. Studies in animal models have suggested that the presence of the H1N1pdm09 M gene may increase transmissibility of the virus [5],[6]. From January 2012 to September 2012, 307 laboratory-confirmed H3N2v-M human infections were reported to Centers for Disease Control and Prevention (CDC) [5] as opposed to 12 throughout 2011 [6]. The majority of cases have been associated with agricultural fairs but there are documented events of human-to-human transmission [7]. The surge in cases observed in summer 2012 raised public health concerns [8]. Threats from zoonoses are not limited to influenza: more than half of all recent emerging infectious disease events were zoonotic [9].
For efficient prevention and control, quantitative and rigorous assessment of the risks associated with emerging zoonoses is desirable—in particular the risk that an emerging pathogen evolves to cause sustained human-to-human transmission. One approach to such risk assessment is by monitoring the reproduction number R of zoonoses in humans, with an alarm being raised if R increases or approaches 1 [9]–[11]. However, until now, estimating R required detailed outbreak investigations of human clusters [10],[11] and suffered from three important limitations: (1) the resources, access, and expertise needed to conduct investigations is not always available; (2) the proportion of cases that are missed during outbreak investigations may vary by setting and be difficult to assess; (3) even if the study is complete, the data collection process can be affected by a selection bias whereby larger outbreaks are more likely to be detected so that estimates of transmissibility may be biased upward. Consider for example a scenario where R = 0.7, where each case has the same detection probability ρ = 1%, and assume that once a cluster is detected, detailed outbreak investigation ensures that all cases in the cluster are detected. With an average size of 18.3 and a 21% probability of 1-case cluster, clusters that are detected are substantially larger than normal ones (average size: 3.3; 65% probability of 1-case cluster) (Figure 1A). As expected, this selection bias leads to R being overestimated as illustrated for methods that use the distribution of detected cluster sizes (Figure 1B) [10].

Here, we present a new approach to estimate R during spillover events, aiming to address many of the limitations of existing methods. We apply our approach to assess the human-to-human transmissibility of swine-origin influenza A variant (H1N1v, H1N2v, and H3N2v) virus, in particular that of the H3N2v-M virus, from US surveillance data for the period December 2005–December 2011. We also present applications to another zoonotic virus (Nipah virus in Malaysia and Bangladesh) as well as to a non-zoonotic pathogen (Vibrio Cholerae in the Dominican Republic).
Materials and Methods
Ethics Statement
This investigation was determined to be part of public health response and was not considered to be human research in accordance with federal human individual protection regulations. Thus, approval from an institutional review board was not required.
Definitions
We define a “chain of transmission” as a single reservoir-to-human transmission event followed by subsequent human-to-human transmission events (if any). A “cluster” of related cases is defined as an outbreak that takes place in a specific location and at a specific time (e.g., outbreak in fair X in August 2011), and can be composed of several chains of transmission (i.e., when a number of people are exposed to the same zoonotic source of infection).
Our method relies on the observation that the proportion of detected cases that are infected by the reservoir is largely determined by the reproduction number for human-to-human transmission, R. For example, for the H1N1pdm09 virus which had R>1 and was therefore generating long and expanding chains of transmission, none of the detected human cases were linked to the original swine reservoir. In contrast, for H5N1 highly pathogenic avian influenza, where R in human populations is considerably below 1, a substantial proportion of cases can be linked to exposure to the animal reservoir.
Surveillance Scenarios
We consider two surveillance scenarios: (1) “Routine sentinel surveillance alone” where we assume that each case has a probability ρ of being detected by routine sentinel surveillance; (2) “Routine sentinel surveillance triggering outbreak investigation”, where we again assume that each case initially has a probability ρ of being detected by routine sentinel surveillance, but that when a case is detected, this may trigger an outbreak investigation. In the latter scenario, once a case is detected in a cluster, the detection probability for other cases in the same cluster may increase.
In both scenarios we assume that there is an investigation of the case detected through routine sentinel surveillance during which the probable source of infection (natural reservoir or human-to-human) is determined. However, only in scenario 2 is there also a full-blown outbreak investigation where other cases are actively searched for. We therefore expect that it is possible to group cases into clusters in scenario 2; but such information is less likely to be available in scenario 1. The US H3N2v-M situation we examine resembles scenario 2, but we also present applications for scenario 1.
We propose to estimate R from two simple summary statistics, the proportion infected by the natural reservoir among detected cases (G) and among the subset of the first detected cases in each cluster (F). We now determine the relationship between G, F, and R and develop a statistical framework to estimate R from G (scenario 1) and from F (scenario 2).
Modelling the Offspring Distribution
We denote the length of a chain of transmission by L. Following Lloyd-Smith et al. [12], the offspring distribution (i.e., number of persons infected by a case) is modelled with a negative binomial distribution with mean R and overdispersion parameter k (parameter k characterizes case-to-case variation in infectiousness). The probability that a chain is of length L is given by [13]:

Relationship between G and R (Scenario 1)
We first consider scenario 1 where each case has the same probability of being detected. In a typical chain of transmission of average length there is, by definition, one reservoir-to-human transmission event and human-to-human infections. The probability G that a case randomly picked up by surveillance was infected by the reservoir is therefore . However, for subcritical outbreaks (0<R<1), branching process theory tells us that the average length of a chain is [14]. We therefore obtain .
Relationship between F and R (Scenario 2)
We now consider surveillance scenario 2 (i.e., detection of a case may trigger an outbreak investigation). We only use data from the first detected case of each cluster to control for the change in surveillance intensity due to the outbreak investigation. We first make the stricter assumption that each cluster is made of one chain of transmission, but this assumption is relaxed in Figure S1 and Text S1. Conditional on detection of the chain, the probability F that the first detected case was the first case of the chain is:



Inference
For scenario 2, we denote the number of clusters that are investigated by M and the number of first detected cases of each cluster that were infected by the reservoir by m.
The likelihood is:

To account for small sample sizes, we compute the bootstrap mean estimate and the bootstrap 95% confidence intervals. We compare these estimates with those obtained under asymptotic conditions (i.e., maximum likelihood and likelihood-ratio confidence intervals) and find that they are similar (Figures S2, S3, S4; Tables S1, S2, S3; Text S1).
For scenario 1, 1−G is an unbiased point estimate of R. A simple binomial likelihood function with probability 1−R can be used to derive confidence intervals for R. These intervals capture uncertainty arising from sampling; but may underestimate other sources of uncertainty if the total number of chains of transmission (both detected and undetected) in the study population remains small (see Text S1).
Data
Novel influenza A virus infections in humans, such as those caused by the H3N2v-M virus, are nationally notifiable conditions in the United States. When US laboratories or health care providers identify a potential infection with a novel influenza A virus they are asked to notify CDC immediately. CDC then collaborates with federal, state, and local human and animal public health colleagues to provide laboratory confirmation of the novel virus and to conduct a case and/or outbreak investigation. Outbreak investigations typically involve case investigation, contact investigations, attempts to determine potential animal or human sources of the infection, and environmental investigations. For each suspected or confirmed case detailed epidemiologic and clinical data are gathered. Notification of these cases is published in the MMWR, and in the weekly FluView national surveillance report.
From August 2011 to December 2011, CDC confirmed 12 human H3N2v-M infections [6],[7],[15],[16]. Eleven of the 12 reported cases occurred in children, most <10 years old, and three of the 12 cases were hospitalized for influenza. These 12 cases were detected in six clusters, with a median number of two cases detected per cluster (range: 1–3 cases). We assumed swine were the source of infection if there were 4 or fewer days from swine contact to onset, and that cases occurring more than 4 days after exposure to swine were likely to have been caused by human-to-human transmission. The potential human-to-human transmission events thus identified were investigated more closely to determine the likely source of infection.
Results
Reproduction Number R, Probabilities G and F
If the detection of a case does not affect detection of other cases from the same cluster (surveillance scenario 1), we find that R can be simply estimated as R = 1−G, where G is the proportion of detected cases that are infected by the reservoir. This is a general result that is independent of the case detection rate ρ and the overdispersion parameter k and that does not require data on clusters. It is valid as long as outbreaks are subcritical (0<R<1).
For situations where detection of a case may increase the probability of detecting other cases in the same cluster (surveillance scenario 2), we estimate R from the proportion F of first detected cases in each cluster that were infected by the reservoir. Figure 2 shows the relationship between the reproduction number R and the proportion F for different values of the detection rate ρ and the overdispersion parameter k (parameter k characterizes case-to-case variation in infectiousness; see [12] and Methods). When the case detection rate ρ is relatively low, at levels similar to those seen in sentinel systems like the US influenza virological surveillance network, the straight line dependence shown in Figure 2 illustrates that R can be estimated as R = 1−F (see Methods and Text S1). We find that it takes relatively high levels of case detection (ρ) or case-to-case variation in infectiousness (k) to cause substantial deviations from this linear relationship, and even then, such deviation only occurs for values of R close to 1 (Figure 2). We also find that the relationship is only weakly sensitive to having multiple chains of transmission per cluster of human cases (Figure S1; Text S1).

Estimating R for Swine Influenza Variants
We apply our method to assess the human-to-human transmissibility of swine-origin influenza A variant (H1N1v, H1N2v, and H3N2v) virus, in particular that of the H3N2v-M virus with data collected up to December 2011. From December 2005 until December 2011, a total of 27 human clusters of swine-origin influenza A variant virus infections were investigated by the CDC in the US. In clusters caused by the H3N2v-M virus, three of six (50%) of the first detected cases were infected by swine compared with 17 of 21 (81%) in clusters caused by the other variant viruses (Table 1). Table 1 gives R estimates for different assumptions about the case detection rate ρ and overdispersion parameter k.

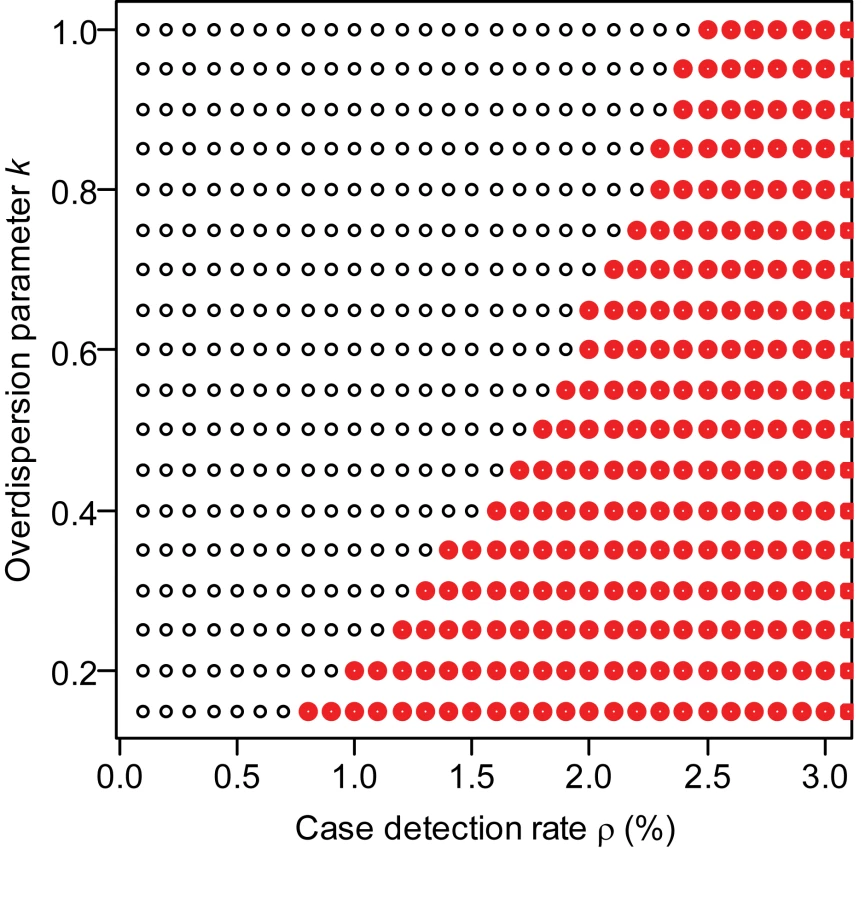
Recent efforts at CDC to evaluate the case detection rate suggest that it is low, of the order of 0.5% of all H3N2v-M-attributable cases (Biggerstaff et al., personal communication). For this likely scenario of low detection rates (≤1%), we estimate the reproduction number of variant viruses other than H3N2v-M to be 0.2 (95% CI 0.1–0.4) (Table 1). For the H3N2v-M virus, the point estimate and the lower bound of the 95% CI of R are 0.5 and 0.2, respectively. The upper bound of the 95% CI of R lies between 0.8 and >1, depending on assumptions about the case detection rate and overdispersion parameter (Table 1). If case-to-case variation in infectiousness is very large (k = 0.16, comparable to that seen in the SARS epidemic [12]), we can rule out the hypothesis R≥1 so long as the case detection rate is ≤0.7%; for a scenario with medium levels of variation (k = 0.5), the case detection rate must be ≤1.7% to rule out R≥1 (Figure 3)—a detection rate of 1.7% is unlikely to be achieved by US sentinel virologic surveillance system. The point estimate of R for H3N2v-M virus is more than double that of other variant viruses, but we cannot reject the hypothesis of equality (p = 0.15), maybe due to a lack of statistical power.
If the case detection rate is substantially higher than expected (10%), the point estimate and 95% lower bound for R for variant viruses other than H3N2v-M remain essentially unchanged but the upper bound increases from 0.4 to 0.4–0.6 (Table 1). The point estimate of R for H3N2v-M virus moves from 0.5 to 0.6–0.7.
Is Smaller Better? A Trade-off between Bias and Precision
If the detection rate ρ is known, it is always possible to invert the relationship shown in Figure 2 to derive an unbiased estimator of R. However, assume that ρ is unknown and that we plan to use estimator 1−F, which is unbiased so long as ρ is small. As the detection rate ρ increases, so does the bias of estimator 1−F. However, for a fixed number of clusters occurring in the study population, larger detection rates increase precision through larger sample sizes. There is therefore a trade-off between the bias and precision of the estimator 1−F. This is illustrated in Figure 4A, for a scenario with reproduction number R = 0.5, overdispersion parameter k = 0.5 and where a total of 10,000 clusters occur. In this scenario, the optimum (i.e., minimizing the root mean square error) trade-off in bias versus precision is obtained for a detection rate of ρ = 1.5%. We find that the optimum detection rate for estimator 1−F is a decreasing function of the reproduction number R and the total number of clusters n, but an increasing function of overdispersion parameter k (Figure 4B). We also find that the absolute bias at the optimum detection rate remains relatively small (<0.06) (Figure 4C). In Figure S5 and Text S1, we show that thinning the data may eliminate the bias of the 1−F estimator, though combining information from F and G (see below) is a simpler and more convenient approach to the same goal.

Uncertainty in the Case Detection Rate and Overdispersion Parameter
In many situations, both the case detection rate ρ and overdispersion parameter k are unknown. Interestingly, 1−F always acts as a lower bound estimate of R. An upper bound for R can be obtained if it is possible to specify an upper bound for the case detection rate and a lower bound for the overdispersion parameter k (see Text S1). We specify that corresponds to the SARS scenario with superspreading events. Figure 5 shows how precision decreases as the upper bound increases. However, even in the scenario , our approach can provide useful insights on transmissibility. For example, R is expected to be in intervals 0.20–0.25, 0.40–0.59, 0.50–0.87 for F = 80%, 60%, and 50%, respectively. For F≤46%, we can only derive a lower bound on R. For example, if F = 20%, we find that R is ≥0.8.

Other Applications
Our simple estimators of transmissibility can be applied generally to study zoonoses. Nipah virus is primarily clinically characterized by fever and encephalitis and was first discovered in a large outbreak in Malaysia in 1998–1999 [17],[18]. During this outbreak (where sick pigs were believed to be the natural reservoir), Parashar et al. [19] conducted a case-control study of the risk factors for infection. They recruited patients who were hospitalized with encephalitis from January through April 1999. Candidate encephalitis patients whose serum specimen tested positive for Nipah antibody were included as cases. Other cases were detected through targeted investigations; but if we restrict analyses to the subset of patients detected through hospital surveillance, this situation resembles surveillance scenario 1 above, where the probability of being detected is independent of cluster allocation. Here, we can therefore estimate the reproduction number as R = 1−G. Probability G can be approximated using the data of Parashar et al. [19] by the proportion of cases who (1) either lived or worked in a farm (G1) or (2) handled a pig, or came within 1 m of a pig and came into contact with pig urine or faeces (G2). Parashar et al. [19] only give estimates for the pooled set of cases (i.e., hospital surveillance+targeted investigations) for which G1 = 95% and G2 = 92%; however, it should be straightforward to derive these statistics for the subset of cases detected through hospital surveillance alone. For G = 0.92–0.95, we estimate R = 0.05–0.08. This suggests low levels of human-to-human transmission in this first outbreak.
Subsequently, outbreaks of Nipah virus occurred in Bangladesh [20],[21]. Luby et al. identified 23 introductions of Nipah virus into human populations in central and northwestern Bangladesh from 2001 to 2007 [21]. They classified 60 of the 122 identified Nipah virus cases as reservoir-to-human infections (G = 49%). Using the simple estimator R = 1−G, we obtain R = 0.51. This is consistent with more detailed contact tracing data that estimated R = 0.48 [21]. However, both these estimators may be biased upwards as the Bengali situation corresponds to surveillance scenario 2 (i.e., detection of a case may trigger an outbreak investigation), which may lead to the selection bias discussed earlier. In such a context, we recommend running our analysis on the subset of first detected cases in each of the 23 introductions and estimating proportion F. Again, it should be possible to derive F from data collected at the time. Since the case detection rate is unknown, we expect 1−F to act as a lower bound for R (Figure 5). From the estimates of G and F, we should therefore be able to derive simple bounds on R: .
Our approach can also be used to evaluate risks associated with non-zoonotic disease. As an example, we consider the cholera outbreak that started in the Dominican Republic in 2010 following the epidemic of that disease in Haiti. We can assess the level of local transmission in the Dominican Republic from the proportion G of cases detected in the Dominican Republic that were linked to Haiti (the latter country here playing the role of the natural reservoir). By December 18, 2010, three of the 59 confirmed cases detected in the Dominican Republic were linked to Haiti (G = 5%) [22]. Our simple estimation method cannot distinguish between R≈1 and R>1; but the observation that G was close to zero is indicative that transmission in the Dominican Republic was close or above levels needed for sustained human-to-human transmission.
Combining Information from F and G
Up to this point, in the scenario where the detection of a case did not affect detection of other cases from the same cluster (scenario 1), we assumed that cluster information was not available, and thus F could not be calculated. Let's now assume that such information is available and therefore both G and F can be estimated; this allows us to estimate both R and the case detection rate ρ, as illustrated in the simulation study shown in Figure 6. First, 1−G gives a point estimate for R. Second, once R is estimated, it is possible to infer the case detection rate by determining by how much proportion F differs from proportion G. If the overdispersion parameter k is known (e.g., from detailed contact tracing data), it is possible to accurately estimate the case detection rate; otherwise we can derive informative bounds (Figure 6).

Discussion
We present simple methods to estimate the reproduction number of emerging zoonoses from routine surveillance data. This research project was initiated to answer a seemingly straightforward question: 50% of the H3N2v-M cases that were detected in the US in 2011 had no contact with swine. What were the implications for the level of human-to-human transmission? At the time, however, the answer did not appear as straightforward as our analysis now shows it to be, with these simple estimators R = 1−G or R = 1−F, depending on the surveillance scenario we are in.
Our approach has specific properties that potentially overcome some of the limitations of existing methods. First, the investigation effort required is less than that for other methods. For example, if there is active case finding (surveillance scenario 2), one only needs to investigate the source of infection of the first case detected in each cluster. Second, the statistical treatment of the data is extremely simple, making it possible for anyone to interpret raw surveillance statistics about the source of infection of cases (statistics G or F) in terms of human-to-human transmissibility (reproduction number R). Third, the method is robust to selection bias (i.e., the fact that larger clusters are more likely to be detected) and under-ascertainment (i.e., ability to detect all cases in a cluster once a cluster is identified).
However, our methods do require that the source of infection (i.e., human or natural reservoir) can be identified for either the first detected cases of a cluster or a random subset of detected cases, depending on the surveillance scenario. In the US H3N2v-M context, where most individuals have no contact with swine, it is usually relatively easy to rule out the natural reservoir as a source of infection. Determining the source of infection might however be harder in situations where a large part of the population has regular contacts with the natural reservoir (e.g., backyard poultry in rural areas). In this case, an in-depth epidemiological investigation of the potential sources of infection is required. Clearly our approach cannot be used if the zoonotic source has not been identified yet. That being said, the examples we have presented show that proxy measures for the source of transmission (e.g., contacts with the natural reservoir) are often available. Although imperfect, these proxy measures are often already used by the scientific and public health community. For example, the World Health Organization fact sheet on Nipah virus states that “in Bangladesh, half of reported cases between 2001 and 2008 were due to human-to-human transmission” [23]. Our methods improve this existing practice in at least three ways: (1) it adds an essential layer of interpretation by providing estimates of the reproduction number, R, which allows assessment of how far transmission is from being sustained (R≥1); (2) it clarifies sources of potential biases; and (3) it provides simple guidelines to reduce these biases (see summary in Box 1).
Box 1. Approach to Assessing the Reproduction Number for Different Surveillance Scenarios
This box summarizes the relationships between G (proportion of detected cases infected by the reservoir), F (proportion of first detected cases in each cluster infected by the reservoir), and the reproduction number R, for the different surveillance scenarios.
-
If detection of a case does not affect detection of other cases from the same cluster (surveillance scenario 1), R can be estimated by R = 1−G. This is a general result that is independent of the case detection rate and the overdispersion parameter.
-
If detection of a case may trigger an outbreak investigation (surveillance scenario 2), the selection bias that arises may lead to R≤1−G. In addition, 1−F always acts as a lower bound to R. An upper bound for R can be obtained if it is possible to specify an upper bound for the case detection rate and a lower bound for the overdispersion parameter k (see Text S1). If the case detection rate is low, R can be estimated by 1−F.
-
If cluster information is available in surveillance scenario 1, R can be estimated by R = 1−G. Furthermore, it is possible to estimate the case detection rate by comparing statistics F and G (see Figure 6).
If the source of infection is unknown for a subset of cases, different options are available. If data are believed to be missing at random, these cases can simply be excluded from the analysis. However, it is possible that certain sources of infection are more likely to generate missing data. In such a scenario, upper bounds for R can be obtained by assuming that all cases with missing data were due to human-to-human transmission and lower bounds by assuming the reverse. If human-to-human cases are prone to be classified as reservoir-to-human transmission, R may be underestimated. If the source of infection is uncertain, the analysis of detailed outbreak data [11],[24] might allow estimation of the probability that the case was infected by the reservoir, which could then be used to estimate F and R using our methods. Phylogenetic analysis might also help resolve uncertainties about the source of infection. Clearly, our approach will start to break down if the proportion of cases with missing data becomes too large or if the classification of cases is too unreliable. We note that other methods, for instance based on contact tracing, would also struggle in these situations.
It is possible that surveillance intensity might change over time due to increasing media attention or health concerns. In such a context, in the surveillance scenario with outbreak investigations (scenario 2), Figure 5 shows that it is possible to derive bounds on R. In surveillance scenario 1, a change in the case detection rate is not expected to impact the summary statistic G or the estimate of R.
A bigger source of concern is if the increase in surveillance intensity focuses disproportionately on those cases that were exposed to the natural reservoir, as this would lead to overestimating F and underestimating R. For example, with the substantial increase in H3N2v-M virus infections during the summer of 2012, CDC changed their recommendations and asked clinicians to obtain respiratory specimens from ill persons with recent swine exposure [25],[26]. Therefore, in the summer 2012, ill persons with recent swine exposure may have been more likely to have been tested for H3N2v-M infection than those without such exposure. For this reason, we cannot use our method to analyze data collected in 2012.
Nonetheless, it is interesting to note that our estimate of R of around 0.5 for H3N2v-M in 2011 seems larger than what is suggested by data collected in summer 2012. We believe that these differences could at least partly be explained by seasonal variations in the ability of the virus to transmit [27]. Indeed, five of the six human-to-human transmission events detected in 2011 occurred in November–December; the remaining one happened in late August.
Our method has been developed for routine surveillance systems where cases are detected independently of each other. It would require modification to be applied to data collected from cluster detection surveillance systems of the type developed after the SARS epidemic, which target unexplained clusters of severe respiratory infection [10].
While a positive property of our approach is that it does not require a full-blown outbreak investigation, there are still many good reasons to target and investigate large outbreaks. Detailed investigations of large outbreaks are indeed needed to estimate essential parameters such as transmission risk factors or the generation time with adequate power.
Our method is designed to estimate R in the context of subcritical outbreaks, i.e., R<1. As illustrated with the cholera in the Dominican Republic example, the method can give a hint that transmission is at levels close or above what is needed for sustained transmission (R≈1 or R>1). However if R≥1, other estimation methods are necessary to derive a point estimate of R.
If transmissibility of a zoonotic infection suddenly increased (for example due to seasonal factors or genetic changes in the virus), it might take time for estimates to adjust (since the method uses data from all clusters detected so far). Future developments of the method could aim to detect sudden changes in the frequency of cases linked to the reservoir, building on methods for the sequential detection of change points in quality control and dynamical systems [28]–[30].
Finally, we hope that simplicity of our method and its limited data requirements will facilitate more robust monitoring of the epidemic potential of many zoonoses known to cause occasional human case clusters (such as Crimean-Congo hemorrhagic fever, Monkeypox virus, E. coli O157∶H7, or Mycobacterium bovis) [12] around the world.
Supporting Information
Zdroje
1. DawoodFS, JainS, FinelliL, ShawMW, LindstromS, et al. (2009) Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 360 : 2605–2615.
2. DonnellyCA, FisherMC, FraserC, GhaniAC, RileyS, et al. (2004) Epidemiological and genetic analysis of severe acute respiratory syndrome. Lancet Infect Dis 4 : 672–683.
3. WHO (2012) WHO Novel coronavirus infection - update. Available: http://www.who.int/csr/don/2012_11_23/en/index.html. Accessed 28 November 2012.
4. AntiaR, RegoesRR, KoellaJC, BergstromCT (2003) The role of evolution in the emergence of infectious diseases. Nature 426 : 658–661.
5. CDC (2011) CDC Case count: Detected U.S. human infections with H3N2v by State since August 2011. Available: http://www.cdc.gov/flu/swineflu/h3n2v-case-count.htm#table1. Accessed 28 November 2012.
6. CDC (2012) Update: Influenza A (H3N2)v transmission and guidelines - five states, 2011. MMWR Morb Mortal Wkly Rep 60 : 1741–1744.
7. CDC (2011) Limited human-to-human transmission of novel influenza A (H3N2) virus–Iowa, November 2011. MMWR Morb Mortal Wkly Rep 60 : 1615–1617.
8. CDC (2012) Evaluation of rapid influenza diagnostic tests for influenza A (H3N2)v virus and updated case count - United States, 2012. MMWR Morb Mortal Wkly Rep 61 : 1–3.
9. Lloyd-SmithJO, GeorgeD, PepinKM, PitzerVE, PulliamJR, et al. (2009) Epidemic dynamics at the human-animal interface. Science 326 : 1362–1367.
10. FergusonNM, FraserC, DonnellyCA, GhaniAC, AndersonRM (2004) Public health. Public health risk from the avian H5N1 influenza epidemic. Science 304 : 968–969.
11. YangY, HalloranME, SugimotoJD, LonginiIM (2007) Detecting human-to-human transmission of avian influenza a (H5N1). Emerg Infect Dis 13 : 1348–1353.
12. Lloyd-SmithJO, SchreiberSJ, KoppPE, GetzWM (2005) Superspreading and the effect of individual variation on disease emergence. Nature 438 : 355–359.
13. NishiuraH, YanP, SleemanCK, ModeCJ (2012) Estimating the transmission potential of supercritical processes based on the final size distribution of minor outbreaks. J Theor Biol 294 : 48–55.
14. De SerresG, GayNJ, FarringtonCP (2000) Epidemiology of transmissible diseases after elimination. Am J Epidemiol 151 : 1039–1048.
15. CDC (2011) Swine-origin influenza A (H3N2) virus infection in two children–Indiana and Pennsylvania, July–August 2011. MMWR Morb Mortal Wkly Rep 60 : 1213–1215.
16. LindstromS, GartenR, BalishA, ShuB, EmeryS, et al. (2012) Human infections with novel reassortant influenza A(H3N2)v viruses, United States, 2011. Emerg Infect Dis 18 : 834–837.
17. ChuaKB (2003) Nipah virus outbreak in Malaysia. J Clin Virol 26 : 265–275.
18. CDC (1999) Update: outbreak of Nipah virus–Malaysia and Singapore, 1999. MMWR Morb Mortal Wkly Rep 48 : 335–337.
19. ParasharUD, SunnLM, OngF, MountsAW, ArifMT, et al. (2000) Case-control study of risk factors for human infection with a new zoonotic paramyxovirus, Nipah virus, during a 1998–1999 outbreak of severe encephalitis in Malaysia. J Infect Dis 181 : 1755–1759.
20. LubySP, GurleyES, HossainMJ (2009) Transmission of human infection with Nipah virus. Clin Infect Dis 49 : 1743–1748.
21. LubySP, HossainMJ, GurleyES, AhmedBN, BanuS, et al. (2009) Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg Infect Dis 15 : 1229–1235.
22. CDC (2010) Update on cholera — Haiti, Dominican Republic, and Florida, 2010. MMWR Morb Mortal Wkly Rep 59 : 1637–1641.
23. WHO (2009) Nipah virus. Fact sheet 262. Available: http://www.who.int/mediacentre/factsheets/fs262/en/. Accessed 9 November 2012.
24. CauchemezS, BhattaraiA, MarchbanksTL, FaganRP, OstroffS, et al. (2011) Role of social networks in shaping disease transmission during a community outbreak of 2009 H1N1 pandemic influenza. P Natl Acad Sci U S A 108 : 2825–2830.
25. CDC (2012) Increase in influenza A H3N2v virus infections in three U.S. states. Available: http://www.bt.cdc.gov/HAN/han00325.asp. Accessed 9 November 2012.
26. CDC (2012) Interim information for clinicians about human infections with H3N2v virus. Available: http://www.cdc.gov/flu/swineflu/h3n2v-clinician.htm. Accessed 9 November 2012.
27. ShamanJ, PitzerVE, ViboudC, GrenfellBT, LipsitchM (2010) Absolute humidity and the seasonal onset of influenza in the continental United States. PLoS Biol 8: e1000316 doi:10.1371/journal.pbio.1000316.
28. LaiTL (1995) Sequential changepoint detection in quality-control and dynamical-systems. J Roy Stat Soc B Met 57 : 613–658.
29. LaiTL (2001) Sequential analysis: some classical problems and new challenges - Rejoinder. Stat Sinica 11 : 388–408.
30. SonessonC, BockD (2003) A review and discussion of prospective statistical surveillance in public health. J Roy Stat Soc A Sta 166 : 5–21.
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2013 Číslo 3
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Rána vizitkou (nejen) chirurga
Nejčtenější v tomto čísle
- Surveillance Programme of IN-patients and Epidemiology (SPINE): Implementation of an Electronic Data Collection Tool within a Large Hospital in Malawi
- Adjunctive Atypical Antipsychotic Treatment for Major Depressive Disorder: A Meta-Analysis of Depression, Quality of Life, and Safety Outcomes
- Strengthening the Expanded Programme on Immunization in Africa: Looking beyond 2015
- The Cost and Impact of Scaling Up Pre-exposure Prophylaxis for HIV Prevention: A Systematic Review of Cost-Effectiveness Modelling Studies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy